Development of Multi-Target Chemometric Models for the Inhibition of Class I PI3K Enzyme Isoforms: A Case Study Using QSAR-Co Tool



Abstract
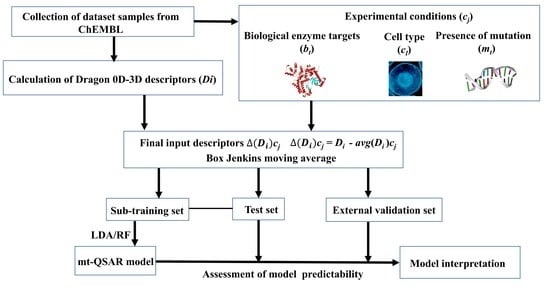
1. Introduction
2. Results and Discussion
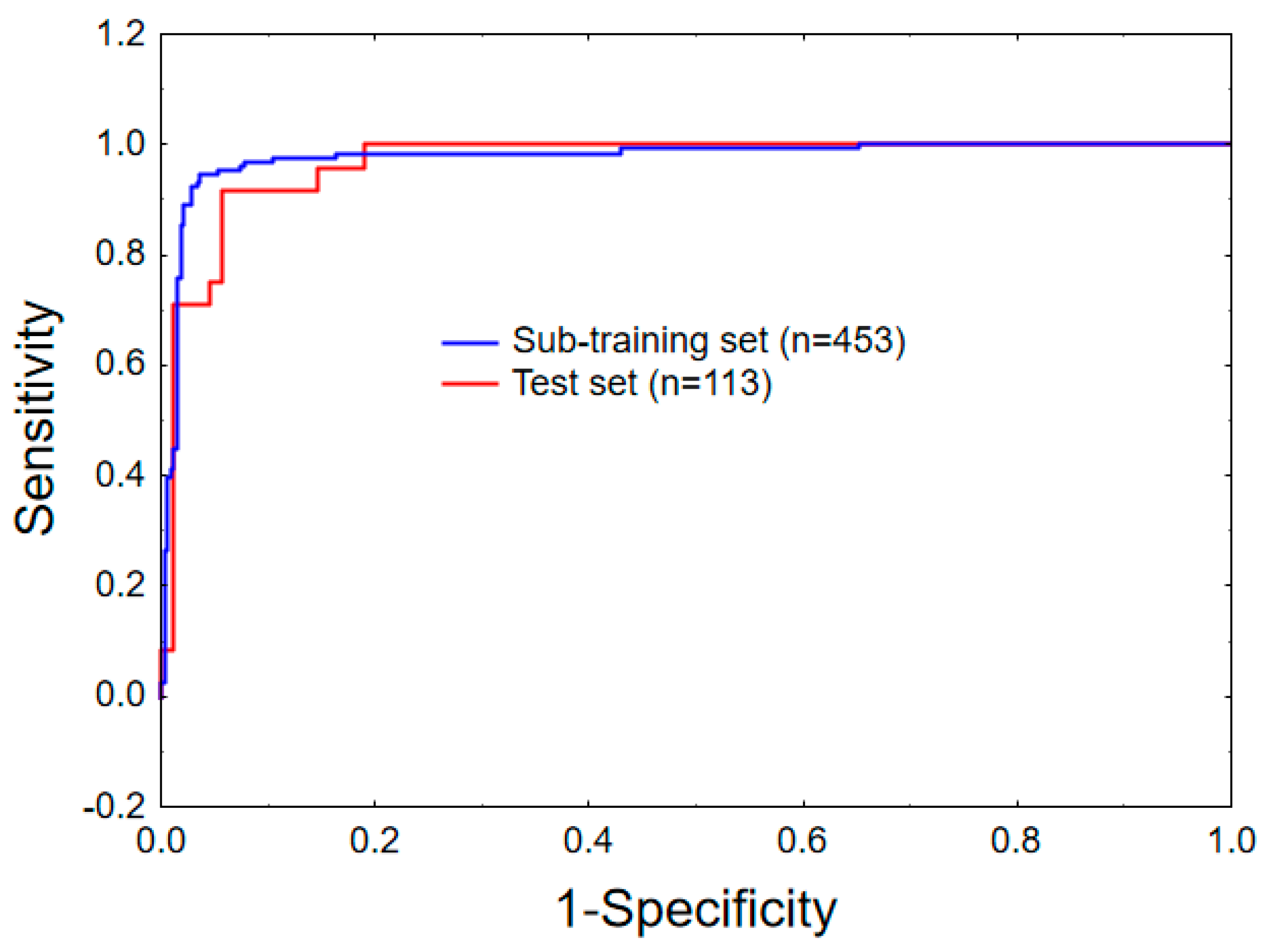
2.1. Linear Mt-QSAR Model Development
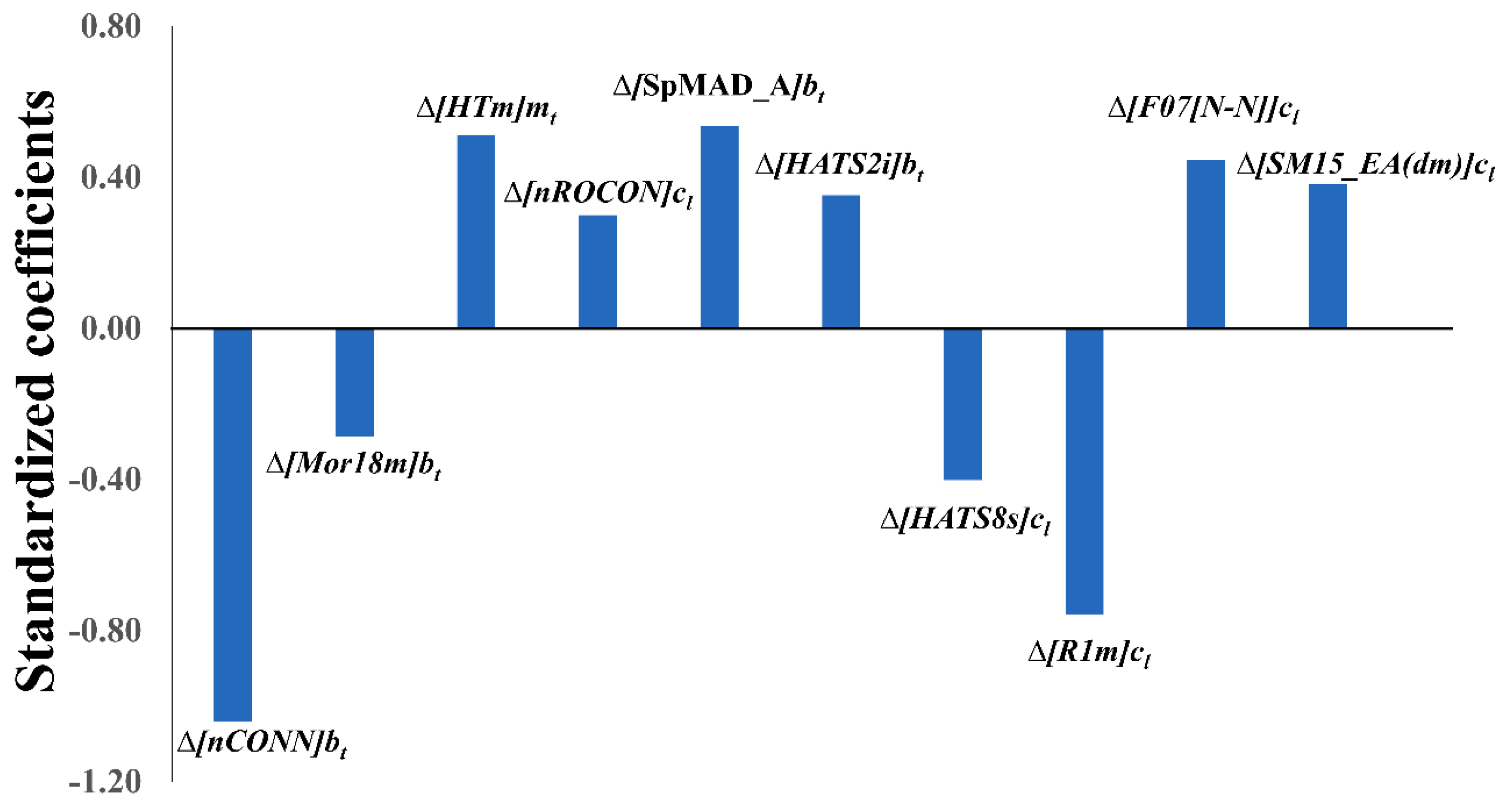
2.2. Physicochemical and Structural Interpretation of the Molecular Descriptors
2.3. Non-Linear Mt-QSAR Model Development
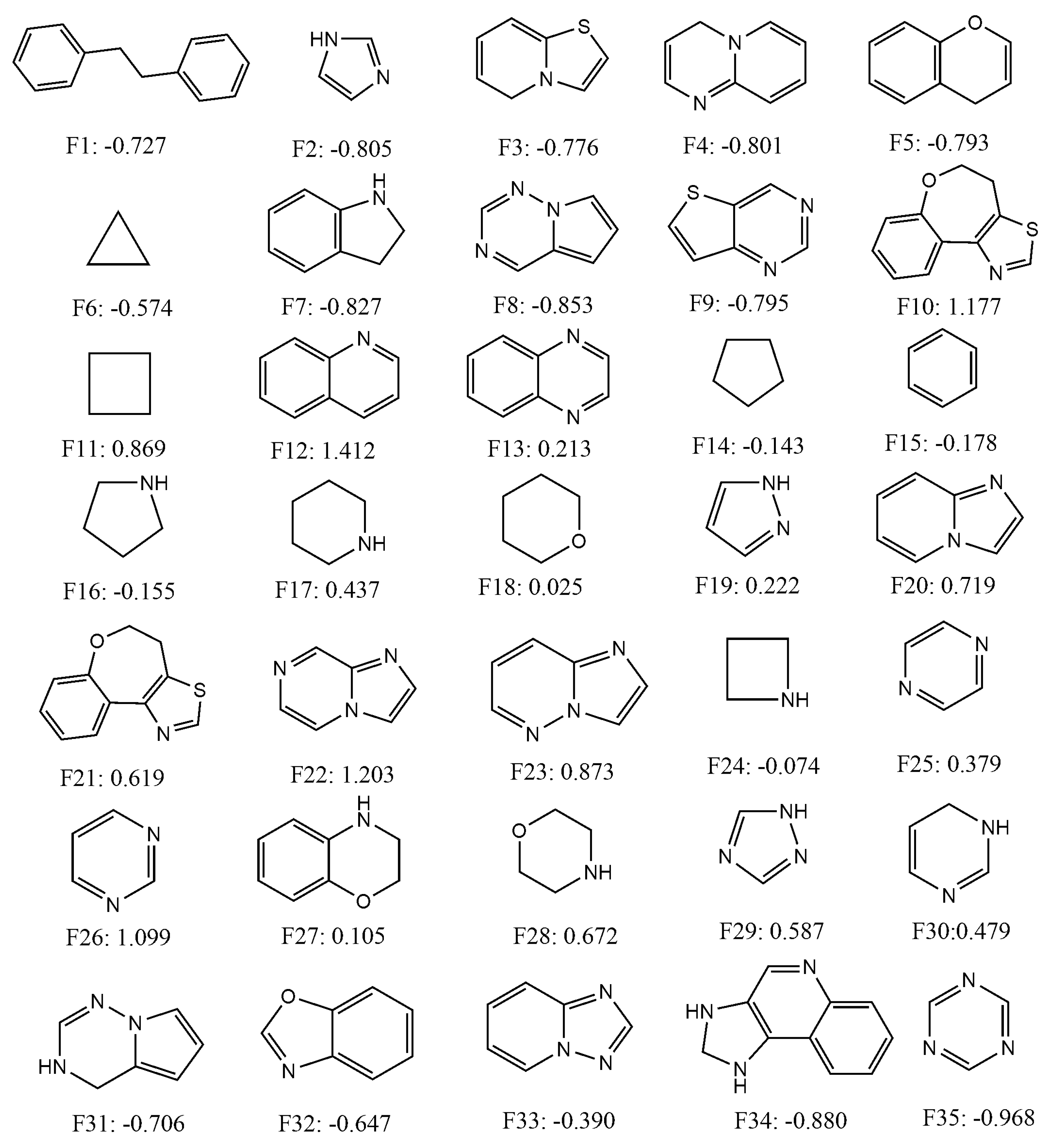
2.4. Quantitative Contributions of the Fragments Towards Inhibitory Activity
3. Materials and Methods
3.1. Dataset Curation and Descriptor Calculation
3.2. Box–Jenkins Approach
3.3. Model Development and Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Searls, D.B. Data integration: Challenges for drug discovery. Nat. Rev. Drug Discov. 2005, 4, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Fan, T.J.; Zhang, N.; Ren, T.; Zhao, L.J.; Zhong, R.G. Identification of the Structural Features of Guanine Derivatives as MGMT Inhibitors Using 3D-QSAR Modeling Combined with Molecular Docking. Molecules 2016, 21, 823. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.J.; Sun, G.H.; Zhao, L.J.; Cui, X.; Zhong, R.G. QSAR and Classification Study on Prediction of Acute Oral Toxicity of N-Nitroso Compounds. Int. J. Mol. Sci. 2018, 19, 3015. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.H.; Fan, T.J.; Sun, X.D.; Hao, Y.X.; Cui, X.; Zhao, L.J.; Ren, T.; Zhou, Y.; Zhong, R.G.; Peng, Y.Z. In Silico Prediction of O-6-Methylguanine-DNA Methyltransferase Inhibitory Potency of Base Analogs with QSAR and Machine Learning Methods. Molecules 2018, 23, 2892. [Google Scholar] [CrossRef]
- Speck-Planche, A. Recent advances in fragment-based computational drug design: Tackling simultaneous targets/biological effects. Future Med. Chem. 2018, 10, 2021–2024. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Luan, F.; Cordeiro, M.N.D.S. Role of Ligand-Based Drug Design Methodologies toward the Discovery of New Anti-Alzheimer Agents: Futures Perspectives in Fragment-Based Ligand Design. Curr. Med. Chem. 2012, 19, 1635–1645. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Multi-target drug discovery in anti-cancer therapy: Fragment-based approach toward the design of potent and versatile anti-prostate cancer agents. Bioorg. Med. Chem. 2011, 19, 6239–6244. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Fragment-based QSAR model toward the selection of versatile anti-sarcoma leads. Eur. J. Med. Chem. 2011, 46, 5910–5916. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. Fragment-based in silico modeling of multi-target inhibitors against breast cancer-related proteins. Mol. Divers. 2017, 21. [Google Scholar]
- Ambure, P.; Halder, A.K.; Gonzalez Diaz, H.; Cordeiro, M. QSAR-Co: An Open Source Software for Developing Robust Multitasking or Multitarget Classification-Based QSAR Models. J. Chem. Inf. Model. 2019, 59, 2538–2544. [Google Scholar] [CrossRef]
- Burke, J.E. Structural Basis for Regulation of Phosphoinositide Kinases and Their Involvement in Human Disease. Mol. Cell 2018, 71, 653–673. [Google Scholar] [CrossRef]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef]
- Yip, P.Y. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 165–176. [Google Scholar] [CrossRef]
- Miller, M.S.; Thompson, P.E.; Gabelli, S.B. Structural Determinants of Isoform Selectivity in PI3K Inhibitors. Biomolecules 2019, 9, 82. [Google Scholar] [CrossRef]
- Maheshwari, S.; Miller, M.S.; O’Meally, R.; Cole, R.N.; Amzel, L.M.; Gabelli, S.B. Kinetic and structural analyses reveal residues in phosphoinositide 3-kinase alpha that are critical for catalysis and substrate recognition. J. Biol. Chem. 2017, 292, 13541–13550. [Google Scholar] [CrossRef]
- De Santis, M.C.; Gulluni, F.; Campa, C.C.; Martini, M.; Hirsch, E. Targeting PI3K signaling in cancer: Challenges and advances. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 361–366. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef]
- Piddock, R.E.; Bowles, K.M.; Rushworth, S.A. The Role of PI3K Isoforms in Regulating Bone Marrow Microenvironment Signaling Focusing on Acute Myeloid Leukemia and Multiple Myeloma. Cancers 2017, 9, 29. [Google Scholar] [CrossRef]
- Jackson, S.P.; Schoenwaelder, S.M.; Goncalves, I.; Nesbitt, W.S.; Yap, C.L.; Wright, C.E.; Kenche, V.; Anderson, K.E.; Dopheide, S.M.; Yuan, Y.; et al. PI 3-kinase p110beta: A new target for antithrombotic therapy. Nat. Med. 2005, 11, 507–514. [Google Scholar] [CrossRef]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef]
- Cushing, T.D.; Metz, D.P.; Whittington, D.A.; McGee, L.R. PI3Kdelta and PI3Kgamma as targets for autoimmune and inflammatory diseases. J. Med. Chem. 2012, 55, 8559–8581. [Google Scholar] [CrossRef]
- Yoo, E.J.; Ojiaku, C.A.; Sunder, K.; Panettieri, R.A., Jr. Phosphoinositide 3-Kinase in Asthma: Novel Roles and Therapeutic Approaches. Am. J. Respir. Cell Mol. Biol. 2017, 56, 700–707. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Miller, B.W.; Przepiorka, D.; de Claro, R.A.; Lee, K.; Nie, L.; Simpson, N.; Gudi, R.; Saber, H.; Shord, S.; Bullock, J.; et al. FDA approval: Idelalisib monotherapy for the treatment of patients with follicular lymphoma and small lymphocytic lymphoma. Clin. Cancer Res. 2015, 21, 1525–1529. [Google Scholar] [CrossRef]
- Sanchez, V.E.; Nichols, C.; Kim, H.N.; Gang, E.J.; Kim, Y.M. Targeting PI3K Signaling in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2019, 20, 412. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Wee, S.; Wiederschain, D.; Maira, S.M.; Loo, A.; Miller, C.; deBeaumont, R.; Stegmeier, F.; Yao, Y.M.; Lengauer, C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 13057–13062. [Google Scholar] [CrossRef]
- Curigliano, G.; Shah, R.R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019, 42, 247–262. [Google Scholar] [CrossRef]
- Evans, C.A.; Liu, T.; Lescarbeau, A.; Nair, S.J.; Grenier, L.; Pradeilles, J.A.; Glenadel, Q.; Tibbitts, T.; Rowley, A.M.; DiNitto, J.P.; et al. Discovery of a Selective Phosphoinositide-3-Kinase (PI3K)-gamma Inhibitor (IPI-549) as an Immuno-Oncology Clinical Candidate. ACS Med. Chem. Lett. 2016, 7, 862–867. [Google Scholar] [CrossRef]
- Greenwell, I.B.; Ip, A.; Cohen, J.B. PI3K Inhibitors: Understanding Toxicity Mechanisms and Management. Oncol. 2017, 31, 821–828. [Google Scholar]
- Bharate, S.B.; Singh, B.; Bharate, J.B.; Jain, S.K.; Meena, S.; Vishwakarma, R.A. QSAR and pharmacophore modeling of N-acetyl-2-aminobenzothiazole class of phosphoinositide-3-kinase-alpha inhibitors. Med. Chem. Res. 2013, 22, 890–899. [Google Scholar] [CrossRef]
- Chadha, N.; Jasuja, H.; Kaur, M.; Bahia, M.S.; Silakari, O. Imidazo[1,2-a]pyrazine inhibitors of phosphoinositide 3-kinase alpha (PI3K alpha): 3D-QSAR analysis utilizing the Hybrid Monte Carlo algorithm to refine receptor-ligand complexes for molecular alignment. Sar Qsar Env. Res. 2014, 25, 221–247. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Silakari, O. Identification of new dual spleen tyrosine kinase (Syk) and phosphoionositide-3-kinase (PI3K) inhibitors using ligand and structure-based integrated ideal pharmacophore models. Sar Qsar Env. Res. 2016, 27, 469–499. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Zhang, F. Pharmacophore modeling and 3D-QSAR analysis of phosphoinositide 3-kinase p110alpha inhibitors. J. Mol. Model. 2010, 16, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.Y.; Ma, X.H.; Yap, C.W. Consensus model for identification of novel PI3K inhibitors in large chemical library. J. Comput. Aided Mol. Des. 2010, 24, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Oluic, J.; Nikolic, K.; Vucicevic, J.; Gagic, Z.; Filipic, S.; Agbaba, D. 3D-QSAR, Virtual Screening, Docking and Design of Dual PI3K/mTOR Inhibitors with Enhanced Antiproliferative Activity. Comb. Chem. High. Throughput Screen. 2017, 20, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Peddi, S.R.; Sivan, S.K.; Manga, V. Discovery and design of new PI3K inhibitors through pharmacophore-based virtual screening, molecular docking, and binding free energy analysis. Struct. Chem. 2018, 29, 1753–1766. [Google Scholar] [CrossRef]
- Peng, X.X.; Feng, K.R.; Ren, Y.J. Molecular modeling studies of quinazolinone derivatives as novel PI3K delta selective inhibitors. RSC Adv. 2017, 7, 56344–56358. [Google Scholar] [CrossRef]
- Ran, T.; Lu, T.; Yuan, H.; Liu, H.; Wang, J.; Zhang, W.; Leng, Y.; Lin, G.; Zhuang, S.; Chen, Y. A selectivity study on mTOR/PI3Kalpha inhibitors by homology modeling and 3D-QSAR. J. Mol. Model. 2012, 18, 171–186. [Google Scholar] [CrossRef]
- Safavi-Sohi, R.; Ghasemi, J.B. Quasi 4D-QSAR and 3D-QSAR study of the pan class I phosphoinositide-3-kinase (PI3K) inhibitors. Med. Chem. Res. 2013, 22, 1587–1596. [Google Scholar] [CrossRef]
- Sharma, P.; Shukla, A.; Kalani, K.; Dubey, V.; Luqman, S.; Srivastava, S.K.; Khan, F. In-silico & In-vitro Identification of Structure-Activity Relationship Pattern of Serpentine & Gallic Acid Targeting PI3Kgamma as Potential Anticancer Target. Curr. Cancer Drug Targets 2017, 17, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.O.; Al-Sha’er, M.A.; Khanfar, M.A.; Al-Nadaf, A.H. Discovery of nanomolar phosphoinositide 3-kinase gamma (PI3K gamma) inhibitors using ligand-based modeling and virtual screening followed by in vitro analysis. Eur. J. Med. Chem. 2014, 84, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Wang, Y.; Bryant, S.H. Structural insights of a PI3K/mTOR dual inhibitor with the morpholino-triazine scaffold. J. Comput. Aided Mol. Des. 2016, 30, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.X.; Chen, Y.D. Pharmacophore models generation by catalyst and phase consensus-based virtual screening protocol against PI3K alpha inhibitors. Mol. Simul. 2013, 39, 529–544. [Google Scholar] [CrossRef]
- Wang, J.; Wang, F.; Xiao, Z.; Sheng, G.; Li, Y.; Wang, Y. Molecular simulation of a series of benzothiazole PI3Kalpha inhibitors: Probing the relationship between structural features, anti-tumor potency and selectivity. J. Mol. Model. 2012, 18, 2943–2958. [Google Scholar] [CrossRef]
- Wu, F.; Hou, X.Y.; Luo, H.; Zhou, M.; Zhang, W.J.; Ding, Z.Y.; Li, R. Exploring the selectivity of PI3K alpha and mTOR inhibitors by 3D-QSAR, molecular dynamics simulations and MM/GBSA binding free energy decomposition. Medchemcomm 2013, 4, 1482–1496. [Google Scholar] [CrossRef]
- Yang, W.J.; Shu, M.; Wang, Y.Q.; Wang, R.; Hu, Y.; Meng, L.X.; Lin, Z.H. 3D-QSAR and docking studies of 3-Pyridine heterocyclic derivatives as potent PI3K/mTOR inhibitors. J. Mol. Struct. 2013, 1054, 107–116. [Google Scholar] [CrossRef]
- Yu, M.; Gu, Q.; Xu, J. Discovering new PI3K alpha inhibitors with a strategy of combining ligand-based and structure-based virtual screening. J. Comput. Aided Mol. Des. 2018, 32, 347–361. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Cordeiro, M.N.D.S. De novo computational design of compounds virtually displaying potent antibacterial activity and desirable in vitro ADMET profiles. Med. Chem. Res. 2017, 26, 2345–2356. [Google Scholar] [CrossRef]
- Rogers, D.; Hopfinger, A.J. Application of Genetic Function Approximation to Quantitative Structure-Activity-Relationships and Quantitative Structure-Property Relationships. J. Chem. Inf. Comput. Sci. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Snedecor, G.W.; Cochran, W.G. Statistical methods, 8th ed.; Iowa State University Press: Ames, IA, USA, 1989; p. 503. [Google Scholar]
- Ambure, P.; Aher, R.B.; Gajewicz, A.; Puzyn, T.; Roy, K. “NanoBRIDGES” software: Open access tools to perform QSAR and nano-QSAR modeling. Chemom. Intell. Lab. Syst. 2015, 147, 1–13. [Google Scholar] [CrossRef]
- Papadatos, G.; Gaulton, A.; Hersey, A.; Overington, J.P. Activity, assay and target data curation and quality in the ChEMBL database. J. Comput. Aided Mol. Des. 2015, 29, 885–896. [Google Scholar] [CrossRef]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef]
- Wilks, S.S. Certain generalizations in the analysis of variance. Biometrika 1932, 24, 471–494. [Google Scholar] [CrossRef]
- Boughorbel, S.; Jarray, F.; El-Anbari, M. Optimal classifier for imbalanced data using Matthews Correlation Coefficient metric. PloS ONE 2017, 12. [Google Scholar] [CrossRef]
- Hanczar, B.; Hua, J.; Sima, C.; Weinstein, J.; Bittner, M.; Dougherty, E.R. Small-sample precision of ROC-related estimates. Bioinformatics 2010, 26, 822–830. [Google Scholar] [CrossRef]
- Rucker, C.; Rucker, G.; Meringer, M. y-Randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Halder, A.K.; Natalia, M.; Cordeiro, D.S. Probing the Environmental Toxicity of Deep Eutectic Solvents and Their Components: An In Silico Modeling Approach. Acs Sustain. Chem. Eng. 2019, 7, 10649–10660. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Consonni, V.; Todeschini, R.; Pavan, M.; Gramatica, P. Structure/Response Correlations and Similarity/Diversity Analysis by GETAWAY Descriptors. 2. Application of the Novel 3D Molecular Descriptors to QSAR/QSPR Studies. J. Chem. Inf. Comput. Sci. 2002, 42, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Consonni, V.; Todeschini, R.; Pavan, M. Structure/Response Correlations and Similarity/Diversity Analysis by GETAWAY Descriptors. 1. Theory of the Novel 3D Molecular Descriptors. J. Chem. Inf. Comput. Sci. 2002, 42, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V.; Mannhold, R.; Kubinyi, H.; Folkers, G.; Wiley Online Library. Molecular Descriptors for Chemoinformatics. Vol. I & II. In Methods and Principles in Medicinal Chemistry Ser 82; Wiley-VCH Imprint. John Wiley & Sons, Incorporated: Hoboken, NJ, USA, 2010; p 1 online resource. [Google Scholar]
- Consonni, V.; Todeschini, R.; Wiley Online Library. Handbook of molecular descriptors. In Methods and principles in medicinal chemistry 11; Wiley-VCH: Weinheim, Germany; New York, NY, USA, 2000; p 1 online resource. [Google Scholar]
- Estrada, E.; Ramirez, A. Edge adjacency relationships and molecular topographic descriptors. Definition and QSAR applications. J. Chem. Inf. Comput. Sci. 1996, 36, 837–843. [Google Scholar] [CrossRef]
- Devinyak, O.; Havrylyuk, D.; Lesyk, R. 3D-MoRSE descriptors explained. J. Mol. Graph. Model. 2014, 54, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Fawagreh, K.; Gaber, M.M.; Elyan, E. Random forests: From early developments to recent advancements. Syst. Sci. Control. Eng. 2014, 2, 602–609. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Nizami, B.; Tetko, I.V.; Koorbanally, N.A.; Honarparvar, B. QSAR models and scaffold-based analysis of non-nucleoside HIV RT inhibitors. Chemom. Intell Lab. 2015, 148, 134–144. [Google Scholar] [CrossRef]
- Halder, A.K. Finding the structural requirements of diverse HIV-1 protease inhibitors using multiple QSAR modelling for lead identification. Sar Qsar Env. Res. 2018, 29, 911–933. [Google Scholar] [CrossRef]
- Marchese Robinson, R.L.; Palczewska, A.; Palczewski, J.; Kidley, N. Comparison of the Predictive Performance and Interpretability of Random Forest and Linear Models on Benchmark Data Sets. J. Chem. Inf. Model. 2017, 57, 1773–1792. [Google Scholar] [CrossRef]
- Guha, R. On the interpretation and interpretability of quantitative structure-activity relationship models. J. Comput. Aid. Mol. Des. 2008, 22, 857–871. [Google Scholar] [CrossRef]
- Ishwaran, H. Variable importance in binary regression trees and forests. Electron. J. Stat. 2007, 1, 519–537. [Google Scholar] [CrossRef]
- Nawar, S.; Mouazen, A.M. Comparison between Random Forests, Artificial Neural Networks and Gradient Boosted Machines Methods of On-Line Vis-NIR Spectroscopy Measurements of Soil Total Nitrogen and Total Carbon. Sensors 2017, 17, 2428. [Google Scholar] [CrossRef]
- Lee, K.; Lee, M.; Kim, D. Utilizing random Forest QSAR models with optimized parameters for target identification and its application to target-fishing server. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 2011, 25, 533–554. [Google Scholar] [CrossRef]
- Sadowski, J.; Gasteiger, J.; Klebe, G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-ray Structures. J. Chem. Inf. Comput. Sci. 1994, 34, 1000–1008. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Herrera-Ibata, D.M.; Duardo-Sanchez, A.; Munteanu, C.R.; Orbegozo-Medina, R.A.; Pazos, A. ANN multiscale model of anti-HIV drugs activity vs AIDS prevalence in the US at county level based on information indices of molecular graphs and social networks. J. Chem. Inf. Model. 2014, 54, 744–755. [Google Scholar] [CrossRef]
- Alonso, N.; Caamano, O.; Romero-Duran, F.J.; Luan, F.; Cordeiro, M.N.D.S.; Yanez, M.; Gonzalez-Diaz, H.; Garcia-Mera, X. Model for high-throughput screening of multitarget drugs in chemical neurosciences: Synthesis, assay, and theoretic study of rasagiline carbamates. ACS Chem. Neurosci. 2013, 4, 1393–1403. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Scotti, M.T. BET bromodomain inhibitors: Fragment-based in silico design using multi-target QSAR models. Mol. Divers. 2018. [Google Scholar] [CrossRef]
- Gore, P.A. 11 - Cluster Analysis. In Handbook of Applied Multivariate Statistics and Mathematical Modeling; Tinsley, H.E.A., Brown, S.D., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 297–321. [Google Scholar] [CrossRef]
- Statsoft-Team (2001) STATISTICA. Data analysis software system. v6.0, Tulsa. Available online: http://www.statsoft.com/Products/STATISTICA-Features (accessed on 22 May 2019).
- Brown, M.T.; Wicker, L.R. 8 - Discriminant Analysis. In Handbook of Applied Multivariate Statistics and Mathematical Modeling; Tinsley, H.E.A., Brown, S.D., Eds.; Academic Press: San Diego, CA, USA, 2000; pp. 209–235. [Google Scholar] [CrossRef]
- Speck-Planche, A. Combining Ensemble Learning with a Fragment-Based Topological Approach To Generate New Molecular Diversity in Drug Discovery: In Silico Design of Hsp90 Inhibitors. Acs Omega 2018, 3, 14704–14716. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classification a | Sub-Training Set | Test Set |
|---|---|---|
| NDTotal | 453 | 113 |
| NDactive | 324 | 89 |
| CCDactive | 310 | 84 |
| Sensitivity (%) | 95.68 | 94.38 |
| NDinactive | 129 | 24 |
| CCDinactive | 122 | 22 |
| Specificity (%) | 94.57 | 91.67 |
| F-measure | 0.967 | 0.960 |
| Accuracy (%) | 95.36 | 93.80 |
| MCC | 0.889 | 0.825 |
| Descriptors | Δ[nCONN]bt | Δ[Mor18m]bt | Δ[HTm]mt | Δ[nROCON]cl | Δ[SpMAD_A]bt | Δ[HATS2i]bt | Δ[HATS8s]cl | Δ[R1m]cl | Δ[F07[N-N]]cl | Δ[SM15_EA(dm)]cl |
|---|---|---|---|---|---|---|---|---|---|---|
| Δ[nCONN]bt | 1.00 | 0.14 | 0.08 | −0.20 | −0.18 | −0.29 | −0.23 | −0.17 | 0.00 | 0.07 |
| Δ[Mor18m]bt | 0.14 | 1.00 | −0.37 | −0.18 | 0.15 | 0.11 | 0.21 | 0.14 | −0.07 | 0.11 |
| Δ[HTm]mt | 0.08 | -0.37 | 1.00 | 0.09 | −0.14 | −0.29 | −0.21 | −0.07 | 0.14 | 0.13 |
| D[nROCON]cl | −0.20 | -0.18 | 0.09 | 1.00 | 0.01 | 0.17 | 0.00 | 0.02 | 0.23 | 0.12 |
| D[SpMAD_A]bt | −0.18 | 0.15 | −0.14 | 0.01 | 1.00 | −0.06 | −0.11 | 0.01 | 0.15 | −0.26 |
| D[HATS2i]bt | −0.29 | 0.11 | −0.29 | 0.17 | −0.06 | 1.00 | 0.18 | 0.02 | −0.04 | 0.01 |
| D[HATS8s]cl | −0.23 | 0.21 | −0.21 | 0.00 | −0.11 | 0.18 | 1.00 | 0.74 | −0.09 | 0.29 |
| D[R1m]cl | −0.17 | 0.14 | −0.07 | 0.02 | 0.01 | 0.02 | 0.74 | 1.00 | −0.02 | 0.36 |
| D[F07[N-N]]cl | 0.00 | −0.07 | 0.14 | 0.23 | 0.15 | −0.04 | −0.09 | −0.02 | 1.00 | 0.17 |
| D[SM15_EA(dm)]cl | 0.07 | 0.11 | 0.13 | 0.12 | −0.26 | 0.01 | 0.29 | 0.36 | 0.17 | 1.00 |
| Name | Description | Descriptor Type |
|---|---|---|
| Δ[nCONN]bt | Number of urea (-thio) fragment, depending on the chemical structure and enzyme target | Functional group counts |
| Δ[R1m]cl | R autocorrelation of lag 1/weighted by mass, depending on the chemical structure and cell type | GETAWAY indices |
| Δ[SpMAD_A]bt | Spectral mean absolute deviation from the adjacency matrix, depending on the chemical structure and biological target enzyme | 2D matrix-based adjacency matrix descriptors |
| Δ[HTm]mt | H total index/weighted by mass, depending on the cell mutation and chemical structure | GETAWAY H-indices |
| Δ[F07[N–N]]cl | Frequency of N-N at topological distance 7, depending on the chemical structure and cell type | 2D Atom Pairs |
| Δ[HATS8s]cl | Leverage-weighted autocorrelation of lag 8/weighted by I-state, depending on the chemical structure and cell type | GETAWAY H-indices |
| Δ[SM15_EA(dm)]cl | Spectral moment of order 15 from edge adjacency matrix weighted by dipole moment, depending on the chemical structure and cell type | Edge adjacency indices |
| Δ[HATS2i]bt | Leverage-weighted autocorrelation of lag 2/weighted by ionization potential, depending on the chemical structure and biological target enzyme | GETAWAY H-indices |
| Δ[nROCON]cl | Number of (thio-) carbamates (aliphatic), depending on the chemical structure and cell type | Functional group counts |
| Δ[Mor18m]bt | Signal 18/weighted by mass, depending on the chemical structure and biological target enzyme | 3D-MoRSE, weighted by mass |
| Classification a | Sub-training Set (10-fold CV) b | Test Set |
|---|---|---|
| NDTotal | 453 | 113 |
| NDactive | 324 | 89 |
| CCDactive | 313 | 87 |
| Sensitivity (%) | 96.6 | 97.75 |
| NDinactive | 129 | 24 |
| CCDinactive | 117 | 21 |
| Specificity (%) | 90.7 | 87.5 |
| F-measure | 0.965 | 0.972 |
| Accuracy (%) | 94.92 | 95.57 |
| MCC | 0.875 | 0.866 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halder, A.K.; Cordeiro, M.N.D.S. Development of Multi-Target Chemometric Models for the Inhibition of Class I PI3K Enzyme Isoforms: A Case Study Using QSAR-Co Tool. Int. J. Mol. Sci. 2019, 20, 4191. https://doi.org/10.3390/ijms20174191
Halder AK, Cordeiro MNDS. Development of Multi-Target Chemometric Models for the Inhibition of Class I PI3K Enzyme Isoforms: A Case Study Using QSAR-Co Tool. International Journal of Molecular Sciences. 2019; 20(17):4191. https://doi.org/10.3390/ijms20174191
Chicago/Turabian StyleHalder, Amit Kumar, and M. Natália Dias Soeiro Cordeiro. 2019. "Development of Multi-Target Chemometric Models for the Inhibition of Class I PI3K Enzyme Isoforms: A Case Study Using QSAR-Co Tool" International Journal of Molecular Sciences 20, no. 17: 4191. https://doi.org/10.3390/ijms20174191
APA StyleHalder, A. K., & Cordeiro, M. N. D. S. (2019). Development of Multi-Target Chemometric Models for the Inhibition of Class I PI3K Enzyme Isoforms: A Case Study Using QSAR-Co Tool. International Journal of Molecular Sciences, 20(17), 4191. https://doi.org/10.3390/ijms20174191