Epigenetic Mechanisms in Hirschsprung Disease

Abstract
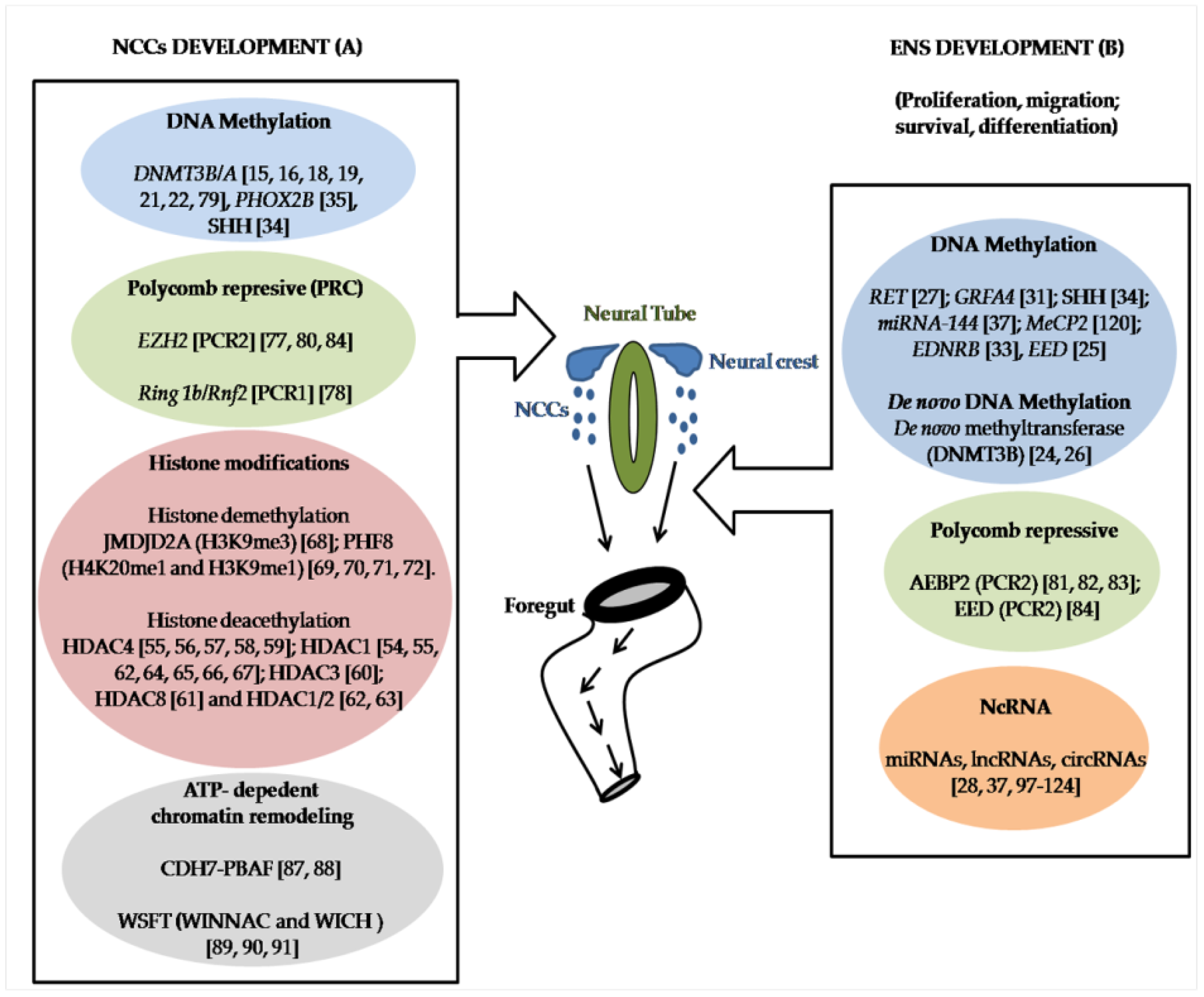
1. Introduction
2. DNA Methylation
3. Histone Modifications
4. Polycomb Repressive Complex (PRC)
5. ATP-Dependent Chromatin Remodeling
6. NcRNA
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chakravarti, A.; Lyonnet, S. The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Beaudet, A.R., Scriver, C.R., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Amiel, J.; Sproat-Emison, E.; Garcia-Barcelo, M.; Lantieri, F.; Burzynski, G.; Borrego, S.; Pelet, A.; Arnold, S.; Miao, X.; Griseri, P.; et al. Hirschsprung disease, associated syndromes and genetics: A review. J. Med. Genet. 2008, 45, 1–14. [Google Scholar] [CrossRef]
- Lake, J.I.; Heuckeroth, R.O. Enteric nervous system development: Migration, differentiation, and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G1–G24. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Calvanese, V.; Lara, E.; Fraga, M.F. Epigenetic code and self-identity. Adv. Exp. Med. Biol. 2012, 738, 236–255. [Google Scholar] [CrossRef]
- Liu, Y.; Xiao, A. Epigenetic regulation in neural crest development. Birth Defects Res. Part AClin. Mol. Teratol. 2011, 91, 788–796. [Google Scholar] [CrossRef]
- Fujita, K.; Ogawa, R.; Kawawaki, S.; Ito, K. Roles of chromatin remodelers in maintenance mechanisms of multipotency of mouse trunk neural crest cells in the formation of neural crest-derived stem cells. Mech. Dev. 2014, 133, 126–145. [Google Scholar] [CrossRef]
- Sergi, C.M.; Caluseriu, O.; McColl, H.; Eisenstat, D.D. Hirschsprung’s disease: Clinical dysmorphology, genes, micro-RNAs, and future perspectives. Pediatric Res. 2017, 81, 177–191. [Google Scholar] [CrossRef]
- Rogers, J.M. Search for the missing lncs: Gene regulatory networks in neural crest development and long non-coding RNA biomarkers of Hirschsprung’s disease. Neurogastroenterol. Motil. 2016, 28, 161–166. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Ooi, S.K.; O’Donnell, A.H.; Bestor, T.H. Mammalian cytosine methylation at a glance. J. Cell Sci. 2009, 122, 2787–2791. [Google Scholar] [CrossRef]
- Hu, N.; Strobl-Mazzulla, P.H.; Bronner, M.E. Epigenetic regulation in neural crest development. Dev. Biol. 2014, 396, 159–168. [Google Scholar] [CrossRef]
- Roellig, D.; Bronner, M.E. The epigenetic modifier DNMT3A is necessary for proper otic placode formation. Dev. Biol. 2016, 411, 294–300. [Google Scholar] [CrossRef]
- Ehrlich, M.; Sanchez, C.; Shao, C.; Nishiyama, R.; Kehrl, J.; Kuick, R.; Kubota, T.; Hanash, S.M. ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity 2008, 41, 253–271. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Linhart, H.G.; Lin, H.; Yamada, Y.; Moran, E.; Steine, E.J.; Gokhale, S.; Lo, G.; Cantu, E.; Ehrich, M.; He, T.; et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007, 21, 3110–3122. [Google Scholar] [CrossRef]
- Yan, X.J.; Xu, J.; Gu, Z.H.; Pan, C.M.; Lu, G.; Shen, Y.; Shi, J.Y.; Zhu, Y.M.; Tang, L.; Zhang, X.W.; et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat. Genet. 2011, 43, 309–315. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Hu, N.; Strobl-Mazzulla, P.; Sauka-Spengler, T.; Bronner, M.E. DNA methyltransferase3A as a molecular switch mediating the neural tube-to-neural crest fate transition. Genes Dev. 2012, 26, 2380–2385. [Google Scholar] [CrossRef]
- Adams, M.S.; Gammill, L.S.; Bronner-Fraser, M. Discovery of transcription factors and other candidate regulators of neural crest development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2008, 237, 1021–1033. [Google Scholar] [CrossRef]
- Jin, B.; Tao, Q.; Peng, J.; Soo, H.M.; Wu, W.; Ying, J.; Fields, C.R.; Delmas, A.L.; Liu, X.; Qiu, J.; et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum. Mol. Genet. 2008, 17, 690–709. [Google Scholar] [CrossRef]
- Lui, V.C.; Cheng, W.W.; Leon, T.Y.; Lau, D.K.; Garcia-Barcelo, M.M.; Miao, X.P.; Kam, M.K.; So, M.T.; Chen, Y.; Wall, N.A.; et al. Perturbation of hoxb5 signaling in vagal neural crests down-regulates ret leading to intestinal hypoganglionosis in mice. Gastroenterology 2008, 134, 1104–1115. [Google Scholar] [CrossRef]
- Torroglosa, A.; Enguix-Riego, M.V.; Fernandez, R.M.; Roman-Rodriguez, F.J.; Moya-Jimenez, M.J.; de Agustin, J.C.; Antinolo, G.; Borrego, S. Involvement of DNMT3B in the pathogenesis of Hirschsprung disease and its possible role as a regulator of neurogenesis in the human enteric nervous system. Genet. Med. 2014, 16, 703–710. [Google Scholar] [CrossRef]
- Villalba-Benito, L.; Torroglosa, A.; Fernandez, R.M.; Ruiz-Ferrer, M.; Moya-Jimenez, M.J.; Antinolo, G.; Borrego, S. Overexpression of DNMT3b target genes during Enteric Nervous System development contribute to the onset of Hirschsprung disease. Sci. Rep. 2017, 7, 6221. [Google Scholar] [CrossRef]
- Torroglosa, A.; Villalba-Benito, L.; Fernandez, R.M.; Moya-Jimenez, M.J.; Antinolo, G.; Borrego, S. Dnmt3b knock-down in enteric precursors reveals a possible mechanism by which this de novo methyltransferase is involved in the enteric nervous system development and the onset of Hirschsprung disease. Oncotarget 2017, 8, 106443–106453. [Google Scholar] [CrossRef]
- Munnes, M.; Patrone, G.; Schmitz, B.; Romeo, G.; Doerfler, W. A 5′-CG-3′-rich region in the promoter of the transcriptionally frequently silenced RET protooncogene lacks methylated cytidine residues. Oncogene 1998, 17, 2573–2583. [Google Scholar] [CrossRef]
- Wang, G.; Guo, F.; Wang, H.; Liu, W.; Zhang, L.; Cui, M.; Wu, X. Downregulation of microRNA-483-5p Promotes Cell Proliferation and Invasion by Targeting GFRA4 in Hirschsprung’s Disease. DNA Cell Biol. 2017, 36, 930–937. [Google Scholar] [CrossRef]
- Yang, J.; Runeberg-Roos, P.; Leppanen, V.M.; Saarma, M. The mouse soluble GFRalpha4 receptor activates RET independently of its ligand persephin. Oncogene 2007, 26, 3892–3898. [Google Scholar] [CrossRef]
- Jing, S.; Wen, D.; Yu, Y.; Holst, P.L.; Luo, Y.; Fang, M.; Tamir, R.; Antonio, L.; Hu, Z.; Cupples, R.; et al. GDNF-induced activation of the ret protein tyrosine kinase is mediated by GDNFR-alpha, a novel receptor for GDNF. Cell 1996, 85, 1113–1124. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, L.; Wang, H.; Cui, M.; Liu, W.; Liu, Y.; Wu, X. Demethylation of GFRA4 Promotes Cell Proliferation and Invasion in Hirschsprung Disease. DNA Cell Biol. 2018, 37, 316–324. [Google Scholar] [CrossRef]
- Sanchez-Mejias, A.; Fernandez, R.M.; Lopez-Alonso, M.; Antinolo, G.; Borrego, S. New roles of EDNRB and EDN3 in the pathogenesis of Hirschsprung disease. Genet. Med. Off. J. Am. Coll. Med. Genet. 2010, 12, 39–43. [Google Scholar] [CrossRef]
- Tang, W.; Li, B.; Tang, J.; Liu, K.; Qin, J.; Wu, W.; Geng, Q.; Zhang, J.; Chen, H.; Xu, X.; et al. Methylation analysis of EDNRB in human colon tissues of Hirschsprung’s disease. Pediatric Surg. Int. 2013, 29, 683–688. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, P.; Zheng, S.; Dong, R. Hypermethylation of SHH in the pathogenesis of congenital anorectal malformations. J. Pediatric Surg. 2014, 49, 1400–1404. [Google Scholar] [CrossRef]
- de Pontual, L.; Trochet, D.; Bourdeaut, F.; Thomas, S.; Etchevers, H.; Chompret, A.; Minard, V.; Valteau, D.; Brugieres, L.; Munnich, A.; et al. Methylation-associated PHOX2B gene silencing is a rare event in human neuroblastoma. Eur. J. Cancer 2007, 43, 2366–2372. [Google Scholar] [CrossRef][Green Version]
- Du, Y.; Xu, Y.; Ding, L.; Yao, H.; Yu, H.; Zhou, T.; Si, J. Down-regulation of miR-141 in gastric cancer and its involvement in cell growth. J. Gastroenterol. 2009, 44, 556–561. [Google Scholar] [CrossRef]
- Tang, W.; Qin, J.; Tang, J.; Zhang, H.; Zhou, Z.; Li, B.; Geng, Q.; Wu, W.; Xia, Y.; Xu, X. Aberrant reduction of MiR-141 increased CD47/CUL3 in Hirschsprung’s disease. Cell. Physiol. Biochem. 2013, 32, 1655–1667. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef]
- Kouzarides, T. SnapShot: Histone-modifying enzymes. Cell 2007, 128, 802. [Google Scholar] [CrossRef]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Marks, P.A.; Miller, T.; Richon, V.M. Histone deacetylases. Curr. Opin. Pharmacol. 2003, 3, 344–351. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Bonn, S.; Zinzen, R.P.; Girardot, C.; Gustafson, E.H.; Perez-Gonzalez, A.; Delhomme, N.; Ghavi-Helm, Y.; Wilczynski, B.; Riddell, A.; Furlong, E.E. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet. 2012, 44, 148–156. [Google Scholar] [CrossRef]
- Cotney, J.; Leng, J.; Oh, S.; DeMare, L.E.; Reilly, S.K.; Gerstein, M.B.; Noonan, J.P. Chromatin state signatures associated with tissue-specific gene expression and enhancer activity in the embryonic limb. Genome Res. 2012, 22, 1069–1080. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef]
- Ahringer, J. NuRD and SIN3 histone deacetylase complexes in development. Trends Genet. 2000, 16, 351–356. [Google Scholar] [CrossRef]
- Murko, C.; Lagger, S.; Steiner, M.; Seiser, C.; Schoefer, C.; Pusch, O. Histone deacetylase inhibitor Trichostatin A induces neural tube defects and promotes neural crest specification in the chicken neural tube. Differ. Res. Biol. Divers. 2013, 85, 55–66. [Google Scholar] [CrossRef]
- Hatta, K.; Takagi, S.; Fujisawa, H.; Takeichi, M. Spatial and temporal expression pattern of N-cadherin cell adhesion molecules correlated with morphogenetic processes of chicken embryos. Dev. Biol. 1987, 120, 215–227. [Google Scholar] [CrossRef]
- Nakagawa, S.; Takeichi, M. Neural crest cell-cell adhesion controlled by sequential and subpopulation-specific expression of novel cadherins. Development 1995, 121, 1321–1332. [Google Scholar] [PubMed]
- Taneyhill, L.A.; Coles, E.G.; Bronner-Fraser, M. Snail2 directly represses cadherin6B during epithelial-to-mesenchymal transitions of the neural crest. Development 2007, 134, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, M.S.; Unal Eroglu, A.; Malireddy, S.; Gallagher, G.; Nambiar, R.M.; Henion, P.D. Distinct functional and temporal requirements for zebrafish Hdac1 during neural crest-derived craniofacial and peripheral neuron development. PloS ONE 2013, 8, e63218. [Google Scholar] [CrossRef] [PubMed]
- Ignatius, M.S.; Moose, H.E.; El-Hodiri, H.M.; Henion, P.D. colgate/hdac1 Repression of foxd3 expression is required to permit mitfa-dependent melanogenesis. Dev. Biol. 2008, 313, 568–583. [Google Scholar] [CrossRef] [PubMed]
- DeLaurier, A.; Nakamura, Y.; Braasch, I.; Khanna, V.; Kato, H.; Wakitani, S.; Postlethwait, J.H.; Kimmel, C.B. Histone deacetylase-4 is required during early cranial neural crest development for generation of the zebrafish palatal skeleton. BMC Dev. Biol. 2012, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Cai, J.; McIntosh, I.; Jabs, E.W.; Fallin, M.D.; Ingersoll, R.; Hetmanski, J.B.; Vekemans, M.; Attie-Bitach, T.; Lovett, M.; et al. High throughput SNP and expression analyses of candidate genes for non-syndromic oral clefts. J. Med. Genet. 2006, 43, 598–608. [Google Scholar] [CrossRef]
- Alsdorf, R.; Wyszynski, D.F. Teratogenicity of sodium valproate. Expert Opin. Drug Saf. 2005, 4, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.R.; Aldred, M.A.; Der Kaloustian, V.M.; Halal, F.; Gowans, G.; McLeod, D.R.; Zondag, S.; Toriello, H.V.; Magenis, R.E.; Elsea, S.H. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am. J. Hum. Genet. 2010, 87, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Trivedi, C.M.; Lu, M.; Mullican, S.E.; Lazar, M.A.; Epstein, J.A. Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract. Circ. Res. 2011, 109, 1240–1249. [Google Scholar] [CrossRef]
- Haberland, M.; Mokalled, M.H.; Montgomery, R.L.; Olson, E.N. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009, 23, 1625–1630. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Lotscher, P.; Engler, S.; Baggiolini, A.; Varum Tavares, S.; Brugger, V.; John, N.; Buchmann-Moller, S.; Snider, P.L.; Conway, S.J.; et al. HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. J. Neurosci. 2014, 34, 6112–6122. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, V.T. Histone deacetylase 1 is required to repress Notch target gene expression during zebrafish neurogenesis and to maintain the production of motoneurones in response to hedgehog signalling. Development 2004, 131, 2983–2995. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, R.M.; Henion, P.D. Sequential antagonism of early and late Wnt-signaling by zebrafish colgate promotes dorsal and anterior fates. Dev. Biol. 2004, 267, 165–180. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stadler, J.A.; Shkumatava, A.; Norton, W.H.; Rau, M.J.; Geisler, R.; Fischer, S.; Neumann, C.J. Histone deacetylase 1 is required for cell cycle exit and differentiation in the zebrafish retina. Dev. Dyn. 2005, 233, 883–889. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Tonou-Fujimori, N.; Komori, A.; Maeda, R.; Nojima, Y.; Li, H.; Okamoto, H.; Masai, I. Histone deacetylase 1 regulates retinal neurogenesis in zebrafish by suppressing Wnt and Notch signaling pathways. Development 2005, 132, 3027–3043. [Google Scholar] [CrossRef]
- Strobl-Mazzulla, P.H.; Sauka-Spengler, T.; Bronner-Fraser, M. Histone demethylase JmjD2A regulates neural crest specification. Dev. Cell 2010, 19, 460–468. [Google Scholar] [CrossRef]
- Maxson, R.; Ishii, M. The Bmp pathway in skull vault development. Front. Oral Biol. 2008, 12, 197–208. [Google Scholar] [CrossRef]
- Monsoro-Burq, A.H.; Wang, E.; Harland, R. Msx1 and Pax3 cooperate to mediate FGF8 and WNT signals during Xenopus neural crest induction. Dev. Cell 2005, 8, 167–178. [Google Scholar] [CrossRef]
- Phillips, B.T.; Kwon, H.J.; Melton, C.; Houghtaling, P.; Fritz, A.; Riley, B.B. Zebrafish msxB, msxC and msxE function together to refine the neural-nonneural border and regulate cranial placodes and neural crest development. Dev. Biol. 2006, 294, 376–390. [Google Scholar] [CrossRef][Green Version]
- Takahashi, K.; Nuckolls, G.H.; Takahashi, I.; Nonaka, K.; Nagata, M.; Ikura, T.; Slavkin, H.C.; Shum, L. Msx2 is a repressor of chondrogenic differentiation in migratory cranial neural crest cells. Dev. Dyn. 2001, 222, 252–262. [Google Scholar] [CrossRef] [PubMed]
- McGinty, J.F. The many faces of MeCP2. Neuropsychopharmacology 2012, 37, 313–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhou, Z.; Qin, J.; Tang, J.; Li, B.; Geng, Q.; Jiang, W.; Wu, W.; Rehan, V.; Tang, W.; Xu, X.; et al. Down-regulation of MeCP2 in Hirschsprung’s disease. J. Pediatric Surg. 2013, 48, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Schuettengruber, B.; Chourrout, D.; Vervoort, M.; Leblanc, B.; Cavalli, G. Genome regulation by polycomb and trithorax proteins. Cell 2007, 128, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.; Varum, S.; Zemke, M.; Scholer, A.; Baggiolini, A.; Draganova, K.; Koseki, H.; Schubeler, D.; Sommer, L. Ezh2 is required for neural crest-derived cartilage and bone formation. Development 2014, 141, 867–877. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, Y.U.; Wang, L.; Querol Cano, L.; Haramis, A.P. The polycomb group protein ring1b/rnf2 is specifically required for craniofacial development. Plos ONE 2013, 8, e73997. [Google Scholar] [CrossRef] [PubMed]
- Martins-Taylor, K.; Schroeder, D.I.; LaSalle, J.M.; Lalande, M.; Xu, R.H. Role of DNMT3B in the regulation of early neural and neural crest specifiers. Epigenetics 2012, 7, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, K.; Kim, J. AEBP2 as a potential targeting protein for Polycomb Repression Complex PRC2. Nucleic Acids Res. 2009, 37, 2940–2950. [Google Scholar] [CrossRef] [PubMed]
- Ahola, J.A.; Koivusalo, A.; Sairanen, H.; Jokinen, E.; Rintala, R.J.; Pakarinen, M.P. Increased incidence of Hirschsprung’s disease in patients with hypoplastic left heart syndrome--a common neural crest-derived etiology? J. Pediatric Surg. 2009, 44, 1396–1400. [Google Scholar] [CrossRef]
- Inoue, K.; Shilo, K.; Boerkoel, C.F.; Crowe, C.; Sawady, J.; Lupski, J.R.; Agamanolis, D.P. Congenital hypomyelinating neuropathy, central dysmyelination, and Waardenburg-Hirschsprung disease: Phenotypes linked by SOX10 mutation. Ann. Neurol. 2002, 52, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kang, K.; Ekram, M.B.; Roh, T.Y.; Kim, J. Aebp2 as an epigenetic regulator for neural crest cells. Plos ONE 2011, 6, e25174. [Google Scholar] [CrossRef] [PubMed]
- Tien, C.L.; Jones, A.; Wang, H.; Gerigk, M.; Nozell, S.; Chang, C. Snail2/Slug cooperates with Polycomb repressive complex 2 (PRC2) to regulate neural crest development. Development 2015, 142, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.S.; Wagner, D. Unwinding chromatin for development and growth: A few genes at a time. Trends Genet. 2007, 23, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.I.; Lessard, J.; Crabtree, G.R. Understanding the words of chromatin regulation. Cell 2009, 136, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Muchardt, C.; Yaniv, M. When the SWI/SNF complex remodels...the cell cycle. Oncogene 2001, 20, 3067–3075. [Google Scholar] [CrossRef]
- Bajpai, R.; Chen, D.A.; Rada-Iglesias, A.; Zhang, J.; Xiong, Y.; Helms, J.; Chang, C.P.; Zhao, Y.; Swigut, T.; Wysocka, J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010, 463, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Barnett, C.; Krebs, J.E. WSTF does it all: A multifunctional protein in transcription, repair, and replication. Biochem. Cell Biol. = Biochim. Et Biol. Cell. 2011, 89, 12–23. [Google Scholar] [CrossRef]
- Barnett, C.; Yazgan, O.; Kuo, H.C.; Malakar, S.; Thomas, T.; Fitzgerald, A.; Harbour, W.; Henry, J.J.; Krebs, J.E. Williams Syndrome Transcription Factor is critical for neural crest cell function in Xenopus laevis. Mech. Dev. 2012, 129, 324–338. [Google Scholar] [CrossRef]
- Yoshimura, K.; Kitagawa, H.; Fujiki, R.; Tanabe, M.; Takezawa, S.; Takada, I.; Yamaoka, I.; Yonezawa, M.; Kondo, T.; Furutani, Y.; et al. Retraction for Yoshimura et al., Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF). Proc. Natl. Acad. Sci. USA 2014, 111, 2398. [Google Scholar] [CrossRef]
- Hausser, J.; Zavolan, M. Identification and consequences of miRNA-target interactions--beyond repression of gene expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.S. MicroRNA function: Multiple mechanisms for a tiny RNA? RNA 2005, 11, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. The functional role of long non-coding RNAs and epigenetics. Biol. Proced. Online 2014, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015, 22, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Cai, P.; Li, H.; Huo, W.; Zhu, H.; Xu, C.; Zang, R.; Lv, W.; Xia, Y.; Tang, W. Aberrant expression of LncRNA-MIR31HG regulates cell migration and proliferation by affecting miR-31 and miR-31* in Hirschsprung’s disease. J. Cell. Biochem. 2018, 119, 8195–8203. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Du, C.; Shen, Z.; Peng, L.; Xie, H.; Zang, R.; Li, H.; Xia, Y.; Tang, W. MicroRNA-939 inhibits cell proliferation via targeting LRSAM1 in Hirschsprung’s disease. Aging 2017, 9, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Peng, L.; Zhu, Z.; Du, C.; Shen, Z.; Zang, R.; Su, Y.; Xia, Y.; Tang, W. LncRNA AFAP1-AS Functions as a Competing Endogenous RNA to Regulate RAP1B Expression by sponging miR-181a in the HSCR. Int. J. Med. Sci. 2017, 14, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Shen, Z.; Zang, R.; Xie, H.; Li, H.; Chen, P.; Hang, B.; Xu, X.; Tang, W.; Xia, Y. Negative feedback circuitry between MIR143HG and RBM24 in Hirschsprung disease. Biochim. Et Biophys. Acta 2016, 1862, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Xie, H.; Zang, R.; Shen, Z.; Li, H.; Chen, P.; Xu, X.; Xia, Y.; Tang, W. Apoptotic neuron-secreted HN12 inhibits cell apoptosis in Hirschsprung’s disease. Int. J. Nanomed. 2016, 11, 5871–5881. [Google Scholar] [CrossRef]
- Budi, N.Y.P.; Kalim, A.S.; Santiko, W.; Musthofa, F.D.; Iskandar, K.; Makhmudi, A. Aberrant expressions of miRNA-206 target, FN1, in multifactorial Hirschsprung disease. Orphanet J. Rare Dis. 2019, 14, 5. [Google Scholar] [CrossRef]
- Lei, H.; Li, H.; Xie, H.; Du, C.; Xia, Y.; Tang, W. Role of MiR-215 in Hirschsprung’s Disease Pathogenesis by Targeting SIGLEC-8. Cell. Physiol. Biochem. 2016, 40, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Tang, J.; Li, H.; Zhang, H.; Lu, C.; Chen, H.; Li, W.; Xia, Y.; Tang, W. MiR-195 affects cell migration and cell proliferation by down-regulating DIEXF in Hirschsprung’s disease. BMC Gastroenterol. 2014, 14, 123. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, B.; Zhu, D.; Xie, H.; Du, C.; Xia, Y.; Tang, W. Downregulation of lncRNA MEG3 and miR-770-5p inhibit cell migration and proliferation in Hirschsprung’s disease. Oncotarget 2017, 8, 69722–69730. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tang, J.; Lei, H.; Cai, P.; Zhu, H.; Li, B.; Xu, X.; Xia, Y.; Tang, W. Decreased MiR-200a/141 suppress cell migration and proliferation by targeting PTEN in Hirschsprung’s disease. Cell. Physiol. Biochem. 2014, 34, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, L.; Lu, C.; Shen, Q.; Su, Y.; Zhi, Z.; Wu, F.; Zhang, H.; Wen, Z.; Chen, G.; et al. Long non-coding RNA FAL1 functions as a ceRNA to antagonize the effect of miR-637 on the down-regulation of AKT1 in Hirschsprung’s disease. Cell Prolif. 2018, 51, e12489. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Luo, Y.; Li, S.; Wang, S.; Wang, N.; Jin, X. Expression profiles of HA117 and its neighboring gene DPF3 in different colon segments of Hirschsprung’s disease. Int. J. Clin. Exp. Pathol. 2014, 7, 3966–3974. [Google Scholar]
- Luo, Y.; Li, S.; Teng, Y.; Wang, N.; Li, L.; Liu, H.; Jin, X. Differential expression of FOXA1, DUSP6, and HA117 in colon segments of Hirschsprung’s disease. Int. J. Clin. Exp. Pathol. 2015, 8, 3979–3986. [Google Scholar]
- Pan, W.; Yu, H.; Zheng, B.; Gao, Y.; Li, P.; Huang, Q.; Xie, C.; Ge, X. Upregulation of MiR-369-3p suppresses cell migration and proliferation by targeting SOX4 in Hirschsprung’s disease. J. Pediatric Surg. 2017, 52, 1363–1370. [Google Scholar] [CrossRef]
- Peng, L.; Chen, G.; Zhu, Z.; Shen, Z.; Du, C.; Zang, R.; Su, Y.; Xie, H.; Li, H.; Xu, X.; et al. Circular RNA ZNF609 functions as a competitive endogenous RNA to regulate AKT3 expression by sponging miR-150-5p in Hirschsprung’s disease. Oncotarget 2017, 8, 808–818. [Google Scholar] [CrossRef]
- Sharan, A.; Zhu, H.; Xie, H.; Li, H.; Tang, J.; Tang, W.; Zhang, H.; Xia, Y. Down-regulation of miR-206 is associated with Hirschsprung disease and suppresses cell migration and proliferation in cell models. Sci. Rep. 2015, 5, 9302. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Peng, L.; Zhu, Z.; Xie, H.; Zang, R.; Du, C.; Chen, G.; Li, H.; Xia, Y.; Tang, W. Downregulated Expression of Long Non-Coding RNA LOC101926975 Impairs both Cell Proliferation and Cell Cycle and Its Clinical Implication in Hirschsprung Disease Patients. Int. J. Med. Sci. 2016, 13, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Wen, Z.; Shen, Q.; Zhang, H.; Peng, L.; Chen, G.; Zhu, Z.; Du, C.; Xie, H.; Li, H.; et al. Long non-coding RNA LOC100507600 functions as a competitive endogenous RNA to regulate BMI1 expression by sponging miR128-1-3p in Hirschsprung’s disease. Cell Cycle 2018, 17, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Tang, J.; He, J.; Zhou, Z.; Qin, Y.; Qin, J.; Li, B.; Xu, X.; Geng, Q.; Jiang, W.; et al. SLIT2/ROBO1-miR-218-1-RET/PLAG1: A new disease pathway involved in Hirschsprung’s disease. J. Cell. Mol. Med. 2015, 19, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Shen, Q.; Zhang, H.; Su, Y.; Zhu, Z.; Chen, G.; Peng, L.; Li, H.; Du, C.; Xie, H.; et al. Circular RNA CCDC66 targets DCX to regulate cell proliferation and migration by sponging miR-488-3p in Hirschsprung’s disease. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Yuan, W.; Chen, J.; Zhou, Z.; Shu, Y.; Ji, J.; Liu, Z.; Tang, Q.; Zhang, X.; Shu, X. Increased miR-214 expression suppresses cell migration and proliferation in Hirschsprung disease by interacting with PLAGL2. Pediatric Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhu, D.; Xu, C.; Zhu, H.; Chen, P.; Li, H.; Liu, X.; Xia, Y.; Tang, W. Long none coding RNA HOTTIP/HOXA13 act as synergistic role by decreasing cell migration and proliferation in Hirschsprung disease. Biochem. Biophys. Res. Commun. 2015, 463, 569–574. [Google Scholar] [CrossRef]
- Zhi, Z.; Zhu, H.; Lv, X.; Lu, C.; Li, Y.; Wu, F.; Zhou, L.; Li, H.; Tang, W. IGF2-derived miR-483-3p associated with Hirschsprung’s disease by targeting FHL1. J. Cell. Mol. Med. 2018, 22, 4913–4921. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Jiang, W.; Zhang, H.; Wen, Z.; Su, Y.; Wu, F.; Zhi, Z.; Shen, Q.; Li, H.; et al. Down-regulation of circ-PRKCI inhibits cell migration and proliferation in Hirschsprung disease by suppressing the expression of miR-1324 target PLCB1. Cell Cycle 2018, 17, 1092–1101. [Google Scholar] [CrossRef]
- Zhu, D.; Xie, H.; Li, H.; Cai, P.; Zhu, H.; Xu, C.; Chen, P.; Sharan, A.; Xia, Y.; Tang, W. Nidogen-1 is a common target of microRNAs MiR-192/215 in the pathogenesis of Hirschsprung’s disease. J. Neurochem. 2015, 134, 39–46. [Google Scholar] [CrossRef]
- Fitze, G.; Schierz, M.; Kuhlisch, E.; Schreiber, M.; Ziegler, A.; Roesner, D.; Schackert, H.K. Novel intronic polymorphisms in the RET proto-oncogene and their association with Hirschsprung disease. Hum. Mutat. 2003, 22, 177. [Google Scholar] [CrossRef] [PubMed]
- Griseri, P.; Lantieri, F.; Puppo, F.; Bachetti, T.; Di Duca, M.; Ravazzolo, R.; Ceccherini, I. A common variant located in the 3′UTR of the RET gene is associated with protection from Hirschsprung disease. Hum. Mutat. 2007, 28, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.W.; Luo, C.F.; Liu, Z.J.; Li, J.C. RET 3′UTR polymorphisms and its protective role in Hirschsprung disease in southeastern Chinese. J. Pediatric Surg. 2012, 47, 1699–1705. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Role on Cellular Processes | ncRNA/Reference | Expression in HSCR Tissue | Change |
|---|---|---|---|
| proliferation and migration | miR-141 [37] | downregulated | ↑CD47/CUL3 |
| miR-195 [104] | upregulated | ↓DIEXF | |
| miR-200a/141 [106] | downregulated | ↑PTEN | |
| miR-206 [102,112] | downregulated | ↑SDPR/FN1 | |
| miR-192/215 [121] | downregulated | ↑NID1 | |
| miR-218-1 [115] | upregulated | ↑SLIT2 ↓RET/PLAG1 | |
| miR-215 [103] | downregulated | ↓IARS2/↑SIGLEC-8 | |
| miR-369-3p [110] | upregulated | ↓SOX4 | |
| miR-483-3p [119] | downregulated | ↓IGF2 ↑FHL1 | |
| miR-214 [117] | upregulated | ↓PLAGL2 | |
| HOTTIP [118] | downregulated | ↓HOXA13 | |
| miR143HG [100] | upregulated | ↓miR-143/↑RBM24 | |
| AFAP1-AS [99] | downregulated | ↑miR-181a/↓RAP1B | |
| MEG3 [105] | downregulated | ↓miR-770-5p/↑SRGAP1 | |
| FAL1 [107] | downregulated | ↓AKT1 | |
| miR31HG [97] | downregulated | ↓miR-31/31* | |
| LOC100507600 [114] | downregulated | ↑miR128–1-3p/↓BMI1 | |
| cir-ZNF609 [111] | downregulated | ↑miR-150-5p/↓AKT3 | |
| circ-PRKCI [120] | downregulated | ↑miR-1324/↓PLCB1 | |
| cir-CCDC66 [116] | downregulated | ↑miR-488-3p/↓DCX | |
| proliferation and apoptosis | miR-483-5p [28] | upregulated | ↓GFRA4 |
| proliferation | miR-939 [98] | upregulated | ↓LRSAM1 |
| LOC101926975 [113] | downregulated | ↓FGF1 | |
| apoptosis | HN12 [101] | upregulated | - |
| Unknown | HA117 [108,109] | upregulated | ↓DPF3/FOXA1/DUSP6 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torroglosa, A.; Villalba-Benito, L.; Luzón-Toro, B.; Fernández, R.M.; Antiñolo, G.; Borrego, S. Epigenetic Mechanisms in Hirschsprung Disease. Int. J. Mol. Sci. 2019, 20, 3123. https://doi.org/10.3390/ijms20133123
Torroglosa A, Villalba-Benito L, Luzón-Toro B, Fernández RM, Antiñolo G, Borrego S. Epigenetic Mechanisms in Hirschsprung Disease. International Journal of Molecular Sciences. 2019; 20(13):3123. https://doi.org/10.3390/ijms20133123
Chicago/Turabian StyleTorroglosa, Ana, Leticia Villalba-Benito, Berta Luzón-Toro, Raquel María Fernández, Guillermo Antiñolo, and Salud Borrego. 2019. "Epigenetic Mechanisms in Hirschsprung Disease" International Journal of Molecular Sciences 20, no. 13: 3123. https://doi.org/10.3390/ijms20133123
APA StyleTorroglosa, A., Villalba-Benito, L., Luzón-Toro, B., Fernández, R. M., Antiñolo, G., & Borrego, S. (2019). Epigenetic Mechanisms in Hirschsprung Disease. International Journal of Molecular Sciences, 20(13), 3123. https://doi.org/10.3390/ijms20133123