Pulmonary Arterial Hypertension Due to NPR-C Mutation: A Novel Paradigm for Normal and Pathologic Remodeling?


{kind=link}
Abstract
1. Introduction
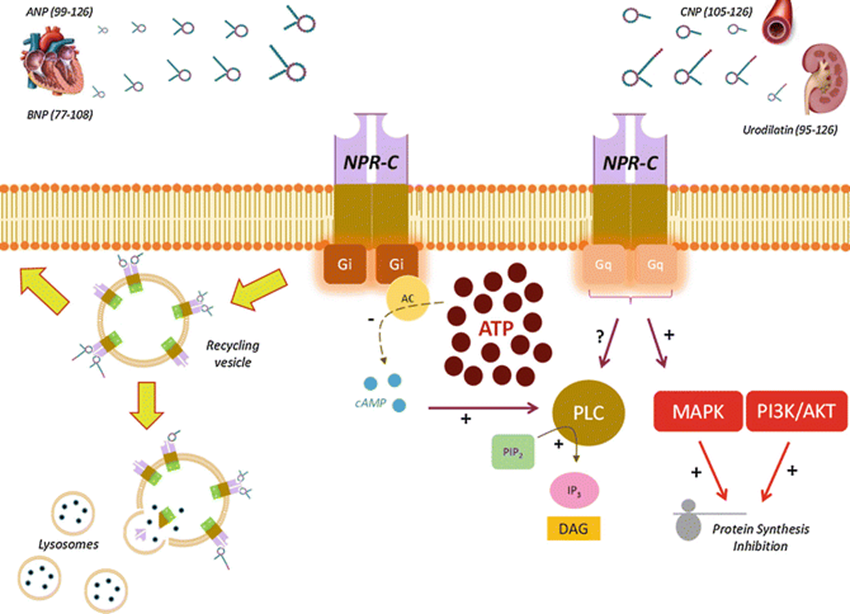
2. Normal NPR-C Signaling
3. Consequences of Npr3 Mutation and/or Alterations in NPR-C Signaling
3.1. Studies in Transgenic and Knockout Mice
3.2. NPR-C signaling in Hypoxia-Induced Pulmonary Hypertension
3.3. Studies implicating NPR-C Signaling in Other Cardiovascular Diseases
4. NPR-C Signaling as a Therapeutic Target in Tissue Remodeling
5. Conclusions
Funding
Conflicts of Interest
References
- Egom, E.E.-A.; Feridooni, T.; Pharithi, R.B.; Khan, B.; Shiwani, H.A.; Maher, V.; El Hiani, Y.; Rose, R.A.; Pasumarthi, K.B.S.; Ribama, H.A. New insights and new hope for pulmonary arterial hypertension: natriuretic peptides clearance receptor as a novel therapeutic target for a complex disease. Int. J. Physiol. Pathophysiol. Pharmacol. 2017, 9, 112–118. [Google Scholar] [CrossRef]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici, A.; Weitzenblum, E.; Cordier, J.-F.; Chabot, F. Pulmonary arterial hypertension in France: results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Badesch, D.B.; Raskob, G.E.; Elliott, C.G.; Krichman, A.M.; Farber, H.W.; Frost, A.E.; Barst, R.J.; Benza, R.L.; Liou, T.G.; Turner, M. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 2010, 137, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Peacock, A.; Murphy, N.; McMurray, J.; Caballero, L.; Stewart, S. An epidemiological study of pulmonary arterial hypertension. Eur. Respir. J. 2007, 30, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S13–S24. [Google Scholar] [CrossRef] [PubMed]
- Pietra, G.G.; Capron, F.; Stewart, S.; Leone, O.; Humbert, M.; Robbins, I.M.; Reid, L.M.; Tuder, R. Pathologic assessment of vasculopathies in pulmonary hypertension. J. Am. Coll. Cardiol. 2004, 43, S25–S32. [Google Scholar] [CrossRef]
- Dupont, M.; Tang, W.W. Right ventricular afterload and the role of nitric oxide metabolism in left-sided heart failure. J. Card. Fail. 2013, 19, 712–721. [Google Scholar] [CrossRef]
- Egom, E.E.; Maher, V.; El Hiani, Y. Evolving use of natriuretic peptide receptor type-C as part of strategies for the treatment of pulmonary hypertension due to left ventricle heart failure. Int. J. Cardiol. 2019, 281, 172–178. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Gibbs, J.S.; Wachter, R.; De Marco, T.; Vonk-Noordegraaf, A.; Vachiery, J.L. Left ventricular heart failure and pulmonary hypertension. Eur. Heart J. 2016, 37, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.; Feridooni, T.; Pharithi, R.; Maher, V.; El Hiani, Y.; Pasumarthi, K.; Ribama, H. New Insights and New Hope for Pulmonary Arterial Hypertension: Natriuretic Peptides Clearance Receptor as a Novel Therapeutic Target for a Complex Disease. J. Am. Coll. Cardiol. 2017, 69 (Suppl. 11), 1902. [Google Scholar] [CrossRef]
- Hobbs, A.; Foster, P.; Prescott, C.; Scotland, R.; Ahluwalia, A. Natriuretic peptide receptor-C regulates coronary blood flow and prevents myocardial ischemia/reperfusion injury: novel cardioprotective role for endothelium-derived C-type natriuretic peptide. Circulation 2004, 110, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E. BNP and Heart Failure: Preclinical and Clinical Trial Data. J. Cardiovasc. Transl. Res. 2015, 8, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.; Feridooni, T.; Hotchkiss, A.; Kruzliak, P.; Pasumarthi, K.B. Mechanisms of renal hyporesponsiveness to BNP in heart failure. Can. J. Physiol. Pharmacol. 2015, 93, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Anand-Srivastava, M.B. Natriuretic peptide receptor-C signaling and regulation. Peptides 2005, 26, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Kouyoumdzian, N.M.; Mikusic, N.L.R.; Lee, H.J.; Fernández, B.E.; Choi, M.R. Natriuretic Peptide Receptor Type C (NPRC). In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Ed. Springer New York: New York, NY, USA, 2017; pp. 1–7. [Google Scholar]
- Cantú, S.M.; Donoso, A.S.; Kouyoumdzian, N.M.; Mikusic, R.; Lucía, N.; Puyó, A.M.; Choi, M.R. Clinical aspects of c-type natriuretic peptide on the cardiovascular system. Int. J. Clin. Endocrinol. Metab. 2015, 1, 3–36. [Google Scholar]
- Li, Y.; Hashim, S.; Anand-Srivastava, M.B. Intracellular peptides of natriuretic peptide receptor-C inhibit vascular hypertrophy via Gqα/MAP kinase signaling pathways. Cardiovasc. Res. 2006, 72, 464–472. [Google Scholar] [CrossRef]
- Jain, A.; Anand-Srivastava, M.B. Natriuretic peptide receptor-C-mediated attenuation of vascular smooth muscle cell hypertrophy involves Gqalpha/PLCbeta1 proteins and ROS-associated signaling. Pharmacol. Res. Perspect. 2018, 6. [Google Scholar] [CrossRef]
- Ventimiglia, M.S.; Najenson, A.C.; Perazzo, J.C.; Carozzo, A.; Vatta, M.S.; Davio, C.A.; Bianciotti, L.G. Blockade of Multidrug Resistance-Associated Proteins Aggravates Acute Pancreatitis and Blunts Atrial Natriuretic Factor’s Beneficial Effect in Rats: Role of MRP4 (ABCC4). Mol. Med. 2015, 21, 58–67. [Google Scholar] [CrossRef]
- Pandey, K.N. Molecular and genetic aspects of guanylyl cyclase natriuretic peptide receptor-A in regulation of blood pressure and renal function. Physiol. Genom. 2018, 50, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Moyes, A.J.; Hobbs, A.J. C-type Natriuretic Peptide: A Multifaceted Paracrine Regulator in the Heart and Vasculature. Int. J. Mol. Sci. 2019, 20, 2281. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.A.; Lomax, A.E.; Kondo, C.S.; Anand-Srivastava, M.B.; Giles, W.R. Effects of C-type natriuretic peptide on ionic currents in mouse sinoatrial node: a role for the NPR-C receptor. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1970–H1977. [Google Scholar] [CrossRef][Green Version]
- Mouawad, R.; Li, Y.; Anand-Srivastava, M.B. Atrial natriuretic peptide-C receptor-induced attenuation of adenylyl cyclase signaling activates phosphatidylinositol turnover in A10 vascular smooth muscle cells. Mol. Pharmacol. 2004, 65, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.N. Guanylyl cyclase / atrial natriuretic peptide receptor-A: role in the pathophysiology of cardiovascular regulation. Can. J. Physiol. Pharmacol. 2011, 89, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Sciarretta, S.; Morriello, A.; Calvieri, C.; Battistoni, A.; Volpe, M. NPR-C: a component of the natriuretic peptide family with implications in human diseases. J. Mol. Med. 2010, 88, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Rose, R.A.; Giles, W.R. Natriuretic peptide C receptor signalling in the heart and vasculature. J. Physiol. 2008, 586, 353–366. [Google Scholar] [CrossRef]
- Dannewitz Prosseda, S.; Tian, X.; Kuramoto, K.; Boehm, M.; Sudheendra, D.; Miyagawa, K.; Zhang, F.; Solow-Cordero, D.; Saldivar, J.C.; Austin, E.D.; et al. FHIT, a Novel Modifier Gene in Pulmonary Arterial Hypertension. Am. J. Respir Crit. Care Med. 2019, 199, 83–98. [Google Scholar] [CrossRef]
- Chen, Y.F. Atrial natriuretic peptide in hypoxia. Peptides 2005, 26, 1068–1077. [Google Scholar] [CrossRef]
- Sun, J.-Z.; Oparil, S.; Lucchesi, P.; Thompson, J.A.; Chen, Y.-F. Tyrosine kinase receptor activation inhibits NPR-C in lung arterial smooth muscle cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2001, 281, L155–L163. [Google Scholar] [CrossRef]
- Adeoye, O.O.; Silpanisong, J.; Williams, J.M.; Pearce, W.J. Role of the sympathetic autonomic nervous system in hypoxic remodeling of the fetal cerebral vasculature. J. Cardiovasc. Pharmacol. 2015, 65, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.; Morrell, N.; Long, L.; Clift, P.; Upton, P.; Polak, J.; Wilkins, M. Vascular remodeling and ET-1 expression in rat strains with different responses to chronic hypoxia. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2000, 278, L981–L987. [Google Scholar] [CrossRef] [PubMed]
- Chassagne, C.; Eddahibi, S.; Adamy, C.; Rideau, D.; Marotte, F.; Dubois-Rande, J.L.; Adnot, S.; Samuel, J.L.; Teiger, E. Modulation of angiotensin II receptor expression during development and regression of hypoxic pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2000, 22, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, N.; Ran, P.; Du, Z. Expression of FGF-b and c-myc in rats lung tissue affected by hypoxia. Zhonghua Jie He He Hu Xi Za Zhi 1997, 20, 22–24. [Google Scholar] [PubMed]
- Myllyharju, J.; Schipani, E. Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res. 2010, 339, 19–29. [Google Scholar] [CrossRef]
- Baron, W.; Metz, B.; Bansal, R.; Hoekstra, D.; de Vries, H. PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol. Cell. Neurosci. 2000, 15, 314–329. [Google Scholar] [CrossRef]
- Bonacina, F.; Baragetti, A.; Catapano, A.L.; Norata, G.D. Long pentraxin 3: experimental and clinical relevance in cardiovascular diseases. Mediat. Inflamm. 2013, 2013, 725102. [Google Scholar] [CrossRef]
- Ehret, G.B.; Munroe, P.B.; Rice, K.M.; Bochud, M.; Johnson, A.D.; Chasman, D.I.; Smith, A.V.; Tobin, M.D.; Verwoert, G.C.; Hwang, S.-J. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011, 478, 103. [Google Scholar]
- Hu, Q.; Liu, Q.; Shasha Wang, X.Z.; Zhang, Z.; Lv, R.; Jiang, G.; Ma, Z.; He, H.; Li, D.; Liu, X. NPR-C gene polymorphism is associated with increased susceptibility to coronary artery disease in Chinese Han population: a multicenter study. Oncotarget 2016, 7, 33662. [Google Scholar] [CrossRef]
- Fox, A.A.; Collard, C.D.; Shernan, S.K.; Seidman, C.E.; Seidman, J.G.; Liu, K.-Y.; Muehlschlegel, J.D.; Perry, T.E.; Aranki, S.F.; Lange, C. Natriuretic peptide system gene variants are associated with ventricular dysfunction after coronary artery bypass grafting. Anesthesiol. 2009, 110, 738–747. [Google Scholar] [CrossRef]
- El Andalousi, J.; Li, Y.; Anand-Srivastava, M.B. Natriuretic peptide receptor-C agonist attenuates the expression of cell cycle proteins and proliferation of vascular smooth muscle cells from spontaneously hypertensive rats: role of Gi proteins and MAPkinase/PI3kinase signaling. PLoS ONE 2013, 8, e76183. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Intracellular mechanisms involved in vascular remodelling of resistance arteries in hypertension: role of angiotensin II. Exp. Physiol. 2005, 90, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Hashim, S.; Li, Y.; Anand-Srivastava, M.B. Small cytoplasmic domain peptides of natriuretic peptide receptor-C attenuate cell proliferation through Giα protein/MAP kinase/PI3-kinase/AKT pathways. Am. J. Physiol. -Heart Circ. Physiol. 2006, 291, H3144–H3153. [Google Scholar] [CrossRef] [PubMed]
- Huntley, B.K.; Sandberg, S.M.; Noser, J.A.; Cataliotti, A.; Redfield, M.M.; Matsuda, Y.; Burnett, J.C., Jr. BNP-induced activation of cGMP in human cardiac fibroblasts: Interactions with fibronectin and natriuretic peptide receptors. J. Cell. Physiol. 2006, 209, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Sangaralingham, S.J.; Huntley, B.K.; Martin, F.L.; McKie, P.M.; Bellavia, D.; Ichiki, T.; Harders, G.E.; Chen, H.H.; Burnett, J.C., Jr. The aging heart, myocardial fibrosis, and its relationship to circulating C-type natriuretic peptide. Hypertension 2011, 57, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.; Vella, K.; Hua, R.; Jansen, H.J.; Moghtadaei, M.; Polina, I.; Bogachev, O.; Hurnik, R.; Mackasey, M.; Rafferty, S.; et al. Impaired sinoatrial node function and increased susceptibility to atrial fibrillation in mice lacking natriuretic peptide receptor C. J. Physiol. 2015, 593, 1127–1146. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; Mackasey, M.; Moghtadaei, M.; Liu, Y.; Kaur, J.; Egom, E.E.; Tuomi, J.M.; Rafferty, S.A.; Kirkby, A.W.; Rose, R.A. NPR-C (Natriuretic Peptide Receptor-C) Modulates the Progression of Angiotensin II-Mediated Atrial Fibrillation and Atrial Remodeling in Mice. Circ. Arrhythmia Electrophysiol. 2019, 12, e006863. [Google Scholar] [CrossRef] [PubMed]
- Mackasey, M.; Egom, E.E.; Jansen, H.J.; Hua, R.; Moghtadaei, M.; Liu, Y.; Kaur, J.; McRae, M.D.; Bogachev, O.; Rafferty, S.A.; et al. Natriuretic Peptide Receptor-C Protects Against Angiotensin II-Mediated Sinoatrial Node Disease in Mice. Jacc. Basic Transl. Sci. 2018, 3, 824–843. [Google Scholar] [CrossRef]
- Rahali, S.; Li, Y.; Anand-Srivastava, M.B. Contribution of oxidative stress and growth factor receptor transactivation in natriuretic peptide receptor C-mediated attenuation of hyperproliferation of vascular smooth muscle cells from SHR. PLoS ONE 2018, 13, e0191743. [Google Scholar] [CrossRef]
- Bubb, K.J.; Aubdool, A.A.; Moyes, A.J.; Lewis, S.; Drayton, J.P.; Tang, O.; Mehta, V.; Zachary, I.C.; Abraham, D.J.; Tsui, J.; et al. Endothelial C-Type Natriuretic Peptide Is a Critical Regulator of Angiogenesis and Vascular Remodeling. Circulation 2019, 139, 1612–1628. [Google Scholar] [CrossRef]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egom, E.E.-A. Pulmonary Arterial Hypertension Due to NPR-C Mutation: A Novel Paradigm for Normal and Pathologic Remodeling? Int. J. Mol. Sci. 2019, 20, 3063. https://doi.org/10.3390/ijms20123063
Egom EE-A. Pulmonary Arterial Hypertension Due to NPR-C Mutation: A Novel Paradigm for Normal and Pathologic Remodeling? International Journal of Molecular Sciences. 2019; 20(12):3063. https://doi.org/10.3390/ijms20123063
Chicago/Turabian StyleEgom, Emmanuel Eroume-A. 2019. "Pulmonary Arterial Hypertension Due to NPR-C Mutation: A Novel Paradigm for Normal and Pathologic Remodeling?" International Journal of Molecular Sciences 20, no. 12: 3063. https://doi.org/10.3390/ijms20123063
APA StyleEgom, E. E.-A. (2019). Pulmonary Arterial Hypertension Due to NPR-C Mutation: A Novel Paradigm for Normal and Pathologic Remodeling? International Journal of Molecular Sciences, 20(12), 3063. https://doi.org/10.3390/ijms20123063