Whole Genome Sequencing and Analysis of Chlorimuron-Ethyl Degrading Bacteria Klebsiella pneumoniae 2N3



Abstract
1. Introduction
2. Results and Discussion
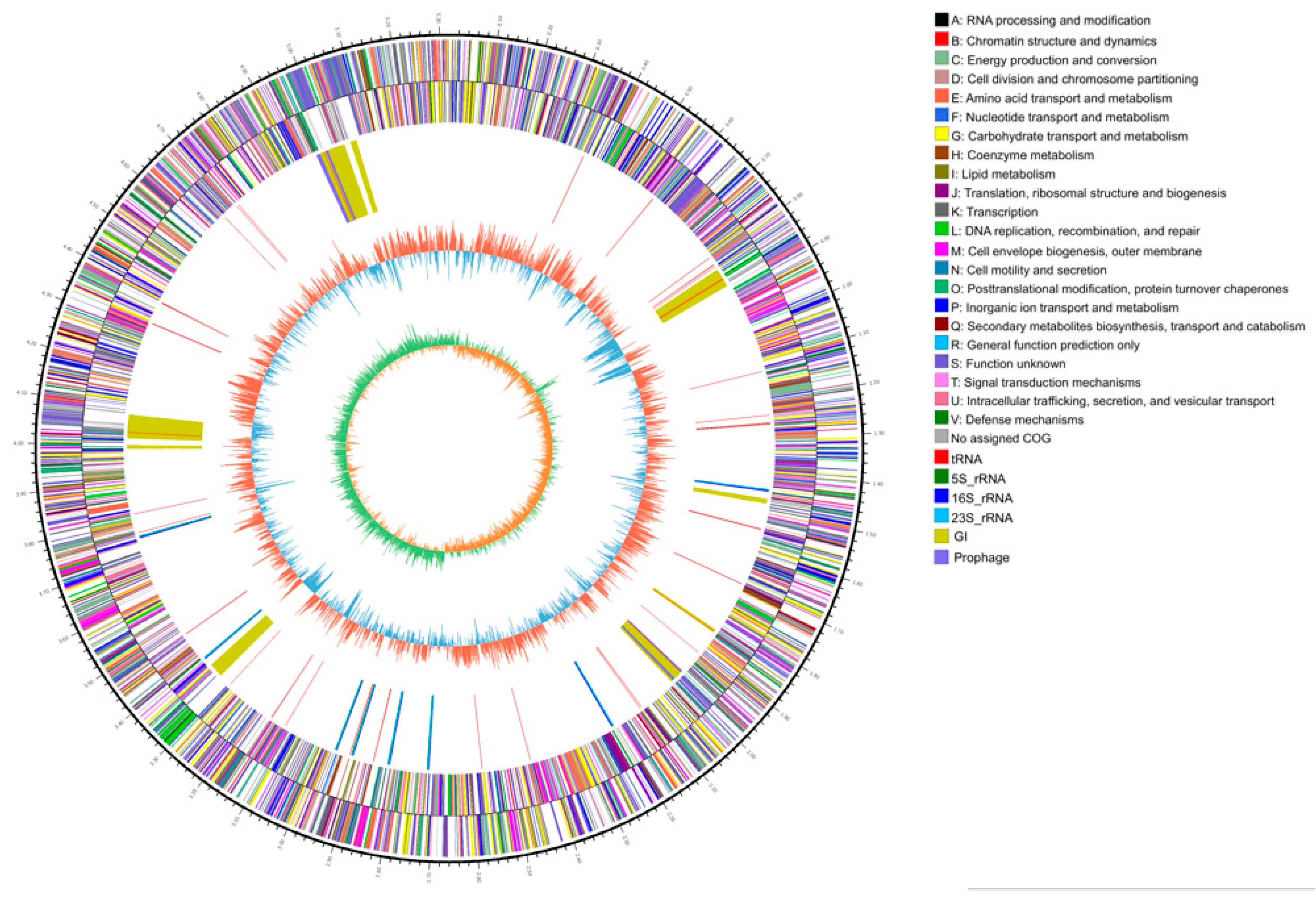
2.1. Genome Sequencing Characteristics
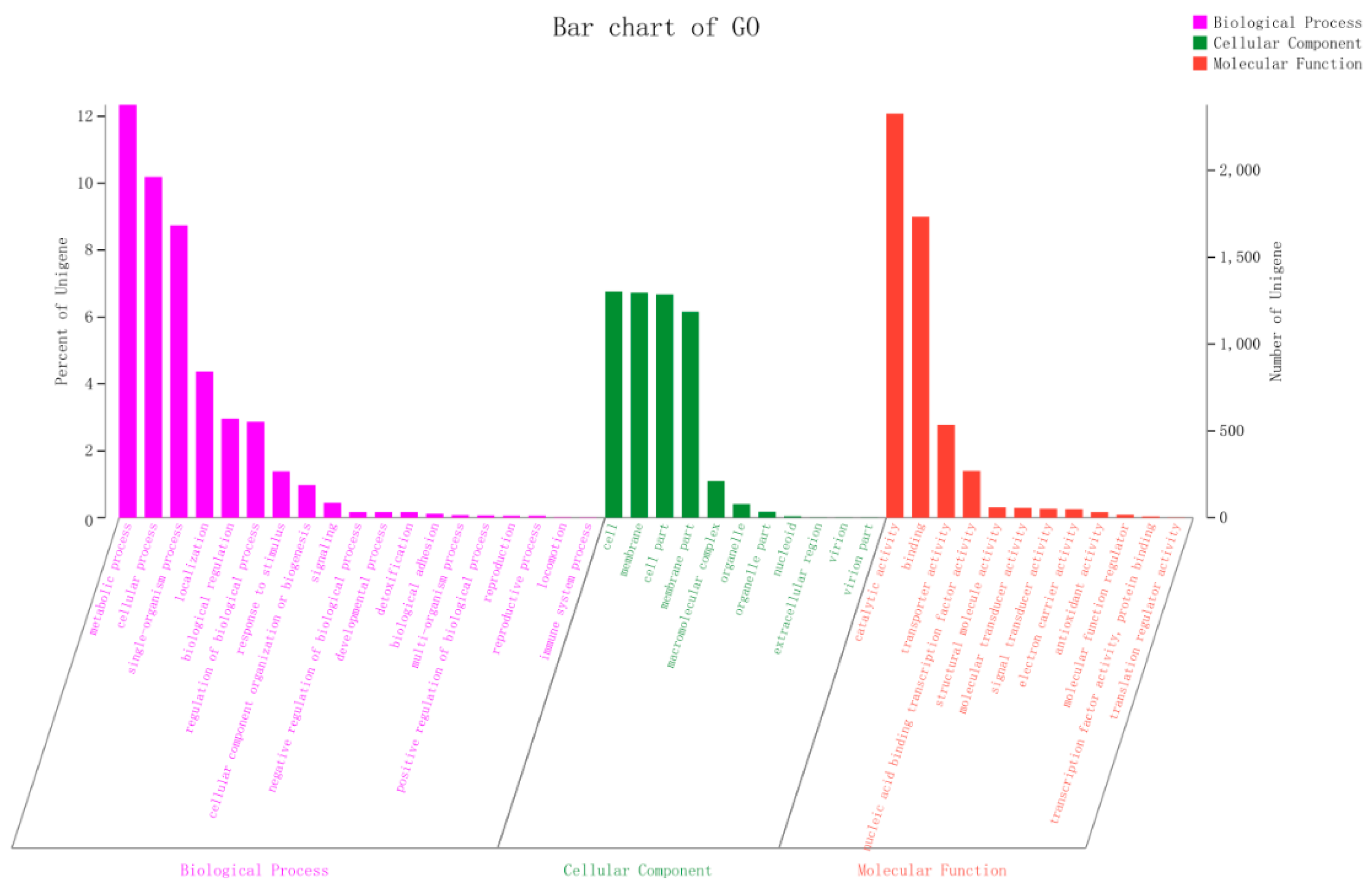
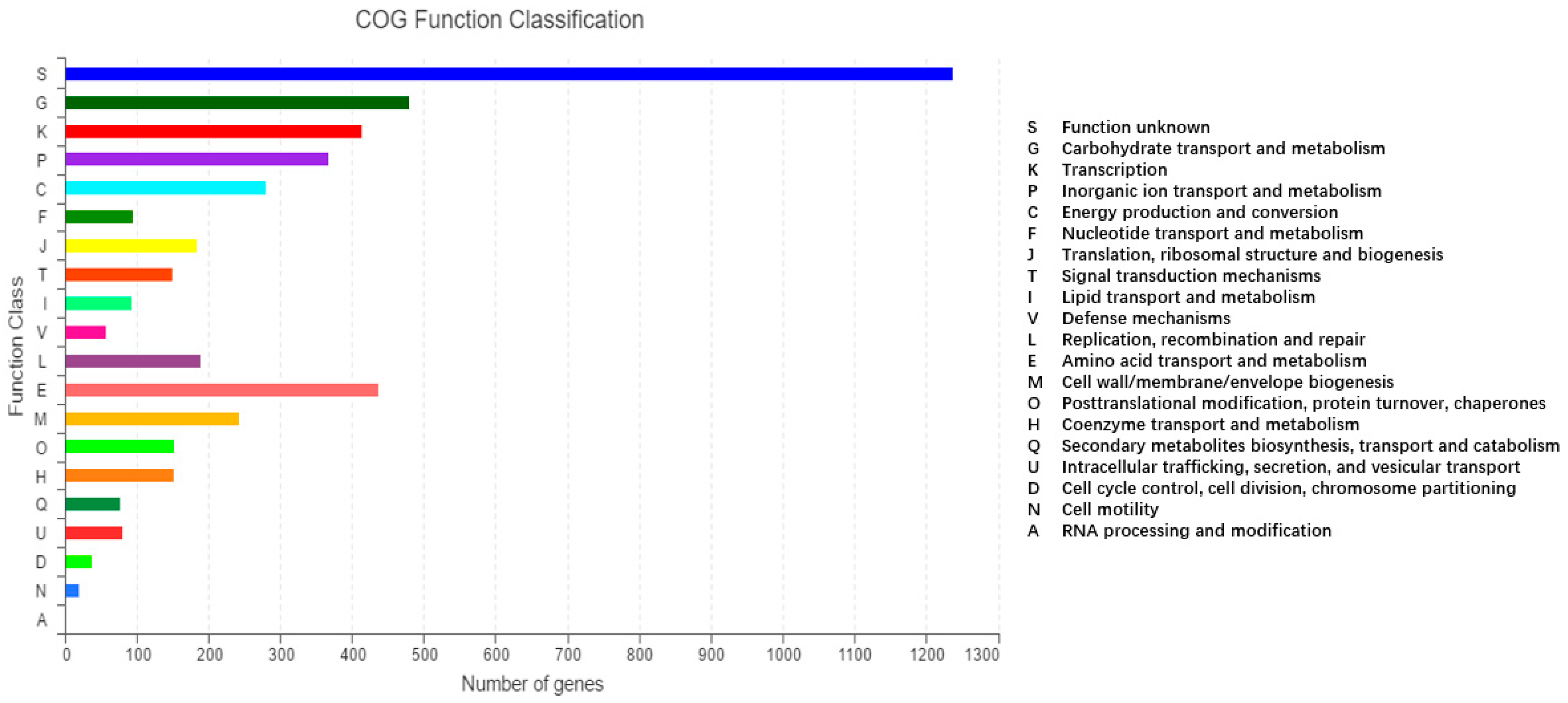
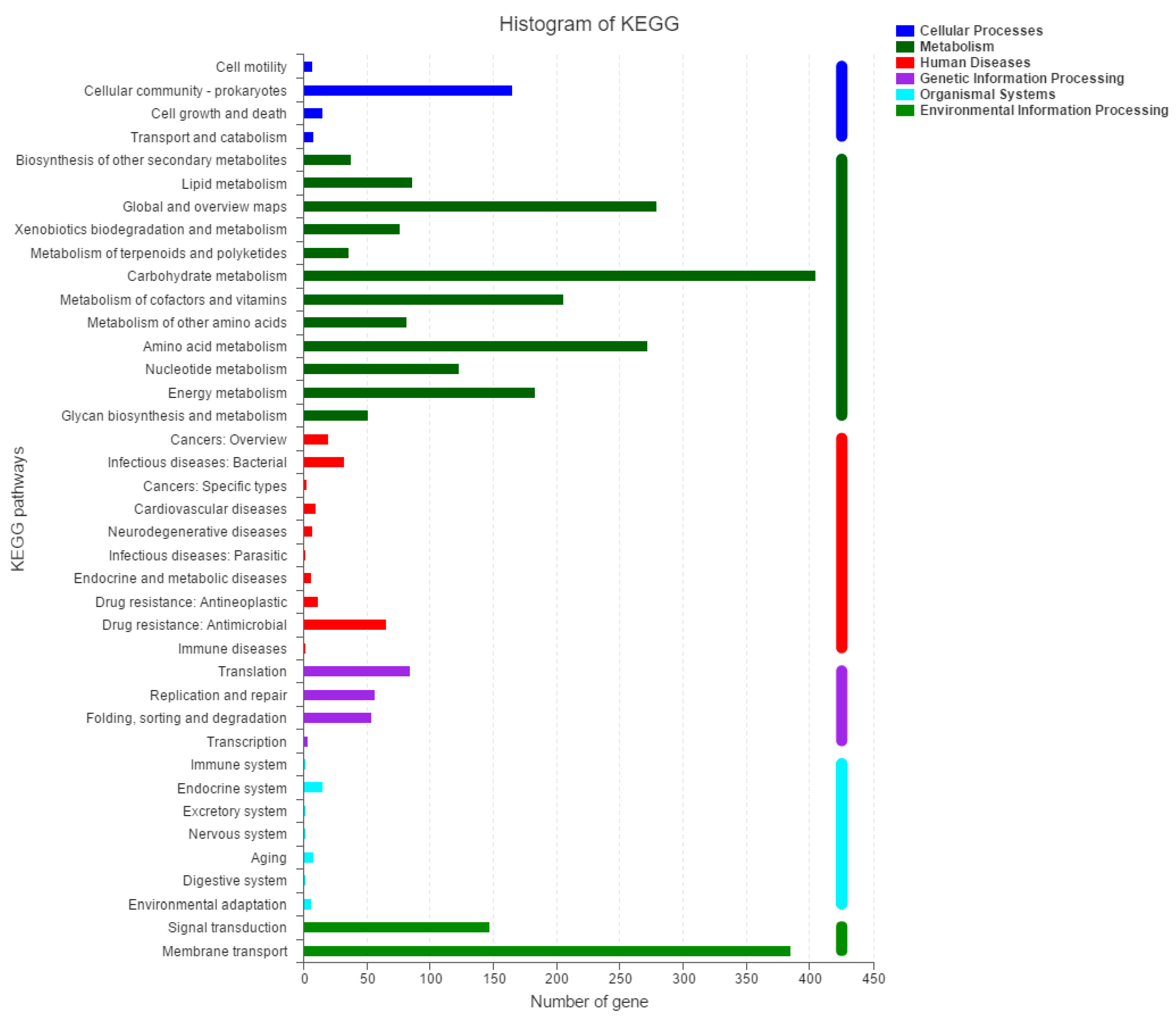
2.2. Molecular Characteristics of the 2N3 Genome
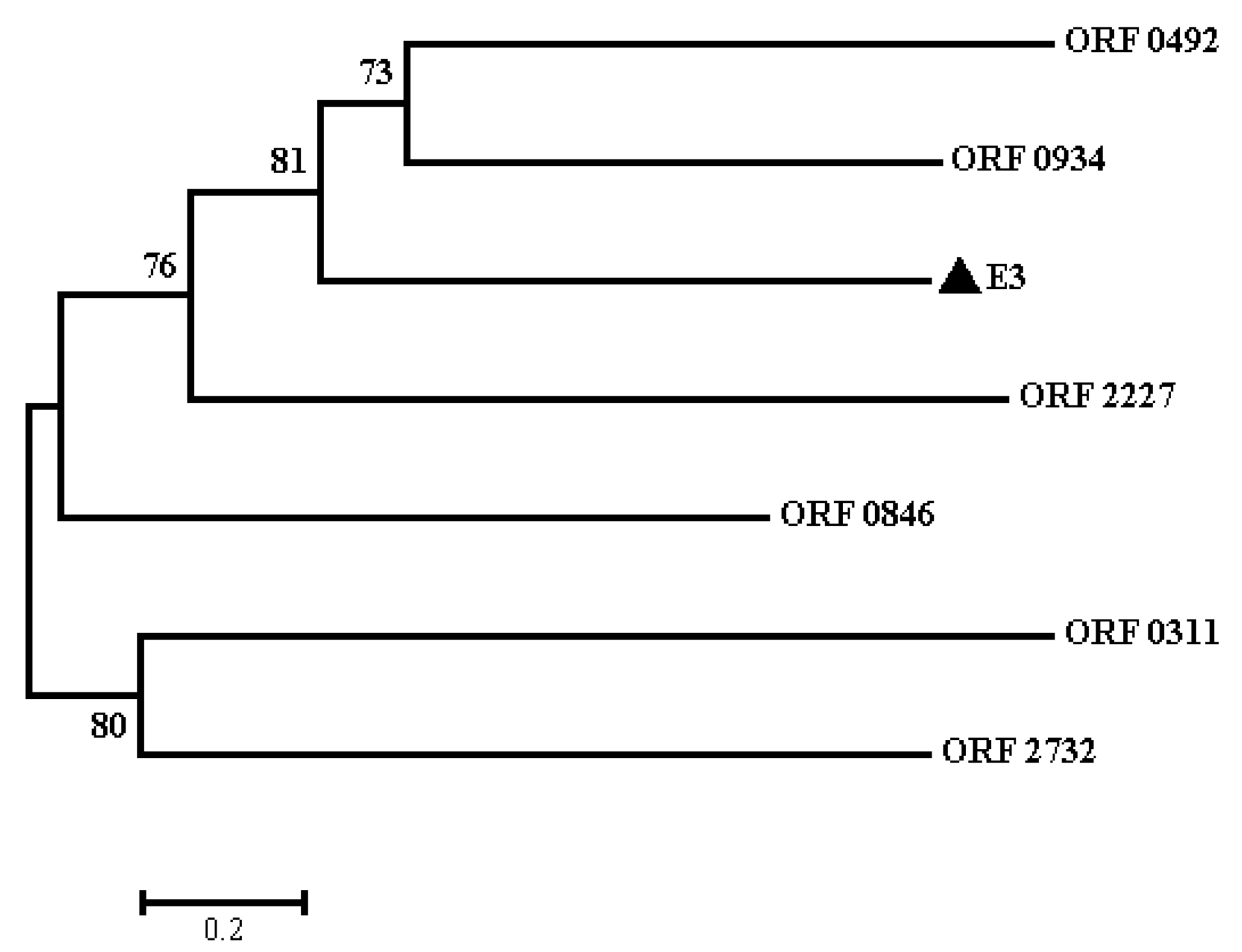
2.3. Protein Sequence and Phylogenetic Analyses of E3
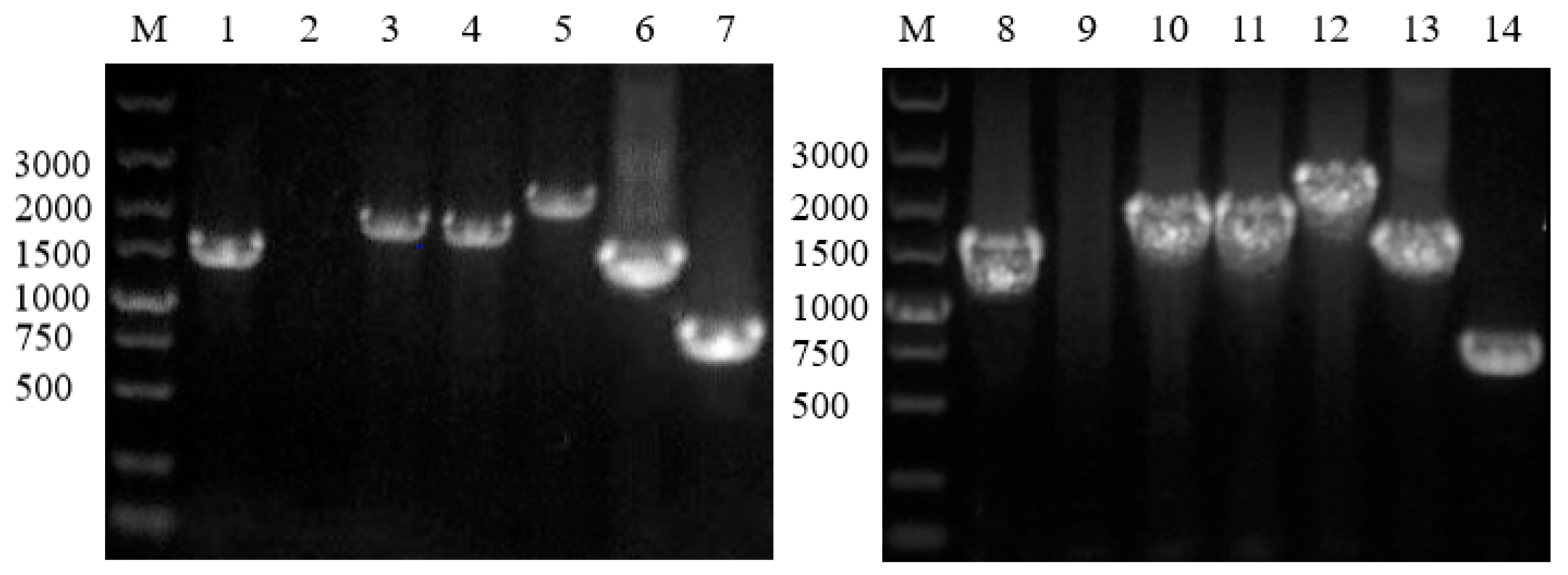
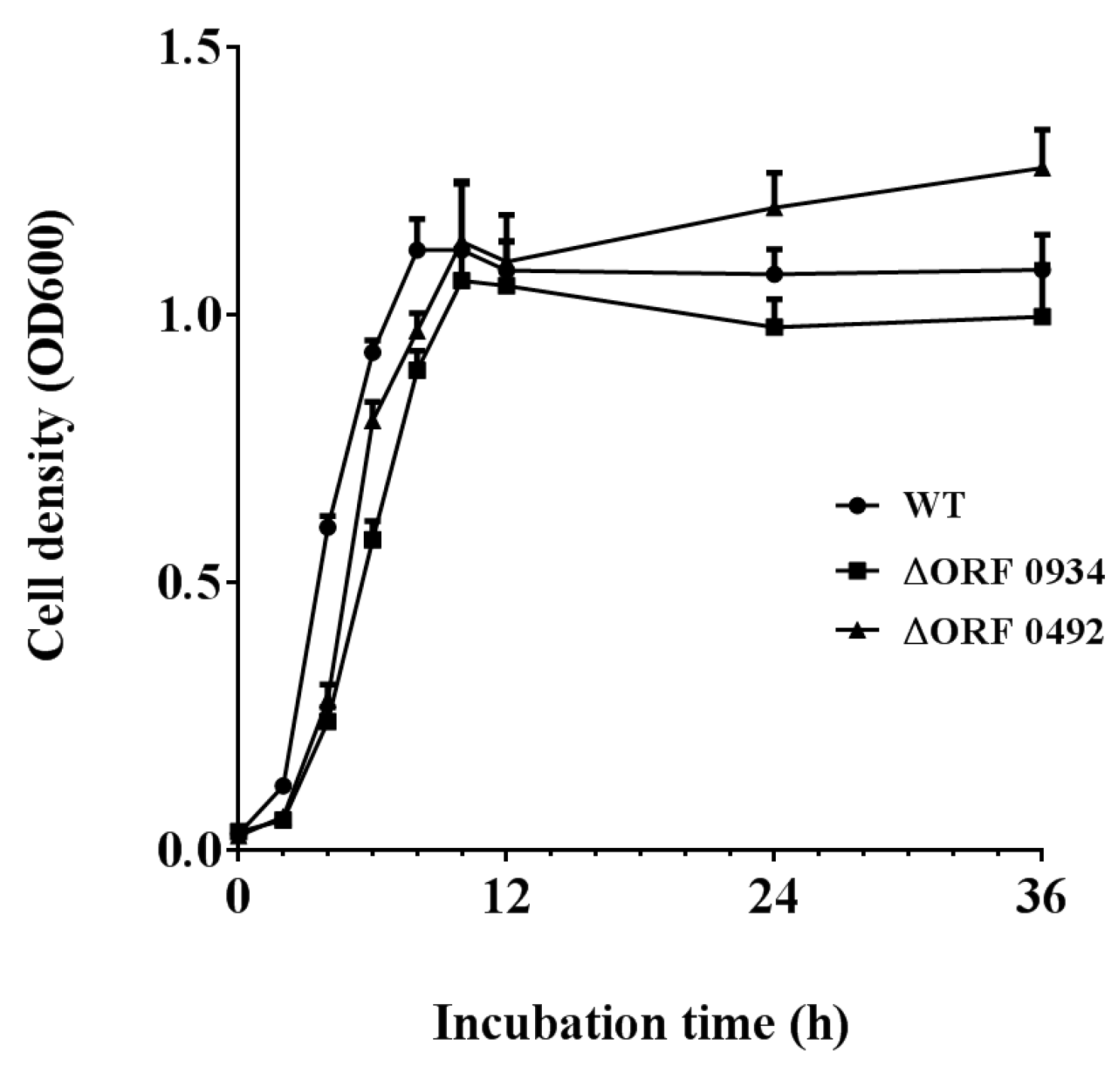
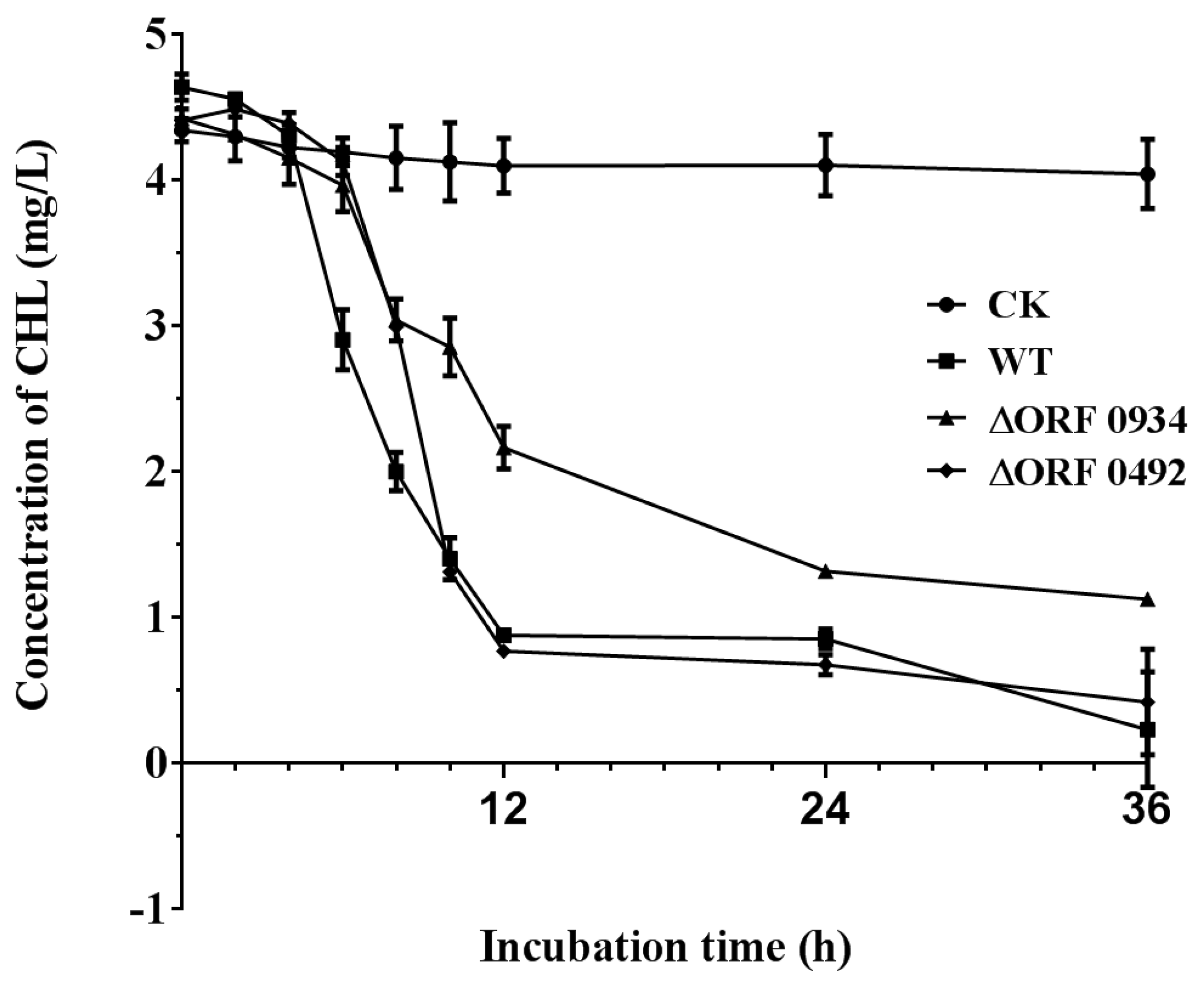
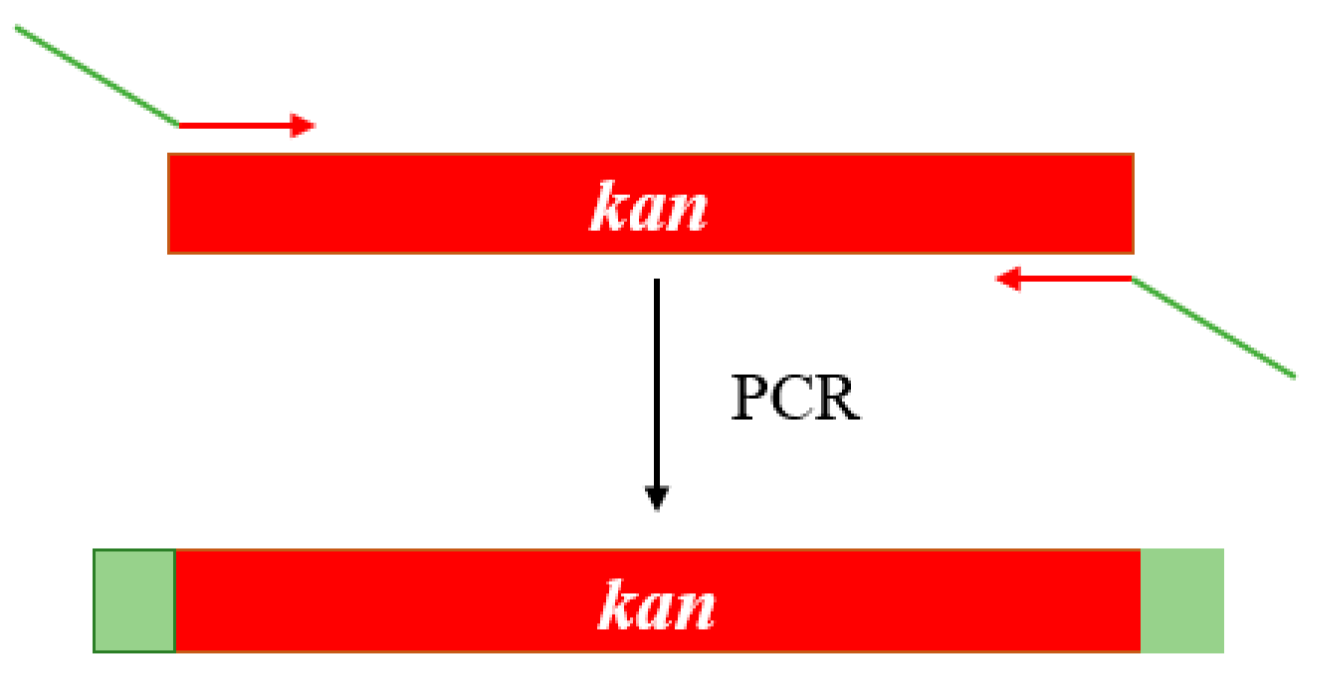
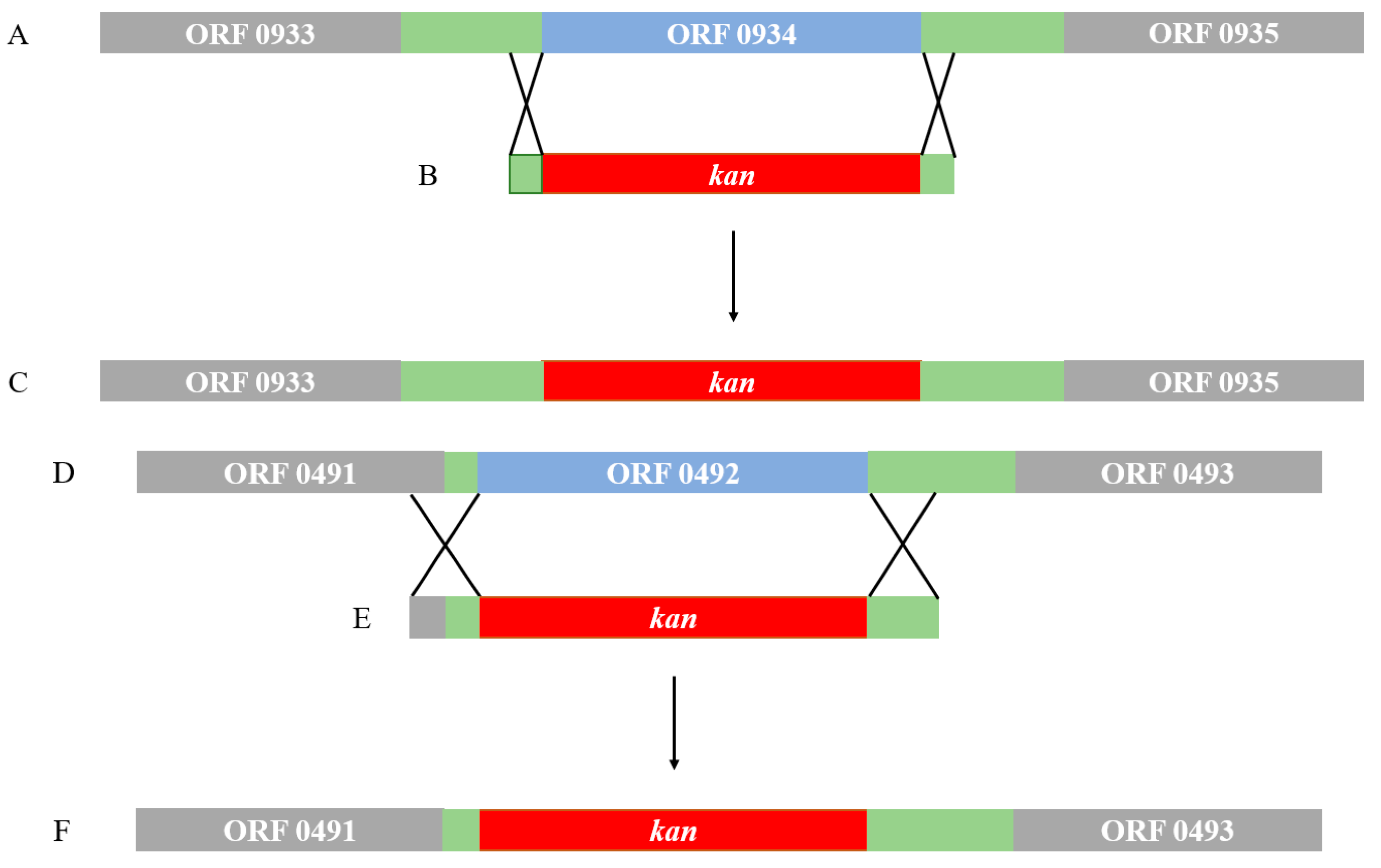
2.4. Gene Knockout and Degradation of Chlorimuron-ethyl
3. Materials and Methods
3.1. Bacterial Strain
3.2. Genome Sequencing, Assembly, and Annotation
3.3. Protein Structural and Phylogenetic Analyses
3.4. Gene Knockout and Degradation of Chlorimuron-Ethyl
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Zhang, X.; Mu, W.; Zhang, H. Bioremediation of chlorimuron-ethyl contaminated soil by strain 2N3. Chin. J. Pestic. Sci. 2010, 12, 49–53. [Google Scholar]
- Afzal, A.M.; Rasool, M.H.; Waseem, M.; Aslam, B. Assessment of heavy metal tolerance and biosorptive potential of Klebsiella variicola isolated from industrial effluents. AMB Express 2017, 7, 184. [Google Scholar] [CrossRef] [PubMed]
- Paczosa, M.K.; Mecsas, J. Klebsiella pneumoniae: Going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 2016, 80, 629–661. [Google Scholar] [CrossRef]
- Wang, S.; Yan, Y. Effects of relA gene knockout on biodegradation chlorpyrifos by Klebsiella sp. CPK. Chin. Biotechnol. 2010, 20, 38–41. [Google Scholar]
- Yang, F.; Yao, K.; Mi, L.; Liu, S.; Jia, D. Optimization of fermentation condition for Klebsiella oxytoca to degrade cypermethrin applying response surface methodology. Chin. Food Ferment. Ind. 2010, 36, 17–20. [Google Scholar]
- Guo, J.; Nie, M.; Xing, J.; Wu, X.; Chen, X. Effect of nitrogen sources on characterization of bioflocculant production by Klebsiella sp. NIII2. Chin. J. Environ. Eng. 2013, 7, 1996–2000. [Google Scholar]
- Li, A.; Pi, S.; Wei, W.; Chen, T.; Yang, J.; Ma, F. Adsorption behavior of tetracycline by extracellular polymeric substrates extracted from Klebsiella sp. J1. Environ. Sci. Pollut. Res. 2016, 23, 25084–25092. [Google Scholar] [CrossRef]
- Xing, J.; Yang, J.X.; Li, A.; Ma, F.; Liu, K.X.; Wu, D.; Wei, W. Removal efficiency and mechanism of Sulfamethoxazole in aqueous solution by bioflocculant MFX. J. Anal. Methods Chem. 2013, 2013, 568614. [Google Scholar] [CrossRef]
- Yang, J.; Wei, W.; Pi, S.; Ma, F.; Li, A.; Wu, D.; Xing, J. Competitive adsorption of heavy metals by extracellular polymeric substances extracted from Klebsiella sp. J1. Bioresour. Technol. 2015, 196, 533–539. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Mu, W.; Wang, J.; Pan, H.; Li, Y. Biodegradation of chlorimuron-ethyl by the bacterium Klebsiella jilinsis 2N3. J. Environ. Sci. Healthpart B 2010, 45, 501–507. [Google Scholar] [CrossRef]
- Pan, X.; Wang, S.; Shi, N.; Hua Fang, H.; Yunlong Yu, Y. Biodegradation and detoxification of chlorimuron-ethyl by Enterobacter ludwigii sp. CE-1. Ecotoxicol. Environ. Saf. 2018, 150, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zang, H.; Wang, H. Global transcriptomic analysis of, Rhodococcus erythropolis, D310-1 in responding to chlorimuron-ethyl. Ecotoxicol. Environ. Saf. 2018, 157, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Banerjee, K.; Choudhury, P.P. Degradation of chlorimuron-ethyl by Aspergillus niger isolated from agricultural soil. FEMS Microbiol. Lett. 2012, 337, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Chen, Y.F.; Fang, T.; Zhou, N.Y. Co-metabolic degradation of tribenuron methyl, a sulfonylurea herbicide, by Pseudomonas sp. strain nyz42. Int. Biodeterior. Biodegrad. 2013, 76, 36–40. [Google Scholar] [CrossRef]
- O’Keefe, D.P.; Harder, P.A. Occurrence and biological function of cytochrome P450 monooxygenases in the actinomycetes. Mol. Microbiol. 1991, 5, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Omer, C.A.; Lenstra, R.; Litle, P.J.; Dean, C.; Tepperman, J.M.; Leto, K.J.; Romesser, J.A.; O’Keefe, D.P. Genes for two herbicide-inducible cytochromes P-450 from Streptomyces griseolus. J. Bacteriol. 1990, 172, 3335–3345. [Google Scholar] [CrossRef] [PubMed]
- Song, J. Isolation and Identification of Nicosulfuron-Degradtive Fungus (Talaromyces flavus) and Study of Degradation Mechanism. Ph.D. Dissertation, Chinese Academy of Agricultural Sciences, Beijing, China, 2013. [Google Scholar]
- Hang, B.J.; Hong, Q.; Xie, X.T.; Huang, X.; Wang, C.H.; He, J.; Li, S.P. SulE, a sulfonylurea herbicide de-esterification esterase from Hansschlegelia zhihuaiae S113. Appl. Environ. Microbiol. 2012, 78, 1962–1968. [Google Scholar] [CrossRef]
- Zhou, S. Isolation, Classification, and Degradation Characterisics Study of Nicosulfuron-Degrading Strain. Ph.D. Dissertation, Huazhong Agricultural University, Wuhan, China, 2015. [Google Scholar]
- Pilato, V.D.; Chiarelli, A.; Boinett, C.J.; Riccobono, E.; Harris, S.R.; D’Andrea, M.M.; Thomson, N.R.; Rossolini, G.M.; Giani, T. Complete genome sequence of the first KPC-Type Carbapenemase-positive Proteus mirabilis strain from a bloodstream infection. Genome Announc. 2016, 4, e00607. [Google Scholar] [CrossRef]
- Zautner, A.E.; Bunk, B.; Pfeifer, Y.; Spröer, C.; Reichard, U.; Eiffert, H.; Scheithauer, S.; Groß, U.; Overmann, J.; Bohne, W. Monitoring microevolution of OXA-48-producing Klebsiella pneumoniae ST147 in a hospital setting by SMRT sequencing. J. Antimicrob. Chemother. 2017, 72, 2737–2744. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Shen, W.; Wang, G.; Zhou, Y. Whole genome sequence of a carbapenem-resistant hypermucoviscous Klebsiella pneumoniae isolate SWU01 with capsular serotype K47 that belongs to ST11 from a patient in China. J. Glob. Antimicrob. Resist. 2017, 11, 87–89. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Qian, Z.; Jiang, P.; Min, T.; Sun, C.; Huang, W. Cloning of an alkaline lipase gene from Penicillium cyclopium and its expression in Escherichia coli. Lipids 2003, 38, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yu, Q.; Lv, T.; Cheng, Y.; Feng, L.; Cheng, X.; Li, C. Insights into the degradation of chlorimuron-ethyl by Stenotrophomonas maltophila D310-3. Chemosphere 2016, 144, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, X.; Li, X.; Su, Z.; Zhang, C.; Zhang, H. Bioremediation of chlorimuron-ethyl-contaminated soil by Hansschlegelia sp. strain CHL1 and the changes of indigenous microbial population and N-cycling function genes during the bioremediation process. J. Hazard. Mater. 2014, 274, 314–321. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A. Third generation DNA sequencing: Pacific biosciences’ single molecule real time technology. Chem. Biol. 2010, 17, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Aboulliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 1, e112963. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopaedia of Genes and Genomes. Nucleic Acids Res. 2002, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | K. pneumoniae 2N3 |
|---|---|
| Length (bp) | 5,319,547 |
| GC content | 57.33% |
| Ns (%) | 0 |
| No. of plasmid | 0 |
| No. of coding genes | 5156 |
| Total bases (bp) | 4,669,044 |
| Length variation (range in bp) | 114–6342 |
| Average length (bp) | 906 |
| Ns (%) | 0 |
| No. of non-coding RNA | 112 |
| No. of ribosomal RNA | 25 |
| No. of transfer RNA | 87 |
| Primers | Sequences |
|---|---|
| FKF0934-F | 5′-gcggcctgataccagacgcggtctggtccaggatcggcgaatcaagctagacagggtaag TGTGTAGGCTGGAGCTG-3′ |
| FKF0934-R | 5′-tggcgcccagggcgctggcggaatgccattaacggcactctgcccggcaaagagggcaga ATCCTCCTTAGTTCCTATTCC-3′ |
| FKF0492-F | 5′-gatcgccggattcggctcgctgagcgccaccaccgagatttgattaaccgggagactaac TGTGTAGGCTGGAGCTG-3′ |
| FKF0492-R | 5′-tgttccgcgtcgcgcagctgccgggccagcgcgtcaagagaaaaagtcatcatttactcc ATCCTCCTTAGTTCCTATTCC-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Hao, Q.; Zhang, Z.; Zhang, X.; Pan, H.; Zhang, J.; Zhang, H.; Sun, F. Whole Genome Sequencing and Analysis of Chlorimuron-Ethyl Degrading Bacteria Klebsiella pneumoniae 2N3. Int. J. Mol. Sci. 2019, 20, 3053. https://doi.org/10.3390/ijms20123053
Zhang C, Hao Q, Zhang Z, Zhang X, Pan H, Zhang J, Zhang H, Sun F. Whole Genome Sequencing and Analysis of Chlorimuron-Ethyl Degrading Bacteria Klebsiella pneumoniae 2N3. International Journal of Molecular Sciences. 2019; 20(12):3053. https://doi.org/10.3390/ijms20123053
Chicago/Turabian StyleZhang, Cheng, Qingkai Hao, Zhengyi Zhang, Xianghui Zhang, Hongyu Pan, Jiahuan Zhang, Hao Zhang, and Fengjie Sun. 2019. "Whole Genome Sequencing and Analysis of Chlorimuron-Ethyl Degrading Bacteria Klebsiella pneumoniae 2N3" International Journal of Molecular Sciences 20, no. 12: 3053. https://doi.org/10.3390/ijms20123053
APA StyleZhang, C., Hao, Q., Zhang, Z., Zhang, X., Pan, H., Zhang, J., Zhang, H., & Sun, F. (2019). Whole Genome Sequencing and Analysis of Chlorimuron-Ethyl Degrading Bacteria Klebsiella pneumoniae 2N3. International Journal of Molecular Sciences, 20(12), 3053. https://doi.org/10.3390/ijms20123053