Proteomics-Metabolomics Combined Approach Identifies Peroxidasin as a Protector against Metabolic and Oxidative Stress in Prostate Cancer

 ,
,
Abstract
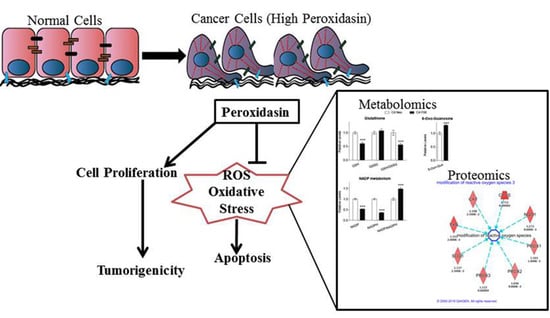
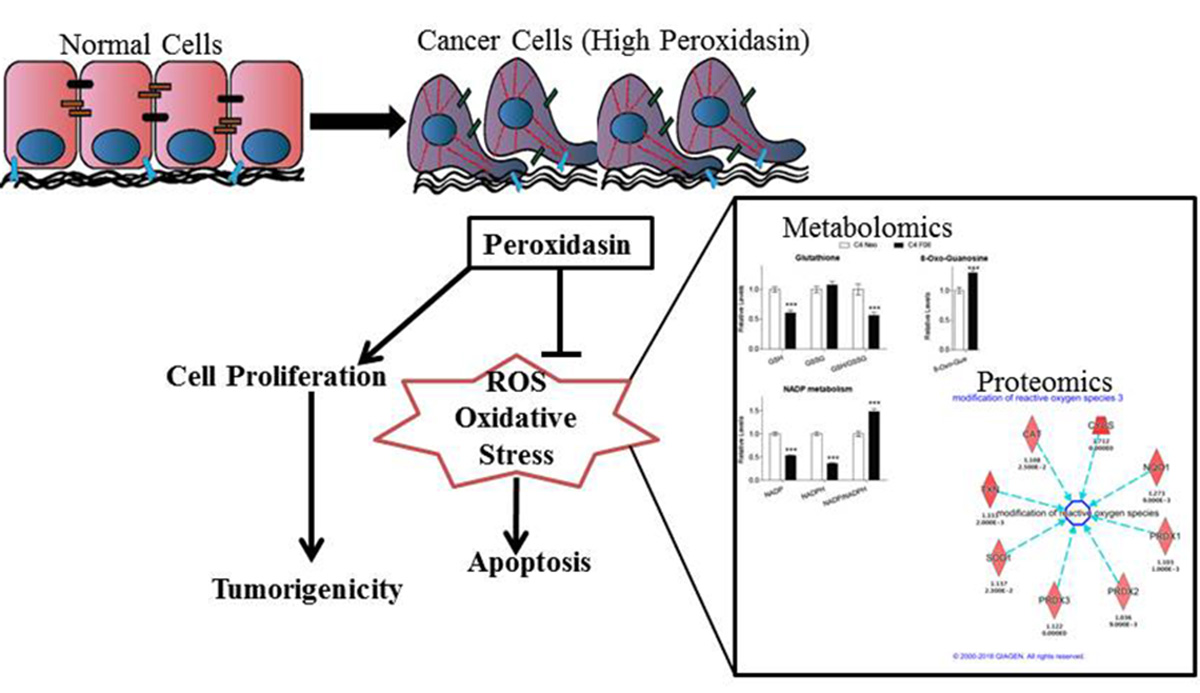
1. Introduction
2. Results
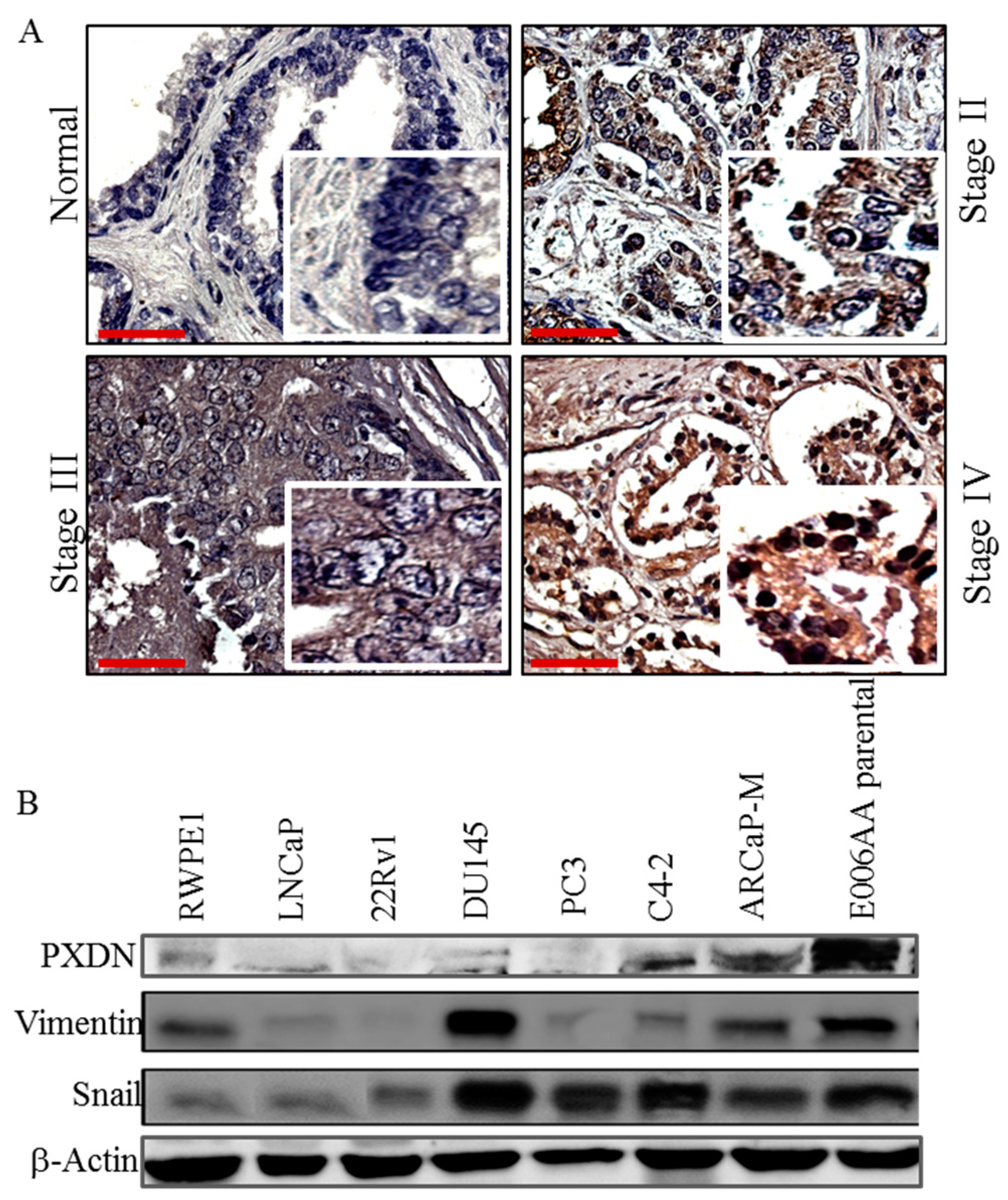
2.1. PXDN Expression Increases with Prostate Cancer Progression
2.2. PXDN Promotes Cell Viability and Tumorigenicity on Soft Agar
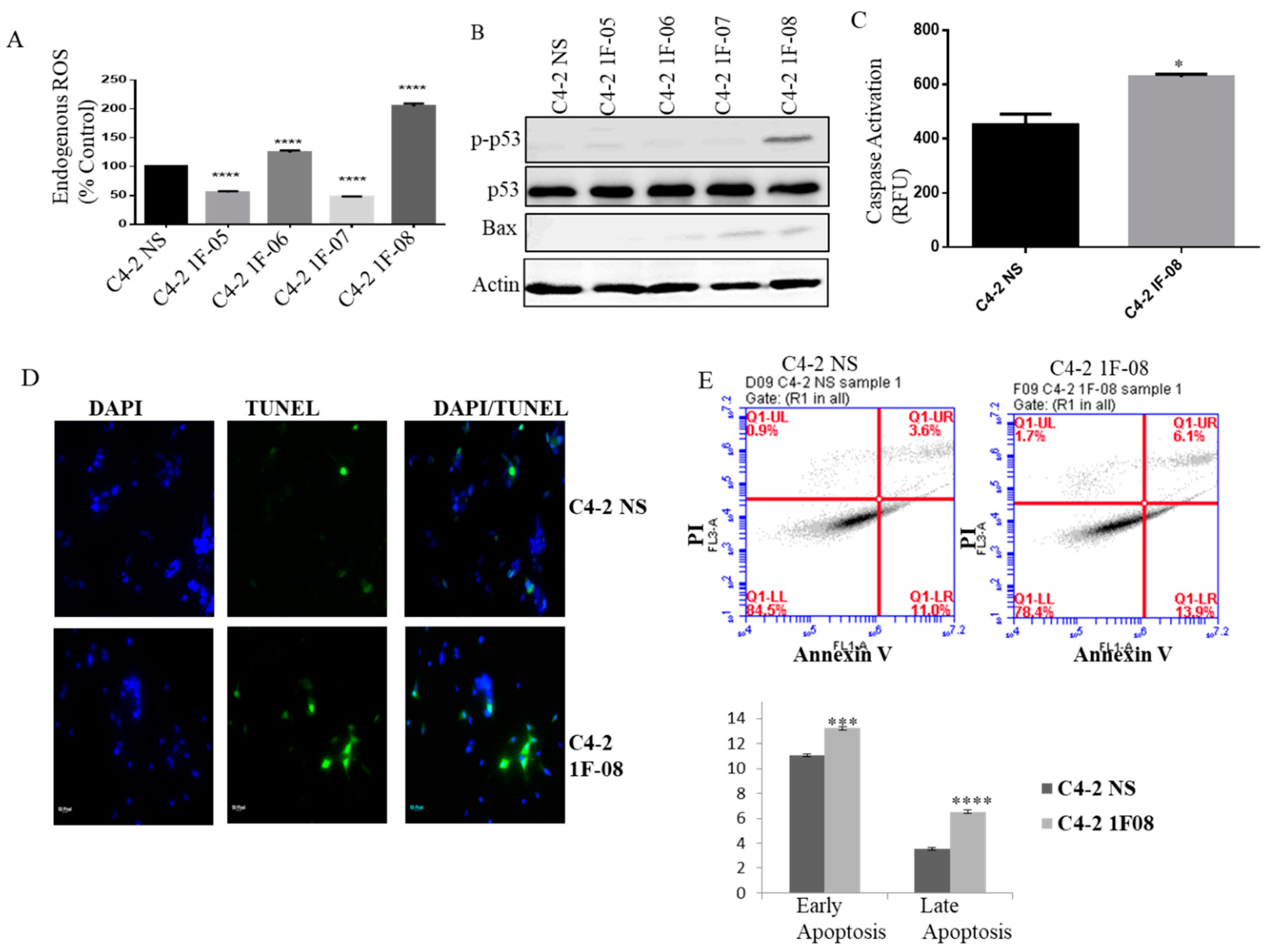
2.3. PXDN Scavenges H2O2 in Prostate Cancer Cells and Inhibits Apoptosis
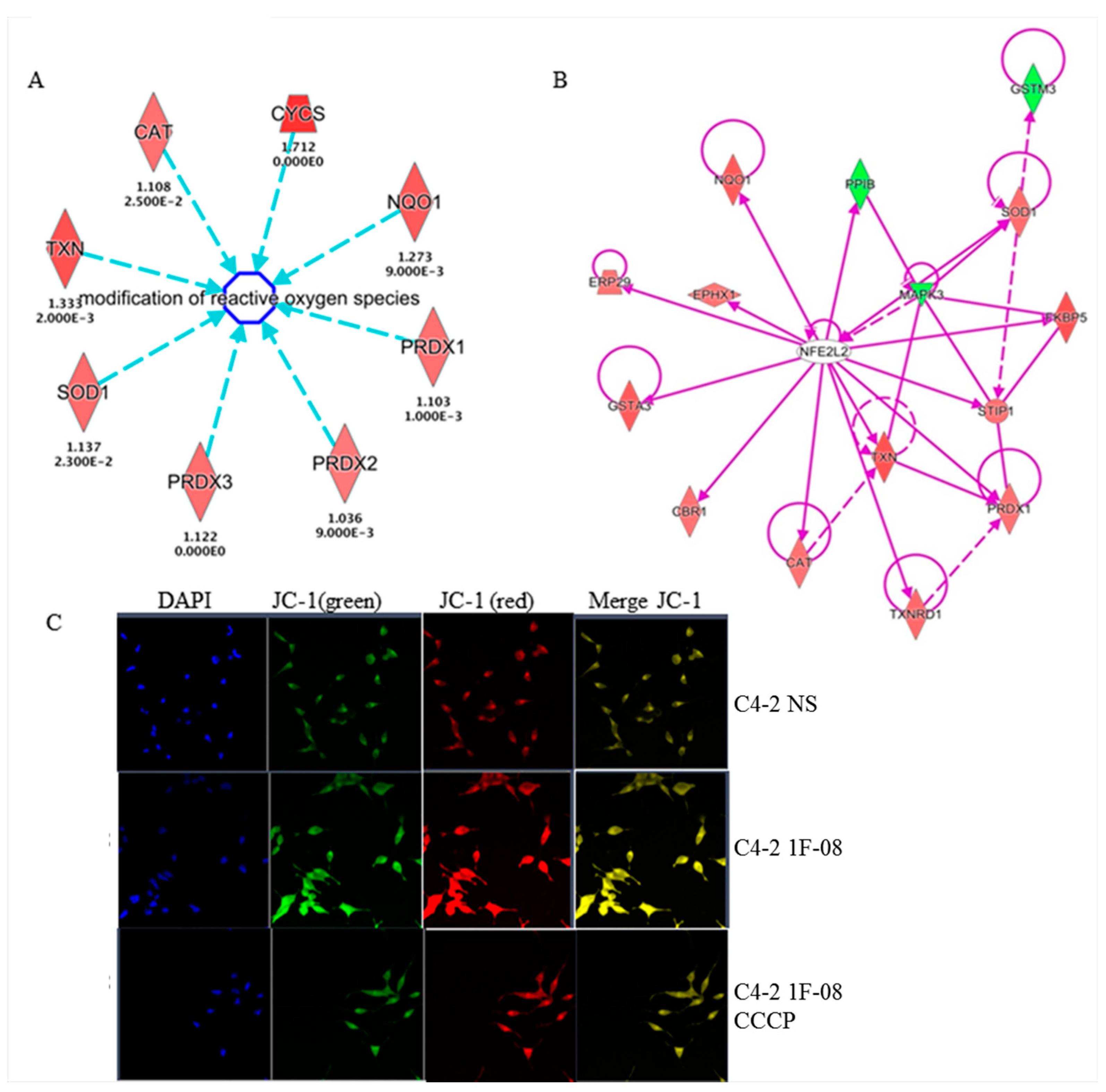
2.4. PXDN Regulates Oxidative Stress Pathways and Decreases Mitochondrial Membrane Potential
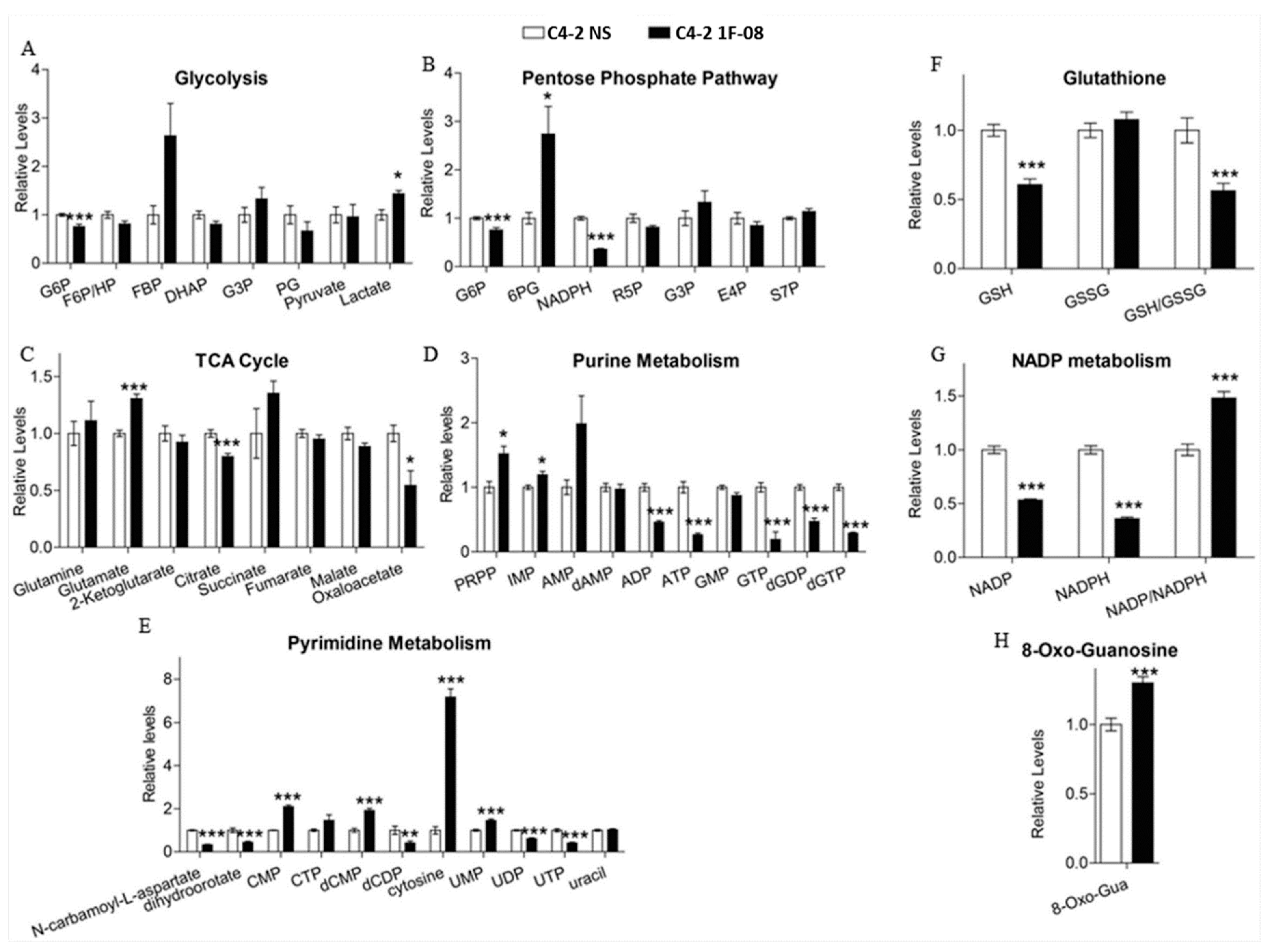
2.5. Metabolomic Analysis Indicates that PXDN Decreases Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Western Blot
4.3. Stable Knockdown of PXDN
4.4. Proteomic Profiling of C4-2 Cells with PXDN Knockdown
4.5. Metabolomics
4.6. Immunohistochemistry
4.7. Cell Viability Assays
4.8. ROS Assay
4.9. Apoptosis Assays
4.10. Detection of Membrane Potential
4.11. MitoCasp Assay
4.12. Soft Agar Colony Formation Assay
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| H2O2 | Hydrogen Peroxide |
| PXDN | Peroxidasin |
| ROS | Reactive Oxygen Species |
| ARCaP | Androgen-Repressed Human Prostate Cancer Cell Line |
| PRPP | Phosphoribosyl Pyrophosphate |
| IMP | Inosine Monophosphate |
| UDP | Uridine Diphosphate |
| UTP | Uridine Triphosphate |
Appendix A
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: Globocan sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Saylor, P.J.; Lee, R.J.; Smith, M.R. Emerging therapies to prevent skeletal morbidity in men with prostate cancer. J. Clin. Oncol. 2011, 29, 3709–3714. [Google Scholar] [CrossRef]
- Otto, S.; Schreyer, C.; Hafner, S.; Mast, G.; Ehrenfeld, M.; Sturzenbaum, S.; Pautke, C. Bisphosphonate-related osteonecrosis of the jaws—Characteristics, risk factors, clinical features, localization and impact on oncological treatment. J. Craniomaxillofac. Surg. 2012, 40, 303–309. [Google Scholar] [CrossRef]
- Dong, L.; Zieren, R.C.; Xue, W.; de Reijke, T.M.; Pienta, K.J. Metastatic prostate cancer remains incurable, why? Asian J. Urol. 2019, 6, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Nelson, R.E.; Fessler, L.I.; Takagi, Y.; Blumberg, B.; Keene, D.R.; Olson, P.F.; Parker, C.G.; Fessler, J.H. Peroxidasin: A novel enzyme-matrix protein of drosophila development. EMBO J. 1994, 13, 3438–3447. [Google Scholar] [CrossRef]
- Ma, Q.L.; Zhang, G.G.; Peng, J. Vascular peroxidase 1: A novel enzyme in promoting oxidative stress in cardiovascular system. Trends Cardiovasc. Med. 2013, 23, 179–183. [Google Scholar] [CrossRef]
- Rees, M.D.; Kennett, E.C.; Whitelock, J.M.; Davies, M.J. Oxidative damage to extracellular matrix and its role in human pathologies. Free Radic. Biol. Med. 2008, 44, 1973–2001. [Google Scholar] [CrossRef]
- Zheng, Y.Z.; Liang, L. High expression of pxdn is associated with poor prognosis and promotes proliferation, invasion as well as migration in ovarian cancer. Ann. Diagn. Pathol. 2018, 34, 161–165. [Google Scholar] [CrossRef]
- Di, Y.; Chen, D.; Yu, W.; Yan, L. Bladder cancer stage-associated hub genes revealed by wgcna co-expression network analysis. Hereditas 2019, 156, 7. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yang, X.; Jin, C.; Li, L.; Cui, Q.; Guo, Y.; Dong, Y.; Yang, X.; Guo, L.; Zhang, M. Identification and verification of differentially expressed micrornas and their target genes for the diagnosis of esophageal cancer. Oncol. Lett. 2018, 16, 3642–3650. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, A.; Prithviraj, P.; Lo, P.H.; Walkiewicz, M.; Anaka, M.; Woods, B.L.; Tan, B.; Behren, A.; Cebon, J.; McKeown, S.J. Identifying and targeting determinants of melanoma cellular invasion. Oncotarget 2016, 7, 41186–41202. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Carson-Walter, E.B.; Cooper, A.; Winans, B.N.; Johnson, M.D.; Walter, K.A. Vascular gene expression patterns are conserved in primary and metastatic brain tumors. J. Neuro-Oncol. 2010, 99, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Barnett, P.; Arnold, R.S.; Mezencev, R.; Chung, L.W.K.; Zayzafoon, M.; Odero-Marah, V. Snail-mediated regulation of reactive oxygen species in arcap human prostate cancer cells. Biochem. Biophys. Res. Commun. 2011, 404, 34–39. [Google Scholar] [CrossRef]
- Gillies, R.J.; Gatenby, R.A. Adaptive landscapes and emergent phenotypes: Why do cancers have high glycolysis? J. Bioenerg. Biomembr. 2007, 39, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Harris, A.L.; Sivridis, E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: A metabolic survival role for tumor-associated stroma. Cancer Res. 2006, 66, 632–637. [Google Scholar] [CrossRef]
- Li, H.; Cao, Z.; Moore, D.R.; Jackson, P.L.; Barnes, S.; Lambeth, J.D.; Thannickal, V.J.; Cheng, G. Microbicidal activity of vascular peroxidase 1 in human plasma via generation of hypochlorous acid. Infect. Immun. 2012, 80, 2528–2537. [Google Scholar] [CrossRef]
- Agarwala, K.L.; Nakamura, S.; Tsutsumi, Y.; Yamakawa, K. Down syndrome cell adhesion molecule dscam mediates homophilic intercellular adhesion. Brain Res. Mol. Brain Res. 2000, 79, 118–126. [Google Scholar] [CrossRef]
- Hardy, S.; Wong, N.N.; Muller, W.J.; Park, M.; Tremblay, M.L. Overexpression of the protein tyrosine phosphatase prl-2 correlates with breast tumor formation and progression. Cancer Res. 2010, 70, 8959–8967. [Google Scholar] [CrossRef]
- Stojnev, S.; Ristic-Petrovic, A.; Jankovic-Velickovic, L. Reactive oxygen species, apoptosis and cancer. Vojnosanit. Pregl. 2013, 70, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhu, X.Y.; Liu, L.M.; Meng, Z.Q. Solute carrier transporters: Potential targets for digestive system neoplasms. Cancer Manag. Res. 2018, 10, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.S.; Kan-Mitchell, J.; Minev, B.; Edman, C.; Deans, R.J. A novel melanoma gene (mg50) encoding the interleukin 1 receptor antagonist and six epitopes recognized by human cytolytic t lymphocytes. Cancer Res. 2000, 60, 6448–6456. [Google Scholar] [PubMed]
- Castronovo, V.; Waltregny, D.; Kischel, P.; Roesli, C.; Elia, G.; Rybak, J.-N.; Neri, D. A chemical proteomics approach for the identification of accessible antigens expressed in human kidney cancer. Mol. Cell. Proteom. 2006, 5, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, K.J.; Dalgleish, A.G. Chronic immune activation and inflammation as the cause of malignancy. Br. J. Cancer 2001, 85, 473–483. [Google Scholar] [CrossRef]
- Ames, B.N. Mutagenesis and carcinogenesis: Endogenous and exogenous factors. Environ. Mol. Mutagenesis 1989, 14, 66–77. [Google Scholar] [CrossRef]
- Frohlich, D.A.; McCabe, M.T.; Arnold, R.S.; Day, M.L. The role of nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 2008, 27, 4353–4362. [Google Scholar] [CrossRef]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. Perk/eif2alpha signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ros. Proc. Natl. Acad. Sci. USA 2013, 110, 4622–4627. [Google Scholar] [CrossRef]
- Lu, N.; Zhou, Z. Membrane trafficking and phagosome maturation during the clearance of apoptotic cells. Int. Rev. Cell Mol. Biol. 2012, 293, 269–309. [Google Scholar]
- He, H.; Kise, Y.; Izadifar, A.; Urwyler, O.; Ayaz, D.; Parthasarthy, A.; Yan, B.; Erfurth, M.L.; Dascenco, D.; Schmucker, D. Cell-intrinsic requirement of dscam1 isoform diversity for axon collateral formation. Science 2014, 344, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Hattori, D.; Chen, Y.; Matthews, B.J.; Salwinski, L.; Sabatti, C.; Grueber, W.B.; Zipursky, S.L. Robust discrimination between self and non-self neurites requires thousands of dscam1 isoforms. Nature 2009, 461, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Jin, Q.; Wang, X.D.; Zhu, H.J.; Ni, Q.C. Aldehyde dehydrogenase 1 expression is correlated with poor prognosis in breast cancer. Medicine 2017, 96, e7171. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, H.; Smeester, L.; Zou, F.; Kesic, M.; Jaspers, I.; Pi, J.; Fry, R.C. The nrf2-mediated oxidative stress response pathway is associated with tumor cell resistance to arsenic trioxide across the nci-60 panel. BMC Med. Genom. 2010, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dong, C. Gluconeogenesis in cancer: Function and regulation of pepck, fbpase, and g6pase. Trends Cancer 2019, 5, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Hu, C.; Yuan, Q.; Yang, T.; Peng, J.; Li, Y.; Bai, Y.; Cao, Z.; Cheng, G.; Zhang, G. Involvement of vascular peroxidase 1 in angiotensin ii-induced vascular smooth muscle cell proliferation. Cardiovasc. Res. 2011, 91, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Sabrautzki, S.; Horsch, M.; Fuchs, H.; Gailus-Durner, V.; Beckers, J.; Hrabe de Angelis, M.; Graw, J. Peroxidasin is essential for eye development in the mouse. Hum. Mol. Genet. 2014, 23, 5597–5614. [Google Scholar] [CrossRef] [PubMed]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686. [Google Scholar] [CrossRef]
- Burton, L.J.; Rivera, M.; Hawsawi, O.; Zou, J.; Hudson, T.; Wang, G.; Zhang, Q.; Cubano, L.; Boukli, N.; Odero-Marah, V. Muscadine grape skin extract induces an unfolded protein response-mediated autophagy in prostate cancer cells: A tmt-based quantitative proteomic analysis. PLoS ONE 2016, 11, e0164115. [Google Scholar] [CrossRef] [PubMed]
- Bajad, S.; Shulaev, V. Lc-ms-based metabolomics. Methods Mol. Biol. 2011, 708, 213–228. [Google Scholar] [PubMed]
- Burton, L.J.; Smith, B.A.; Smith, B.N.; Loyd, Q.; Nagappan, P.; McKeithen, D.; Wilder, C.L.; Platt, M.O.; Hudson, T.; Odero-Marah, V.A. Muscadine grape skin extract can antagonize snail-cathepsin l-mediated invasion, migration and osteoclastogenesis in prostate and breast cancer cells. Carcinogenesis 2015, 36, 1019–1027. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Fold Change |
|---|---|
| DSCAM (Down syndrome cell adhesion molecule); Plays a role in CNS development [19]. | 2.198 |
| PTP4A2 (protein tyrosine phosphatase type IVA, member 2, phosphatase of regenerating liver 2 (PRL2) [20] | 1.941 |
| CYCS (cytochrome c, somatic); Component of electron transport chain in mitochondria, also plays a role in apoptosis initiation [21]. | 1.712 |
| PRDX4 (peroxiredoxin 4); Reduces H202 and alkyl hydroxides to water and alcohol. Upregulated in prostate cancer [22]. | 1.663 |
| PTGES2-AS1 (prostaglandin E synthase 2- antisense RNA 1) | 1.572 |
| SRRM1 (serine/arginine repetitive matrix 1) | 1.57 |
| ALDH1A1 (aldehyde dehydrogenase 1 family member A1); Component of alcohol metabolism pathway. | −1.707 |
| HBA1/HBA2 (hemoglobin subunit alpha ½); Components of hemoglobin. | −1.684 |
| FECH (ferrochelatase); Enzyme involved in heme synthesis. | −1.632 |
| TUBA1B (tubulin alpha 1B); Upregulation associated with poor outcome in hepatocellular carcinoma. | −1.539 |
| SLC25A1 (solute carrier family 25); Regulates transport of citrate across inner membranes of mitochondria. Shown to play a role in inflammation induced by TNFα and IFNγ [23]. | −1.509 |
| Top Canonical Pathways | |||
|---|---|---|---|
| Name | p-Value | Overlap | Molecules from DE Proteins in PXDN KD Experiment in Pathway |
| NRF2-mediated Oxidative Stress Response | 2.49 × 10−8 | 8.3% (15/180) | GSTA3,PPIB,PRDX1,GSTM3,NQO1,SOD1,TXNRD1,ERP29, STIP1,MAPK3,CAT,TXN,FKBP5,CBR1,EPHX1 |
| phagosome maturation | 1.27 × 10−6 | 8.7% (11/127) | NSF,CALR,TUBA1B,PRDX1,TUBB4B,RAB7A,CANX,TUBB4A, TUBB,PRDX6,PRDX2 |
| EIF2 Signaling | 1.71 × 10−6 | 7.0% (13/187) | RPS28,EIF1,RPLP1,RPL13,RPL3,MAPK3,RPS27L,RPLP2,RPS10, RPS3,RPS12,RPL7,EIF3K |
| Mitochondrial Dysfunction | 1.82 × 10−6 | 6.9% (13/188) | SDHA,PRDX3,NDUFA5,NDUFA6,CAT,UQCRC2,CYCS,VDAC1, OGDH,NDUFS3,NDUFAB1,COX4I1,VDAC2 |
| Gluconeogenesis I | 2.63 × 10−6 | 15.2% (7/46) | PGK1,PGAM1,ALDOA,GAPDH,MDH1,MDH2,ALDOC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dougan, J.; Hawsawi, O.; Burton, L.J.; Edwards, G.; Jones, K.; Zou, J.; Nagappan, P.; Wang, G.; Zhang, Q.; Danaher, A.; et al. Proteomics-Metabolomics Combined Approach Identifies Peroxidasin as a Protector against Metabolic and Oxidative Stress in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 3046. https://doi.org/10.3390/ijms20123046
Dougan J, Hawsawi O, Burton LJ, Edwards G, Jones K, Zou J, Nagappan P, Wang G, Zhang Q, Danaher A, et al. Proteomics-Metabolomics Combined Approach Identifies Peroxidasin as a Protector against Metabolic and Oxidative Stress in Prostate Cancer. International Journal of Molecular Sciences. 2019; 20(12):3046. https://doi.org/10.3390/ijms20123046
Chicago/Turabian StyleDougan, Jodi, Ohuod Hawsawi, Liza J. Burton, Gabrielle Edwards, Kia Jones, Jin Zou, Peri Nagappan, Guangdi Wang, Qiang Zhang, Alira Danaher, and et al. 2019. "Proteomics-Metabolomics Combined Approach Identifies Peroxidasin as a Protector against Metabolic and Oxidative Stress in Prostate Cancer" International Journal of Molecular Sciences 20, no. 12: 3046. https://doi.org/10.3390/ijms20123046
APA StyleDougan, J., Hawsawi, O., Burton, L. J., Edwards, G., Jones, K., Zou, J., Nagappan, P., Wang, G., Zhang, Q., Danaher, A., Bowen, N., Hinton, C., & Odero-Marah, V. A. (2019). Proteomics-Metabolomics Combined Approach Identifies Peroxidasin as a Protector against Metabolic and Oxidative Stress in Prostate Cancer. International Journal of Molecular Sciences, 20(12), 3046. https://doi.org/10.3390/ijms20123046