Luminescence- and Fluorescence-Based Complementation Assays to Screen for GPCR Oligomerization: Current State of the Art

 ,
,
Abstract
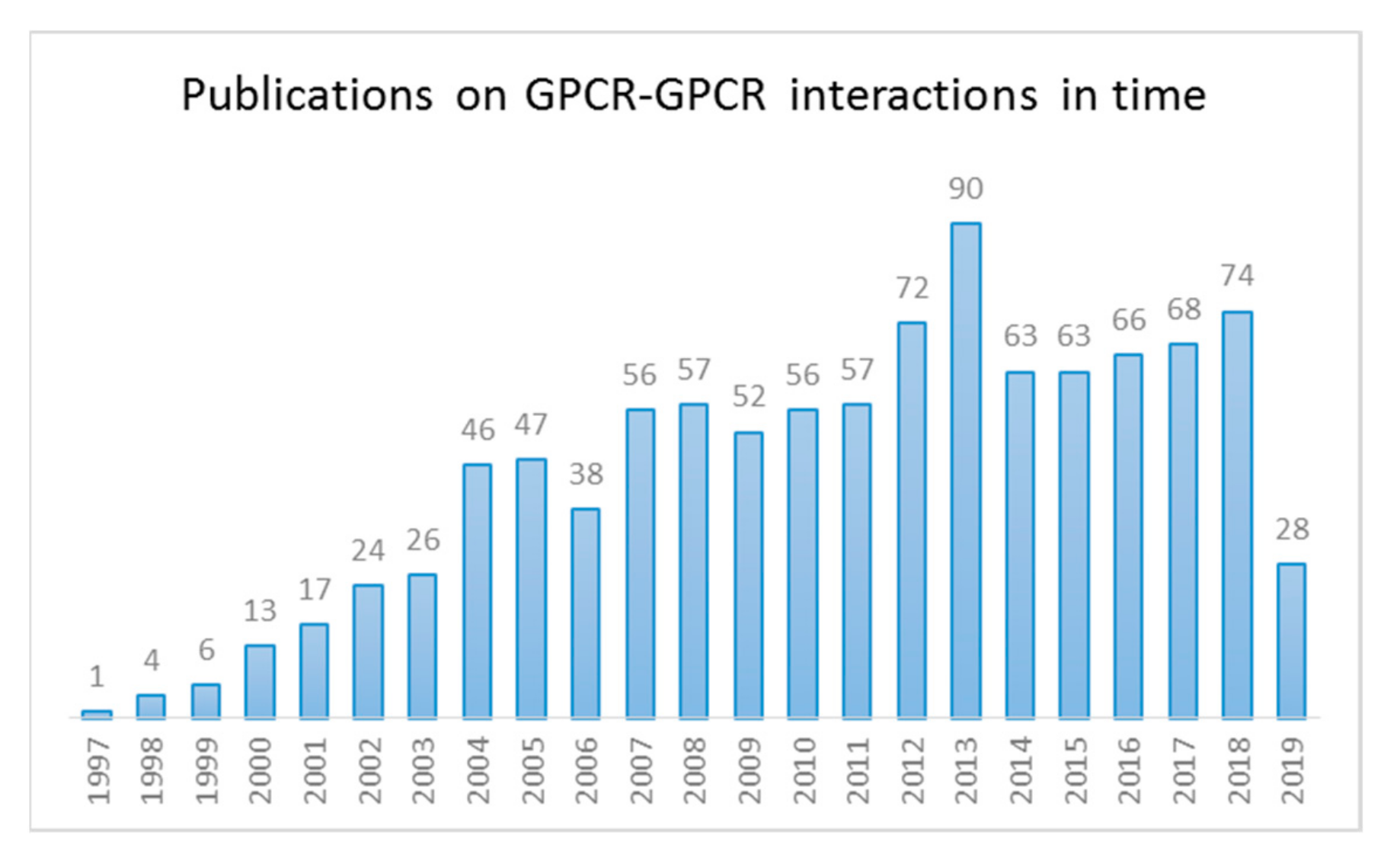
1. Introduction
1.1. GPCR Dimerization
1.2. Studying GPCR–GPCR Interactions: Biochemical Methods
1.3. Protein Complementation Assays
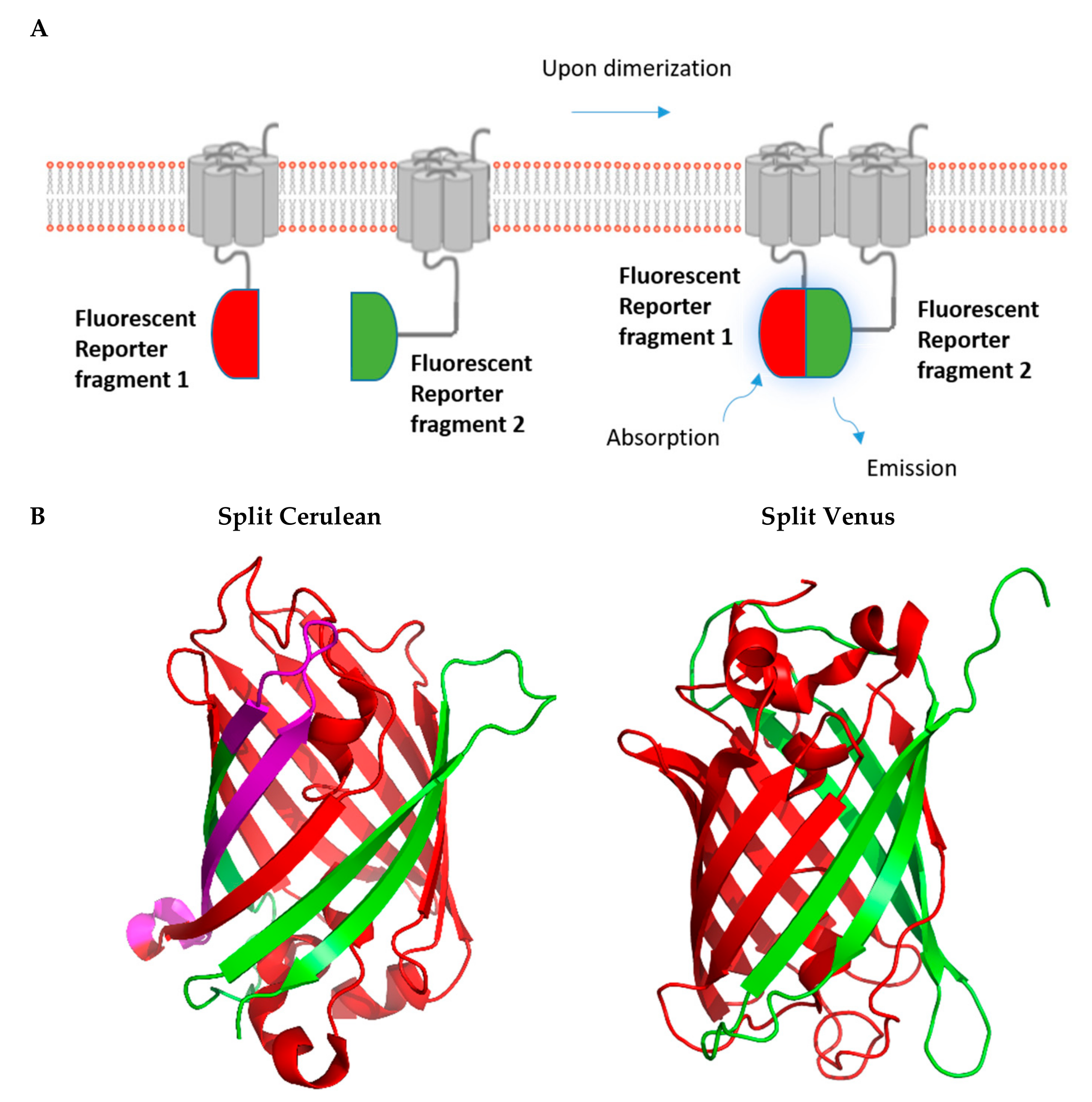
2. Fluorescence-Based Complementation Assays
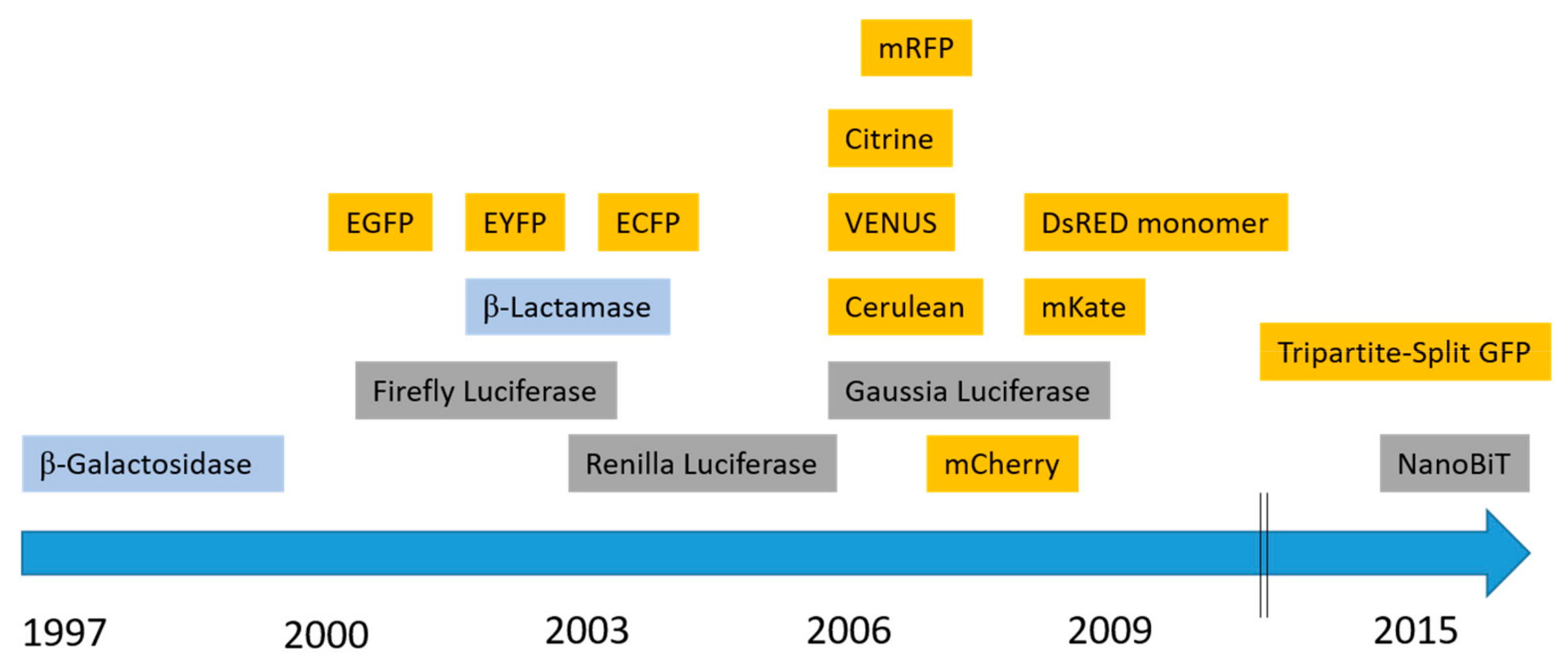
2.1. Fluorescent Proteins
2.1.1. Green Fluorescent Protein (GFP)
2.1.2. Yellow Fluorescent Protein (YFP) Variants
2.1.3. Cyan Fluorescent Protein (CFP)
2.1.4. Red, Far-Red and Near-Infrared Fluorescent Proteins
2.2. BiFC Assays
2.2.1. MBiFC
2.2.2. BiFC-RET
2.3. Ligand-Dependent Modulation of Dimerization
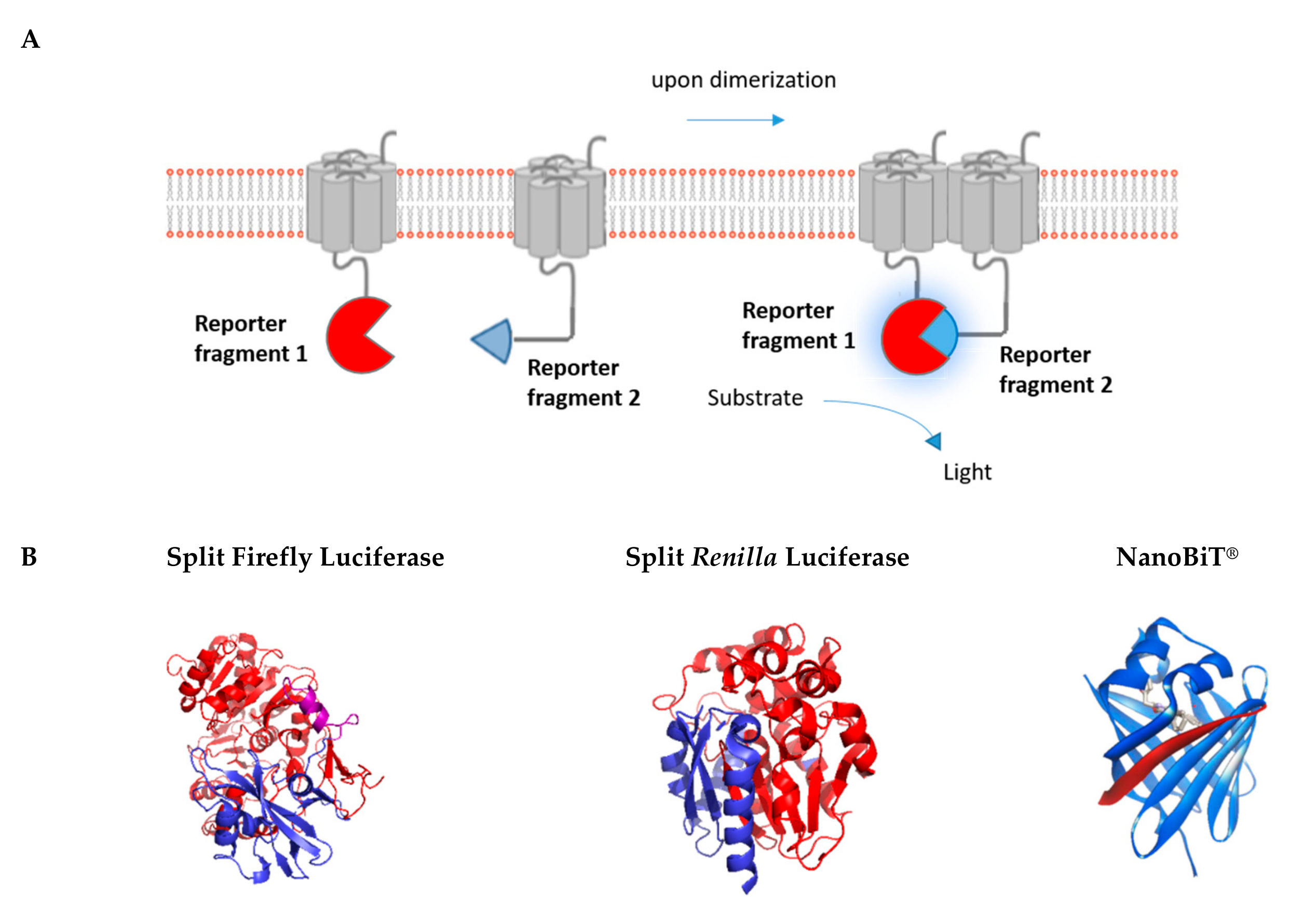
3. Luminescence-Based Complementation Assays
3.1. Renilla/Firefly Luciferase
3.2. NanoLuciferase
3.3. BiLC-RET
4. Combinatorial Assays: BiFC and BiLC
5. Comparison of Split Protein Approaches
5.1. Advantages of PCA
5.2. Limitations of PCA
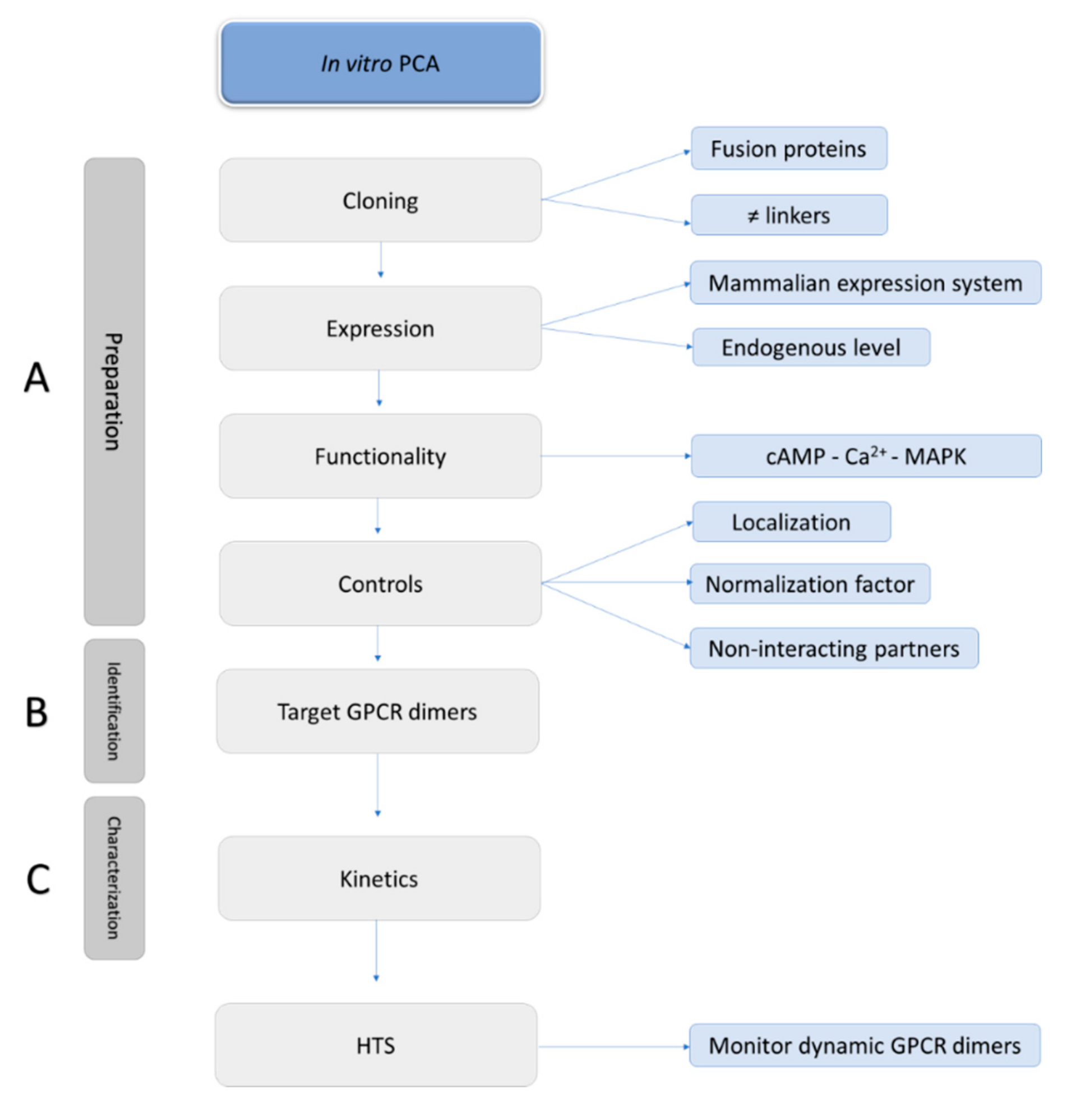
6. Guidelines to Perform Accurate PCA-Based Assays
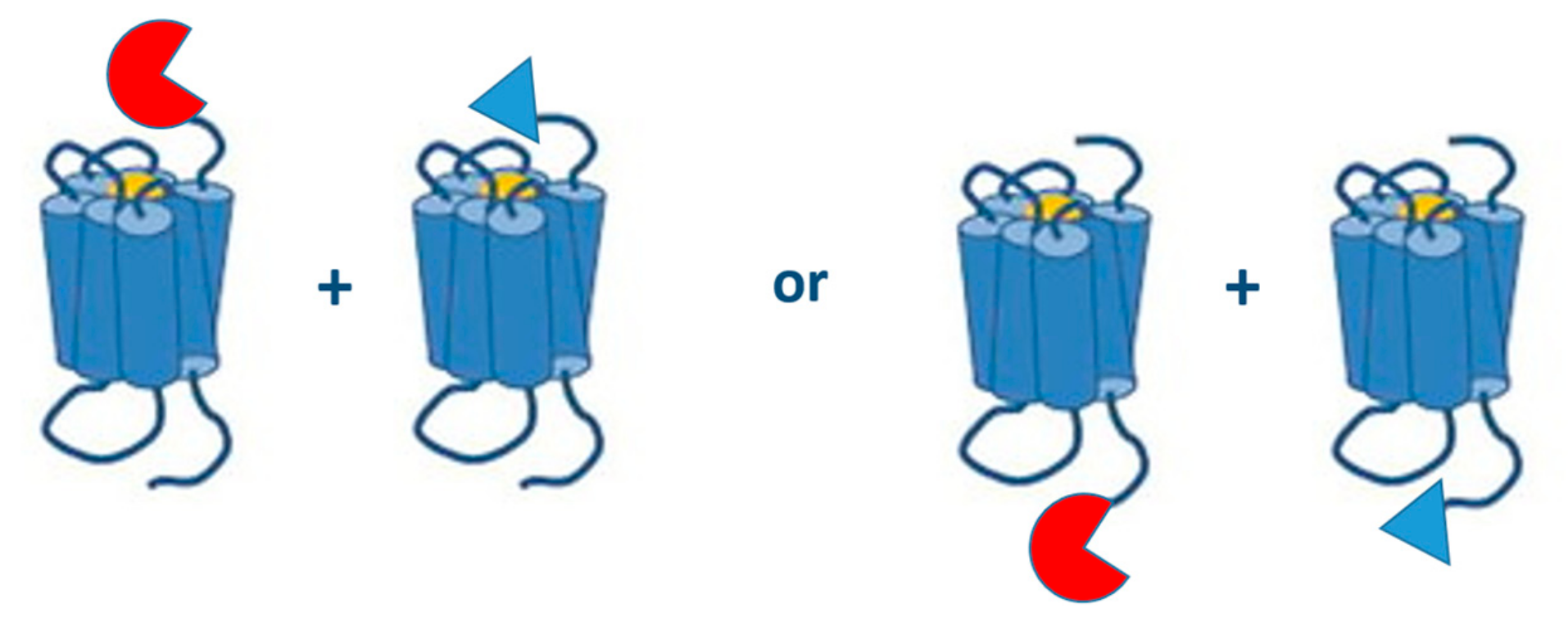
6.1. Possible Fusions
6.1.1. Selection of the Reporter System
6.1.2. (N- or) C-Terminally Tagged GPCRs
6.1.3. Linkers
6.2. Functionality and Localization of the Fusion Proteins
6.3. Non-Interacting Partners
6.4. Normalization Factor
6.5. Endogenous Expression Levels
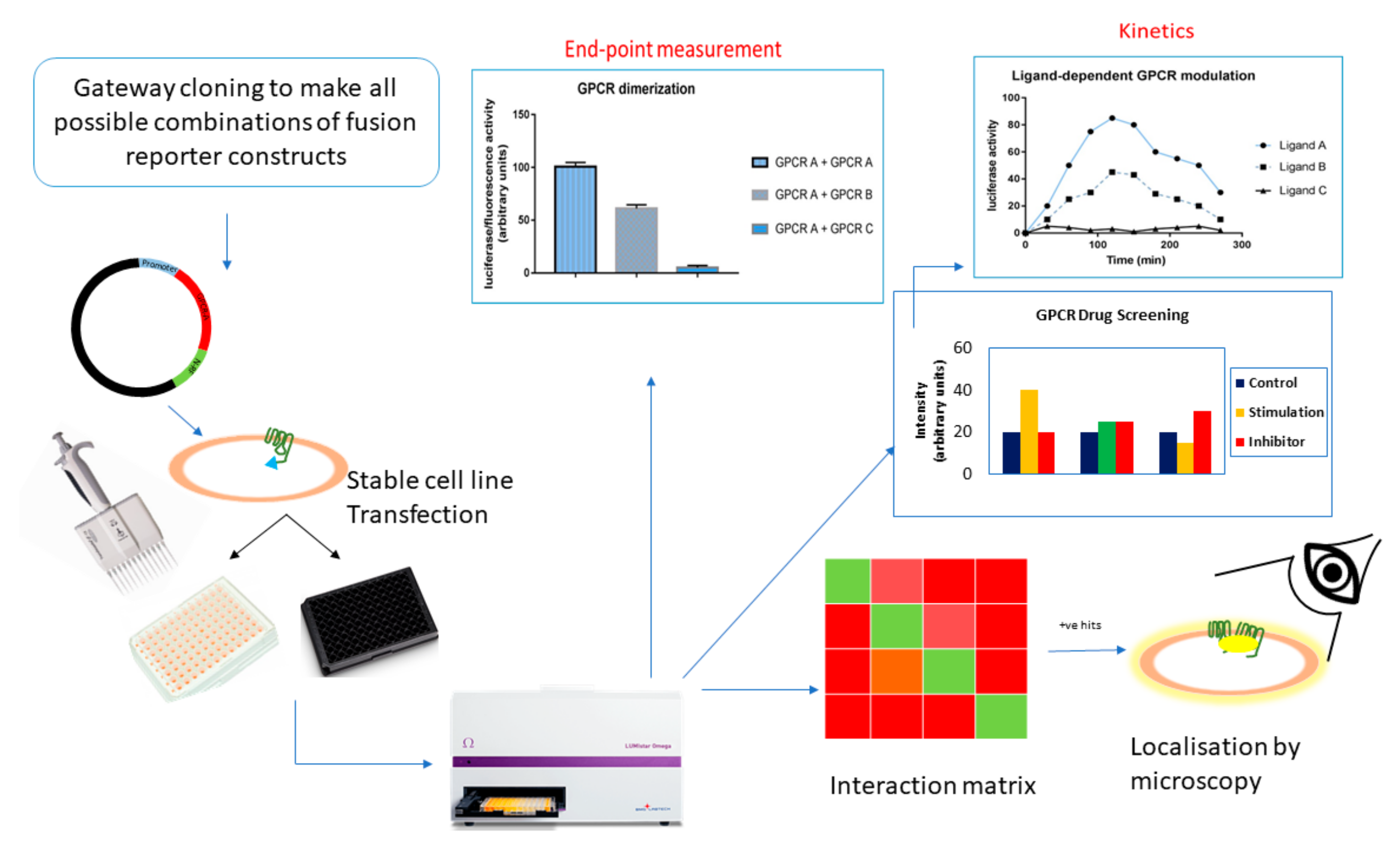
6.6. Kinetics
7. HTS with Cell-Based PCAs
7.1. GPCR Oligomerization Screening
7.2. GPCR Drug Discovery
8. In Vivo Application
9. Conclusions
Funding
Conflicts of Interest
Abbreviations
| A1/A2A | Adenosine receptor type 1/type 2A |
| α1b | α1b adrenergic receptor |
| AT1/AT2 | Angiotensin II receptor type 1/type 2 |
| β2AR | β2 Adrenergic receptor |
| BiFC | Bimolecular fluorescence complementation assay |
| BiLC | Bimolecular luminescence complementation assay |
| BRET | Bioluminescence resonance energy transfer |
| CAD | Cath.-a-differentiated |
| cAMP | Cyclic adenosine monophosphate |
| CB1 | Cannabinoid receptor 1 |
| CC | C-terminal fragment of split Cerulean |
| CFP | Cerulean fluorescent protein |
| CC2 | C-C chemokine receptor type 2 |
| CFLuc | C-terminal fragment of split Firefly luciferase |
| CGFP | C-terminal fragment of split green fluorescent protein |
| CID | Chemically induced dimerization |
| CN | N-terminal fragment of split Cerulean |
| CODA-RET | Complemented donor–acceptor resonance energy transfer |
| co-IP | Co-immunoprecipitation |
| CPA | N6-cyclopentyladenosine |
| CXCR4/CXCR7 | C-X-C chemokine receptor type 4/type 7 |
| D1R/D2R/D3R | Dopamine receptor type 1/type 2/type 3 |
| D2LR | Dopamine receptor type 2 long isoform |
| D2SR | Dopamine receptor type 2 short isoform |
| DsRed | Discosoma Red |
| ECL | Extracellular loop |
| EGFP | Enhanced green fluorescent protein |
| FLuc | Firefly Luciferase |
| FFAR2/3 | Free fatty acid receptor type 2/type 3 |
| FRB | FKBP-rapamycin binding domain of mTOR |
| FKBP12 | FK506- and rapamycin-binding protein |
| FRET | Förster resonance energy transfer |
| GHSR1a | Growth hormone secretagogue receptor 1α |
| GLuc | Gaussia princeps luciferase |
| GPCR | G protein-coupled receptor |
| GRK | G protein-coupled receptor kinase |
| HEK | Human embryonic kidney |
| Homo-FC | Homo-molecular fluorescence complementation |
| 5-HTR | 5-hydroxytryptamine receptor |
| HTS | High-throughput screening |
| L-DOPA | L-3,4-dihydroxyphenylalanine |
| LcBiT | C-terminal fragment of split large BiT |
| LgBiT | Large BiT, large subunit of Nanoluciferase |
| LnBiT | N-terminal fragment of split large BiT |
| M1/M2/M4 | Muscarinic acetylcholine receptor type 1/type 2/type 4 |
| MAPK | Mitogen activated protein kinase |
| MBiFC | Multicolor Bimolecular fluorescence complementation assay |
| MC4R | Melanocortin 4 receptor |
| MOP | Mu-opioid receptor |
| MRAP2 | Melanocortin 2 receptor accessory protein 2 |
| α-MSH | α-Melanocyte- stimulating hormone |
| mGluR2/mGLuR5/mGluR6 | Metabotropic glutamate receptor type 2/type 5/type 6 |
| mRFP | Monomer red fluorescent protein |
| NanoBiT® | NanoLuc Binary Technology® |
| NFLuc | N-terminal fragment of split Firefly luciferase |
| NGFP | N-terminal fragment of split green fluorescent protein |
| NPFF2 | Neuropeptide FF receptor 2 |
| NPY | Neuropeptide Y |
| OX1R | Orexin 1 receptor |
| PCA | Protein complementation assay |
| PLA | Proximity ligation assay |
| PPI | Protein–protein interaction |
| P2RY1 | P2Y purinoceptor 1 |
| RLuc | Renilla Luciferase |
| SDS-PAGE | Sodium dodecyl sulfate–polyacrylamide gel electrophoresis |
| SH3 | SRC Homology 3 Domain |
| SmBiT | Small BiT, small subunit of Nanoluciferase |
| SpIDA | Spatial intensity distribution analysis |
| SRET | Sequential resonance energy transfer |
| TAT | Transactivator of transcription |
| TM | Transmembrane |
| TSHr | Thyrotropin receptor |
| VC | C-terminal fragment of split Venus |
| VN | N-terminal fragment of split Venus |
| YC | C-terminal fragment of split yellow fluorescent protein |
| YFP | Yellow fluorescent protein |
| YN | N-terminal fragment of split yellow fluorescent protein |
References
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Foord, S.M.; Bonner, T.I.; Neubig, R.R.; Rosser, E.M.; Pin, J.P.; Davenport, A.P.; Spedding, M.; Harmar, A.J. International Union of Pharmacology. XLVI. G protein-coupled receptor list. Pharmacol. Rev. 2005, 57, 279–288. [Google Scholar] [CrossRef]
- Bjarnadottir, T.K.; Gloriam, D.E.; Hellstrand, S.H.; Kristiansson, H.; Fredriksson, R.; Schioth, H.B. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 2006, 88, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Resina, I.; Aguinaga, D.; Labandeira-Garcia, J.L.; Lanciego, J.L.; Navarro, G.; Franco, R. Usefulness of identifying G-protein-coupled receptor dimers for diagnosis and therapy of neurodegenerative diseases and of gliomas. Histol. Histopathol. 2018, 11963. [Google Scholar] [CrossRef]
- Zhao, J.; Deng, Y.; Jiang, Z.; Qing, H. G Protein-Coupled Receptors (GPCRs) in Alzheimer’s Disease: A Focus on BACE1 Related GPCRs. Front. Aging Neurosci. 2016, 8, 58. [Google Scholar] [CrossRef] [PubMed]
- Tomita, H.; Ziegler, M.E.; Kim, H.B.; Evans, S.J.; Choudary, P.V.; Li, J.Z.; Meng, F.; Dai, M.; Myers, R.M.; Neal, C.R.; et al. G protein-linked signaling pathways in bipolar and major depressive disorders. Front. Genet. 2013, 4, 297. [Google Scholar] [CrossRef]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef]
- Nieto Gutierrez, A.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell. Signal. 2018, 41, 65–74. [Google Scholar] [CrossRef]
- Huang, Y.H.; Todd, N.; Thathiah, A. The role of GPCRs in neurodegenerative diseases: Avenues for therapeutic intervention. Curr. Opin. Pharmacol. 2017, 32, 96–110. [Google Scholar] [CrossRef]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41.e19–54.e19. [Google Scholar] [CrossRef] [PubMed]
- Waldhoer, M.; Fong, J.; Jones, R.M.; Lunzer, M.M.; Sharma, S.K.; Kostenis, E.; Portoghese, P.S.; Whistler, J.L. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. USA 2005, 102, 9050–9055. [Google Scholar] [CrossRef] [PubMed]
- Vinals, X.; Moreno, E.; Lanfumey, L.; Cordomi, A.; Pastor, A.; de La Torre, R.; Gasperini, P.; Navarro, G.; Howell, L.A.; Pardo, L.; et al. Cognitive Impairment Induced by Delta9-tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLoS Biol. 2015, 13, e1002194. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.C.; Vasudevan, L.; Huysentruyt, J.; Risseeuw, M.D.P.; Stove, C.; Vanderheyden, P.M.L.; Van Craenenbroeck, K.; Van Calenbergh, S. Design, Synthesis, and Biological Evaluation of Bivalent Ligands Targeting Dopamine D-2-Like Receptors and the -Opioid Receptor. Chemmedchem 2018, 13, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Wouters, E.; Dalton, J.A.R.; Risseeuw, M.D.P.; Crans, R.A.J.; Stove, C.; Giraldo, J.; Van Craenenbroeck, K.; Van Calenbergh, S. Synthesis toward Bivalent Ligands for the Dopamine D2 and Metabotropic Glutamate 5 Receptors. J. Med. Chem. 2018, 61, 8212–8225. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, K.; Rual, J.F.; Vazquez, A.; Stelzl, U.; Lemmens, I.; Hirozane-Kishikawa, T.; Hao, T.; Zenkner, M.; Xin, X.F.; Goh, K.I.; et al. An empirical framework for binary interactome mapping. Nat. Methods 2009, 6, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T.; et al. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef] [PubMed]
- AbdAlla, S.; Lother, H.; el Massiery, A.; Quitterer, U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat. Med. 2001, 7, 1003–1009. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P. Dopamine D2 receptors as treatment targets in schizophrenia. Clin. Schizophr. Relat. Psychoses 2010, 4, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Renner, U.; Zeug, A.; Woehler, A.; Niebert, M.; Dityatev, A.; Dityateva, G.; Gorinski, N.; Guseva, D.; Abdel-Galil, D.; Frohlich, M.; et al. Heterodimerization of serotonin receptors 5-HT1A and 5-HT7 differentially regulates receptor signalling and trafficking. J. Cell Sci. 2012, 125, 2486–2499. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Romero-Fernandez, W.; Perez-Alea, M.; Narvaez, M.; Tarakanov, A.O.; Mudo, G.; Agnati, L.F.; Ciruela, F.; Belluardo, N.; Fuxe, K. The existence of FGFR1-5-HT1A receptor heterocomplexes in midbrain 5-HT neurons of the rat: relevance for neuroplasticity. J. Neurosci. 2012, 32, 6295–6303. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Corrales, F.; Narvaez, M.; Oflijan, J.; Agnati, L.F.; Palkovits, M.; Fuxe, K. Dynamic modulation of FGFR1-5-HT1A heteroreceptor complexes. Agonist treatment enhances participation of FGFR1 and 5-HT1A homodimers and recruitment of beta-arrestin2. Biochem. Biophys. Res. Commun. 2013, 441, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Perez-Alea, M.; Narvaez, M.; Tarakanov, A.O.; Mudo, G.; Jimenez-Beristain, A.; Agnati, L.F.; Ciruela, F.; Belluardo, N.; Fuxe, K. Enhancement of the FGFR1 signaling in the FGFR1-5-HT1A heteroreceptor complex in midbrain raphe 5-HT neuron systems. Relevance for neuroplasticity and depression. Biochem. Biophys. Res. Commun. 2015, 463, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Borroto-Escuela, D.O.; Tarakanov, A.O.; Fuxe, K. FGFR1-5-HT1A Heteroreceptor Complexes: Implications for Understanding and Treating Major Depression. Trends Neurosci. 2016, 39, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Millon, C.; Flores-Burgess, A.; Narvaez, M.; Borroto-Escuela, D.O.; Santin, L.; Parrado, C.; Narvaez, J.A.; Fuxe, K.; Diaz-Cabiale, Z. A role for galanin N-terminal fragment (1-15) in anxiety- and depression-related behaviors in rats. Int. J. Neuropsychopharmacol. 2014, 18, pyu064. [Google Scholar] [CrossRef] [PubMed]
- Millon, C.; Flores-Burgess, A.; Narvaez, M.; Borroto-Escuela, D.O.; Santin, L.; Gago, B.; Narvaez, J.A.; Fuxe, K.; Diaz-Cabiale, Z. Galanin (1-15) enhances the antidepressant effects of the 5-HT1A receptor agonist 8-OH-DPAT: involvement of the raphe-hippocampal 5-HT neuron system. Brain Struct. Funct. 2016, 221, 4491–4504. [Google Scholar] [CrossRef] [PubMed]
- Rondard, P.; Huang, S.; Monnier, C.; Tu, H.; Blanchard, B.; Oueslati, N.; Malhaire, F.; Li, Y.; Trinquet, E.; Labesse, G.; et al. Functioning of the dimeric GABA(B) receptor extracellular domain revealed by glycan wedge scanning. EMBO J. 2008, 27, 1321–1332. [Google Scholar] [CrossRef]
- Maggio, R.; Millan, M.J. Dopamine D2-D3 receptor heteromers: Pharmacological properties and therapeutic significance. Curr. Opin. Pharmacol. 2010, 10, 100–107. [Google Scholar] [CrossRef]
- So, C.H.; Varghese, G.; Curley, K.J.; Kong, M.M.; Alijaniaram, M.; Ji, X.; Nguyen, T.; O’Dowd B, F.; George, S.R. D1 and D2 dopamine receptors form heterooligomers and cointernalize after selective activation of either receptor. Mol. Pharmacol. 2005, 68, 568–578. [Google Scholar]
- Petrin, D.; Hebert, T.E. The functional size of GPCRs - monomers, dimers or tetramers? Subcell. Biochem. 2012, 63, 67–81. [Google Scholar]
- Bouvier, M.; Hébert, T.E. CrossTalk proposal: Weighing the evidence for Class A GPCR dimers, the evidence favours dimers. J. Physiol. 2014, 592, 2439–2441. [Google Scholar] [CrossRef] [PubMed]
- Lambert, N.A.; Javitch, J.A. CrossTalk opposing view: Weighing the evidence for class A GPCR dimers, the jury is still out. J. Physiol. 2014, 592, 2443–2445. [Google Scholar] [CrossRef] [PubMed]
- Felce, J.H.; Latty, S.L.; Knox, R.G.; Mattick, S.R.; Lui, Y.; Lee, S.F.; Klenerman, D.; Davis, S.J. Receptor Quaternary Organization Explains G Protein-Coupled Receptor Family Structure. Cell Rep. 2017, 20, 2654–2665. [Google Scholar] [CrossRef] [PubMed]
- Herrick-Davis, K.; Milligan, G.; Di Giovanni, G. G-Protein-Coupled Receptor Dimers; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Marsango, S.; Caltabiano, G.; Jimenez-Roses, M.; Millan, M.J.; Pediani, J.D.; Ward, R.J.; Milligan, G. A Molecular Basis for Selective Antagonist Destabilization of Dopamine D3 Receptor Quaternary Organization. Sci. Rep. 2017, 7, 2134. [Google Scholar] [CrossRef] [PubMed]
- Nenasheva, T.A.; Neary, M.; Mashanov, G.I.; Birdsall, N.J.; Breckenridge, R.A.; Molloy, J.E. Abundance, distribution, mobility and oligomeric state of M(2) muscarinic acetylcholine receptors in live cardiac muscle. J. Mol. Cell. Cardiol. 2013, 57, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hern, J.A.; Baig, A.H.; Mashanov, G.I.; Birdsall, B.; Corrie, J.E.; Lazareno, S.; Molloy, J.E.; Birdsall, N.J. Formation and dissociation of M1 muscarinic receptor dimers seen by total internal reflection fluorescence imaging of single molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 2693–2698. [Google Scholar] [CrossRef]
- Kasai, R.S.; Kusumi, A. Single-molecule imaging revealed dynamic GPCR dimerization. Curr. Opin. Cell Biol. 2014, 27, 78–86. [Google Scholar] [CrossRef]
- Kasai, R.S.; Ito, S.V.; Awane, R.M.; Fujiwara, T.K.; Kusumi, A. The Class-A GPCR Dopamine D2 Receptor Forms Transient Dimers Stabilized by Agonists: Detection by Single-Molecule Tracking. Cell Biochem. Biophys. 2018, 76, 29–37. [Google Scholar] [CrossRef]
- Wouters, E.; Marin, A.R.; Dalton, J.A.R.; Giraldo, J.; Stove, C. Distinct Dopamine D(2) Receptor Antagonists Differentially Impact D(2) Receptor Oligomerization. Int. J. Mol. Sci. 2019, 20, 1686. [Google Scholar] [CrossRef]
- Van Craenenbroeck, K. GPCR oligomerization: contribution to receptor biogenesis. Sub-Cell. Biochem. 2012, 63, 43–65. [Google Scholar]
- Baragli, A.; Alturaihi, H.; Watt, H.L.; Abdallah, A.; Kumar, U. Heterooligomerization of human dopamine receptor 2 and somatostatin receptor 2 Co-immunoprecipitation and fluorescence resonance energy transfer analysis. Cell. Signal. 2007, 19, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Suzuki, T.; Kishimoto, Y.; Hirasawa, N. Biochemical assay of G protein-coupled receptor oligomerization: Adenosine A1 and thromboxane A2 receptors form the novel functional hetero-oligomer. Methods Cell Biol. 2013, 117, 213–227. [Google Scholar] [PubMed]
- Skieterska, K.; Duchou, J.; Lintermans, B.; Van Craenenbroeck, K. Detection of G protein-coupled receptor (GPCR) dimerization by coimmunoprecipitation. Methods Cell Biol. 2013, 117, 323–340. [Google Scholar] [PubMed]
- Ciruela, F.; Vilardaga, J.P.; Fernandez-Duenas, V. Lighting up multiprotein complexes: lessons from GPCR oligomerization. Trends Biotechnol. 2010, 28, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Pediani, J.D.; Godin, A.G.; Milligan, G. Regulation of oligomeric organization of the serotonin 5-hydroxytryptamine 2C (5-HT2C) receptor observed by spatial intensity distribution analysis. J. Biol. Chem. 2015, 290, 12844–12857. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Pediani, J.D.; Harikumar, K.G.; Miller, L.J.; Milligan, G. Spatial intensity distribution analysis quantifies the extent and regulation of homodimerization of the secretin receptor. Biochem. J. 2017, 474, 1879–1895. [Google Scholar] [CrossRef] [PubMed]
- Pediani, J.D.; Ward, R.J.; Godin, A.G.; Marsango, S.; Milligan, G. Dynamic Regulation of Quaternary Organization of the M1 Muscarinic Receptor by Subtype-selective Antagonist Drugs. J. Biol. Chem. 2016, 291, 13132–13146. [Google Scholar] [CrossRef]
- Pediani, J.D.; Ward, R.J.; Marsango, S.; Milligan, G. Spatial Intensity Distribution Analysis: Studies of G Protein-Coupled Receptor Oligomerisation. Trends Pharmacol. Sci. 2018, 39, 175–186. [Google Scholar] [CrossRef]
- Wehr, M.C.; Rossner, M.J. Split protein biosensor assays in molecular pharmacological studies. Drug Discov. Today 2016, 21, 415–429. [Google Scholar] [CrossRef]
- Ghosh, I.; Hamilton, A.D.; Regan, L. Antiparallel leucine zipper-directed protein reassembly: Application to the green fluorescent protein. J. Am. Chem. Soc. 2000, 122, 5658–5659. [Google Scholar] [CrossRef]
- Hu, C.D.; Chinenov, Y.; Kerppola, T.K. Visualization of interactions among bZip and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 2002, 9, 789–798. [Google Scholar] [CrossRef]
- Shyu, Y.J.; Liu, H.; Deng, X.H.; Hu, C.D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques 2006, 40, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Cevheroglu, O.; Kumas, G.; Hauser, M.; Becker, J.M.; Son, C.D. The yeast Ste2p G protein-coupled receptor dimerizes on the cell plasma membrane. Bba-Biomembranes 2017, 1859, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Nguyen, H.B.; Pedelacq, J.D.; Koraichi, F.; Chaudhary, A.; Ganguly, K.; Lockard, M.A.; Favre, G.; Terwilliger, T.C.; Waldo, G.S. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci. Rep. 2013, 3, 2854. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Sawano, A.; Park, E.S.; Miyawaki, A. Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. USA 2001, 98, 3197–3202. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, O.; Baird, G.S.; Campbell, R.E.; Zacharias, D.A.; Tsien, R.Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 2001, 276, 29188–29194. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K.; Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2002, 20, 87–90. [Google Scholar] [CrossRef]
- Vidi, P.A.; Chemel, B.R.; Hu, C.D.; Watts, V.J. Ligand-dependent oligomerization of dopamine D(2) and adenosine A(2A) receptors in living neuronal cells. Mol. Pharmacol. 2008, 74, 544–551. [Google Scholar] [CrossRef]
- Vidi, P.-A.; Chen, J.; Irudayaraj, J.M.K.; Watts, V.J. Adenosine A2A receptors assemble into higher-order oligomers at the plasma membrane. FEBS Lett. 2008, 582, 3985–3990. [Google Scholar] [CrossRef]
- Kilpatrick, L.E.; Humphrys, L.J.; Holliday, N.D. A G protein-coupled receptor dimer imaging assay reveals selectively modified pharmacology of neuropeptide Y Y1/Y5 receptor heterodimers. Mol. Pharmacol. 2015, 87, 718–732. [Google Scholar] [CrossRef]
- Przybyla, J.A.; Watts, V.J. Ligand-induced regulation and localization of cannabinoid CB1 and dopamine D2L receptor heterodimers. J. Pharmacol. Exp. Ther. 2010, 332, 710–719. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Xiong, D.; Wu, M.; Ding, J.L. FFAR2-FFAR3 receptor heteromerization modulates short-chain fatty acid sensing. FASEB J. 2018, 32, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Bai, B.; Ji, B.; Chen, X.; Wang, C.; Wang, P.; Yang, C.; Zhang, R.; Jiang, Y.; Pan, Y.; et al. Ghrelin Through GHSR1a and OX1R Heterodimers Reveals a Galphas-cAMP-cAMP Response Element Binding Protein Signaling Pathway in Vitro. Front. Mol. Neurosci. 2018, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Cordomi, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Perez-Benito, L.; Ferre, S.; Cortes, A.; Casado, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Hinz, S.; Navarro, G.; Borroto-Escuela, D.; Seibt, B.F.; Ammon, Y.C.; de Filippo, E.; Danish, A.; Lacher, S.K.; Cervinkova, B.; Rafehi, M.; et al. Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget 2018, 9, 13593–13611. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Jung, Y. Homo-molecular Fluorescence Complementation for Direct Visualization of Receptor Oligomerization in Living Cells. Angew. Chem. (Int. Ed. Engl.) 2019, 58, 2045–2049. [Google Scholar] [CrossRef] [PubMed]
- Vidi, P.A.; Chemel, B.R.; Watts, V.J. Direct visualization of adenosine A(2A) and dopamine D-2L receptor oligomers in a neuronal cell model. FASEB J. 2008, 22. [Google Scholar]
- Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. USA 2000, 97, 11984–11989. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Y.; Cui, Z.Q.; Wei, H.P.; Zhang, Z.P.; Zhou, Y.F.; Wang, Y.P.; Zhang, X.E. Split mCherry as a new red bimolecular fluorescence complementation system for visualizing protein-protein interactions in living cells. Biochem. Biophys. Res. Commun. 2008, 367, 47–53. [Google Scholar] [CrossRef]
- Jach, G.; Pesch, M.; Richter, K.; Frings, S.; Uhrig, J.F. An improved mRFP1 adds red to bimolecular fluorescence complementation. Nat. Methods 2006, 3, 597–600. [Google Scholar] [CrossRef]
- Shcherbo, D.; Merzlyak, E.M.; Chepurnykh, T.V.; Fradkov, A.F.; Ermakova, G.V.; Solovieva, E.A.; Lukyanov, K.A.; Bogdanova, E.A.; Zaraisky, A.G.; Lukyanov, S.; et al. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods 2007, 4, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Zhang, Z.; Zheng, Y.; Yang, J.; Qin, L.; Lu, J.; Huang, Z.L.; Zeng, S.; Luo, Q. A novel far-red bimolecular fluorescence complementation system that allows for efficient visualization of protein interactions under physiological conditions. Biosens. Bioelectron. 2009, 25, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.D.; Kerppola, T.K. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 2003, 21, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Vidi, P.A.; Przybyla, J.A.; Hu, C.D.; Watts, V.J. Visualization of G protein-coupled receptor (GPCR) interactions in living cells using bimolecular fluorescence complementation (BiFC). Curr. Protoc. Neurosci. 2010, 51, 5–29. [Google Scholar] [CrossRef]
- Vidi, P.A.; Watts, V.J. Fluorescent and bioluminescent protein-fragment complementation assays in the study of G protein-coupled receptor oligomerization and signaling. Mol. Pharmacol. 2009, 75, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Rebois, R.V.; Robitaille, M.; Petrin, D.; Zylbergold, P.; Trieu, P.; Hebert, T.E. Combining protein complementation assays with resonance energy transfer to detect multipartner protein complexes in living cells. Methods (San Diego, Calif.) 2008, 45, 214–218. [Google Scholar] [CrossRef]
- Navarro, G.; Carriba, P.; Gandia, J.; Ciruela, F.; Casado, V.; Cortes, A.; Mallol, J.; Canela, E.I.; Lluis, C.; Franco, R. Detection of heteromers formed by cannabinoid CB1, dopamine D2, and adenosine A2A G-protein-coupled receptors by combining bimolecular fluorescence complementation and bioluminescence energy transfer. Sci. World J. 2008, 8, 1088–1097. [Google Scholar] [CrossRef]
- Shyu, Y.J.; Suarez, C.D.; Hu, C.D. Visualization of AP-1 NF-kappaB ternary complexes in living cells by using a BiFC-based FRET. Proc. Natl. Acad. Sci. USA 2008, 105, 151–156. [Google Scholar] [CrossRef]
- Bagher, A.M.; Laprairie, R.B.; Toguri, J.T.; Kelly, M.E.M.; Denovan-Wright, E.M. Bidirectional allosteric interactions between cannabinoid receptor 1 (CB1) and dopamine receptor 2 long (D2L) heterotetramers. Eur. J. Pharmacol. 2017, 813, 66–83. [Google Scholar] [CrossRef]
- Spencer, D.M.; Wandless, T.J.; Schreiber, S.L.; Crabtree, G.R. Controlling Signal-Transduction with Synthetic Ligands. Science 1993, 262, 1019–1024. [Google Scholar] [CrossRef]
- Cao, X.; Liang, L.; Hadcock, J.R.; Iredale, P.A.; Griffith, D.A.; Menniti, F.S.; Factor, S.; Greenamyre, J.T.; Papa, S.M. Blockade of cannabinoid type 1 receptors augments the antiparkinsonian action of levodopa without affecting dyskinesias in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated rhesus monkeys. J. Pharmacol. Exp. Ther. 2007, 323, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Wood, K.V. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 2007, 5, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Luker, K.E.; Smith, M.C.; Luker, G.D.; Gammon, S.T.; Piwnica-Worms, H.; Piwnica-Worms, D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc. Natl. Acad. Sci. USA 2004, 101, 12288–12293. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Umezawa, Y.; Gambhir, S.S. Noninvasive imaging of protein-protein interactions in living subjects by using reporter protein complementation and reconstitution strategies. Proc. Natl. Acad. Sci. USA 2002, 99, 15608–15613. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Gambhir, S.S. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal. Chem. 2003, 75, 1584–1589. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Massoud, T.F.; Huang, J.; Gambhir, S.S. Molecular imaging of drug-modulated protein-protein interactions in living subjects. Cancer Res. 2004, 64, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Remy, I.; Michnick, S.W. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat. Methods 2006, 3, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Bodle, C.R.; Hayes, M.P.; O’Brien, J.B.; Roman, D.L. Development of a bimolecular luminescence complementation assay for RGS: G protein interactions in cells. Anal. Biochem. 2017, 522, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hashi, K.; Hirata, Y.; Kiuchi, K. SOD1 dimerization monitoring using a novel split NanoLuc, NanoBit. Cell Biochem. Funct. 2016, 34, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, L.G.; Mattioli, M.; Ricci, F.; Li, Y.C.; Wade, M. Multiplex detection of protein-protein interactions using a next generation luciferase reporter. Biochim. Biophys. Acta 2016, 1863, 284–292. [Google Scholar] [CrossRef]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, A.; Primeau, M.; Trudeau, L.E.; Michnick, S.W. beta-Lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein-protein interactions. Nat. Biotechnol. 2002, 20, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Paulmurugan, R.; Gambhir, S.S. Novel fusion protein approach for efficient high-throughput screening of small molecule-mediating protein-protein interactions in cells and living animals. Cancer Res. 2005, 65, 7413–7420. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matthews, J.C.; Hori, K.; Cormier, M.J. Substrate and substrate analogue binding properties of Renilla luciferase. Biochemistry US 1977, 16, 5217–5220. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.C.; Hori, K.; Cormier, M.J. Purification and properties of Renilla reniformis luciferase. Biochemistry US 1977, 16, 85–91. [Google Scholar] [CrossRef]
- Loening, A.M.; Fenn, T.D.; Wu, A.M.; Gambhir, S.S. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng. Des. Sel. 2006, 19, 391–400. [Google Scholar] [CrossRef]
- Luker, K.E.; Gupta, M.; Luker, G.D. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009, 23, 823–834. [Google Scholar] [CrossRef]
- Armando, S.; Quoyer, J.; Lukashova, V.; Maiga, A.; Percherancier, Y.; Heveker, N.; Pin, J.P.; Prezeau, L.; Bouvier, M. The chemokine CXC4 and CC2 receptors form homo- and heterooligomers that can engage their signaling G-protein effectors and betaarrestin. FASEB J. 2014, 28, 4509–4523. [Google Scholar] [CrossRef]
- Guo, W.; Urizar, E.; Kralikova, M.; Mobarec, J.C.; Shi, L.; Filizola, M.; Javitch, J.A. Dopamine D2 receptors form higher order oligomers at physiological expression levels. EMBO J. 2008, 27, 2293–2304. [Google Scholar] [CrossRef]
- Casado-Anguera, V.; Bonaventura, J.; Moreno, E.; Navarro, G.; Cortes, A.; Ferre, S.; Casado, V. Evidence for the heterotetrameric structure of the adenosine A2A-dopamine D2 receptor complex. Biochem. Soc. Trans. 2016, 44, 595–600. [Google Scholar] [CrossRef]
- Hall, M.P.; Unch, J.; Binkowski, B.F.; Valley, M.P.; Butler, B.L.; Wood, M.G.; Otto, P.; Zimmerman, K.; Vidugiris, G.; Machleidt, T.; et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 2012, 7, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- England, C.G.; Ehlerding, E.B.; Cai, W. NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence. Bioconjug. Chem. 2016, 27, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Cannaert, A.; Franz, F.; Auwarter, V.; Stove, C.P. Activity-Based Detection of Consumption of Synthetic Cannabinoids in Authentic Urine Samples Using a Stable Cannabinoid Reporter System. Anal. Chem. 2017, 89, 9527–9536. [Google Scholar] [CrossRef] [PubMed]
- Cannaert, A.; Storme, J.; Franz, F.; Auwarter, V.; Stove, C.P. Detection and Activity Profiling of Synthetic Cannabinoids and Their Metabolites with a Newly Developed Bioassay. Anal. Chem. 2016, 88, 11476–11485. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.; Laschet, C.; Franssen, D.; Szpakowska, M.; Gilissen, J.; Geubelle, P.; Soni, A.; Parent, A.S.; Pirotte, B.; Chevigne, A.; et al. Activation of the Orphan G Protein-Coupled Receptor GPR27 by Surrogate Ligands Promotes beta-Arrestin 2 Recruitment. Mol. Pharmacol. 2017, 91, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.; Cannaert, A.; Linnet, K.; Stove, C.P. Application of an activity-based receptor bioassay to investigate the in vitro activity of selected indole- and indazole-3-carboxamide-based synthetic cannabinoids at CB1 and CB2 receptors. Drug Test. Anal. 2019, 11, 501–511. [Google Scholar] [CrossRef]
- Storme, J.; Cannaert, A.; Van Craenenbroeck, K.; Stove, C.P. Molecular dissection of the human A3 adenosine receptor coupling with beta-arrestin2. Biochem. Pharmacol. 2018, 148, 298–307. [Google Scholar] [CrossRef]
- Wouters, E.; Mogler, L.; Cannaert, A.; Auwärter, V.; Stove, C. Functional evaluation of carboxy metabolites of synthetic cannabinoid receptor agonists featuring scaffolds based on L-valine or L-tert-leucine. Drug Test. Anal. 2019. [Google Scholar] [CrossRef]
- Cannaert, A.; Vasudevan, L.; Friscia, M.; Mohr, A.L.A.; Wille, S.M.R.; Stove, C.P. Activity-Based Concept to Screen Biological Matrices for Opiates and (Synthetic) Opioids. Clin. Chem. 2018, 64, 1221–1229. [Google Scholar] [CrossRef]
- Laschet, C.; Dupuis, N.; Hanson, J. A dynamic and screening-compatible nanoluciferase-based complementation assay enables profiling of individual GPCR-G protein interactions. J. Biol. Chem. 2018. [Google Scholar] [CrossRef]
- Wouters, E.; Vasudevan, L.; Ciruela, F.; Saini, D.K.; Stove, C.; Van Craenenbroeck, K. Assessing GPCR Dimerization in Living Cells: Comparison of the NanoBiT Assay with Related Bioluminescence- and Fluorescence-Based Approaches. In Receptor-Receptor Interactions in the Central Nervous System; Fuxe, K., Borroto-Escuela, D.O., Eds.; Springer: New York, NY, USA, 2018; pp. 239–250. [Google Scholar] [CrossRef]
- Habara, M.; Mori, N.; Okada, Y.; Kawasumi, K.; Nakao, N.; Tanaka, Y.; Arai, T.; Yamamoto, I. Molecular characterization of feline melanocortin 4 receptor and melanocortin 2 receptor accessory protein 2. Gen. Comp. Endocrinol. 2018, 261, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Dixon, A.S.; Kim, S.J.; Baumgartner, B.K.; Krippner, S.; Owen, S.C. A Tri-part Protein Complementation System Using AntibodySmall Peptide Fusions Enables Homogeneous Immunoassays. Sci. Rep.-UK 2017, 7, 8186. [Google Scholar] [CrossRef] [PubMed]
- Ohmuro-Matsuyama, Y.; Ueda, H. Homogeneous Noncompetitive Luminescent Immunodetection of Small Molecules by Ternary Protein Fragment Complementation. Anal. Chem. 2018, 90, 3001–3004. [Google Scholar] [CrossRef] [PubMed]
- Moustaqil, M.; Bhumkar, A.; Gonzalez, L.; Raoul, L.; Hunter, D.J.B.; Carrive, P.; Sierecki, E.; Gambin, Y. A Split-Luciferase Reporter Recognizing GFP and mCherry Tags to Facilitate Studies of Protein-Protein Interactions. Int. J. Mol. Sci. 2017, 18, 2681. [Google Scholar] [CrossRef] [PubMed]
- Sahlholm, K.; Gomez-Soler, M.; Valle-Leon, M.; Lopez-Cano, M.; Taura, J.J.; Ciruela, F.; Fernandez-Duenas, V. Antipsychotic-Like Efficacy of Dopamine D2 Receptor-Biased Ligands is Dependent on Adenosine A2A Receptor Expression. Mol. Neurobiol. 2018, 55, 4952–4958. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, J.; Navarro, G.; Casado-Anguera, V.; Azdad, K.; Rea, W.; Moreno, E.; Brugarolas, M.; Mallol, J.; Canela, E.I.; Lluis, C.; et al. Allosteric interactions between agonists and antagonists within the adenosine A2A receptor-dopamine D2 receptor heterotetramer. Proc. Natl. Acad. Sci. USA 2015, 112, E3609–E3618. [Google Scholar] [CrossRef] [PubMed]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar] [CrossRef]
- Gokhale, R.S.; Khosla, C. Role of linkers in communication between protein modules. Curr. Opin. Chem. Biol. 2000, 4, 22–27. [Google Scholar] [CrossRef]
- Argos, P. An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J. Mol. Biol. 1990, 211, 943–958. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- Carayon, K.; Mouledous, L.; Combedazou, A.; Mazeres, S.; Haanappel, E.; Salome, L.; Mollereau, C. Heterologous regulation of Mu-opioid (MOP) receptor mobility in the membrane of SH-SY5Y cells. J. Biol. Chem. 2014, 289, 28697–28706. [Google Scholar] [CrossRef] [PubMed]
- Stefan, E.; Aquin, S.; Berger, N.; Landry, C.R.; Nyfeler, B.; Bouvier, M.; Michnick, S.W. Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 16916–16921. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Pfleger, K.D.; Seeber, R.M.; Qian, H.; Oro, C.; Abogadie, F.; Delbridge, L.M.; Thomas, W.G. Heteromerization of angiotensin receptors changes trafficking and arrestin recruitment profiles. Cell. Signal. 2011, 23, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, S.; Weise, A.; Walsh, R.P.; Ward, J.M.; Frommer, W.B. Fusion to GFP blocks intercellular trafficking of the sucrose transporter SUT1 leading to accumulation in companion cells. BMC Plant Biol. 2003, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Fromherz, P. Ca2+ activation of hSlo K+ channel is suppressed by N-terminal GFP tag. Eur. J. Neurosci. 1999, 11, 1105–1108. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol 2005, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Ventura, S.; Aviles, F.X. Protein complementation assays: approaches for the in vivo analysis of protein interactions. FEBS Lett. 2009, 583, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Cabello, N.; Gandia, J.; Bertarelli, D.C.; Watanabe, M.; Lluis, C.; Franco, R.; Ferre, S.; Lujan, R.; Ciruela, F. Metabotropic glutamate type 5, dopamine D2 and adenosine A2a receptors form higher-order oligomers in living cells. J. Neurochem. 2009, 109, 1497–1507. [Google Scholar] [CrossRef]
- Wang, M.; Pei, L.; Fletcher, P.J.; Kapur, S.; Seeman, P.; Liu, F. Schizophrenia, amphetamine-induced sensitized state and acute amphetamine exposure all show a common alteration: increased dopamine D2 receptor dimerization. Mol. Brain 2010, 3, 25. [Google Scholar] [CrossRef]
- Zawarynski, P.; Tallerico, T.; Seeman, P.; Lee, S.P.; O’Dowd, B.F.; George, S.R. Dopamine D2 receptor dimers in human and rat brain. FEBS Lett. 1998, 441, 383–386. [Google Scholar] [CrossRef]
- Bonaventura, J.; Rico, A.J.; Moreno, E.; Sierra, S.; Sanchez, M.; Luquin, N.; Farre, D.; Muller, C.E.; Martinez-Pinilla, E.; Cortes, A.; et al. L-DOPA-treatment in primates disrupts the expression of A(2A) adenosine-CB1 cannabinoid-D-2 dopamine receptor heteromers in the caudate nucleus. Neuropharmacology 2014, 79, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.S.; Weaver, D.R.; Rivkees, S.A.; Peterfreund, R.A.; Pollack, A.E.; Adler, E.M.; Reppert, S.M. Molecular-Cloning of the Rat Adenosine-A2 Receptor—Selective Coexpression with D2-Dopamine Receptors in Rat Striatum. Mol. Brain Res. 1992, 14, 186–195. [Google Scholar] [CrossRef]
- Fuxe, K.; Ferre, S.; Canals, M.; Torvinen, M.; Terasmaa, A.; Marcellino, D.; Goldberg, S.R.; Staines, W.; Jacobsen, K.X.; Lluis, C.; et al. Adenosine A(2A) and dopamine D-2 heteromeric receptor complexes and their function. J. Mol. Neurosci. 2005, 26, 209–219. [Google Scholar] [CrossRef]
- Hillion, J.; Canals, M.; Torvinen, M.; Casado, V.; Scott, R.; Terasmaa, A.; Hansson, A.; Watson, S.; Olah, M.E.; Mallol, J.; et al. Coaggregation, cointernalization, and codesensitization of adenosine A(2A) receptors and dopamine D-2 receptors. J. Biol. Chem. 2002, 277, 18091–18097. [Google Scholar] [CrossRef] [PubMed]
- Soriano, A.; Ventura, R.; Molero, A.; Hoen, R.; Casado, V.; Cortes, A.; Fanelli, F.; Albericio, F.; Lluis, C.; Franco, R.; et al. Adenosine A(2A) Receptor-Antagonist/Dopamine D-2 Receptor-Agonist Bivalent Ligands as Pharmacological Tools to Detect A(2A)-D-2 Receptor Heteromers. J. Med. Chem. 2009, 52, 5590–5602. [Google Scholar] [CrossRef]
- Riggleman, A.; Harvey, J.; Baylis, C. Endothelin mediates some of the renal actions of acutely administered angiotensin II. Hypertension 2001, 38, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.Y.; Hopfer, U.; Asico, L.D.; Eisner, G.M.; Felder, R.A.; Jose, P.A. Altered AT(1) receptor regulation of ETB receptors in renal proximal tubule cells of spontaneously hypertensive rats. Hypertension 2005, 46, 926–931. [Google Scholar] [CrossRef]
- Zeng, C.Y.; Wang, Z.; Asico, L.D.; Hopfer, U.; Eisner, G.M.; Felder, R.A.; Jose, P.A. Aberrant ETB receptor regulation of AT(1) receptors in immortalized renal proximal tubule cells of spontaneously hypertensive rats. Kidney Int. 2005, 68, 623–631. [Google Scholar] [CrossRef]
- Ciruela, F.; Casado, V.; Rodrigues, R.J.; Lujan, R.; Burgueno, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A(1)-A(2A) receptor heteromers. J. Neurosci. 2006, 26, 2080–2087. [Google Scholar] [CrossRef]
- Moreno, J.L.; Miranda-Azpiazu, P.; Garcia-Bea, A.; Younkin, J.; Cui, M.; Kozlenkov, A.; Ben-Ezra, A.; Voloudakis, G.; Fakira, A.K.; Baki, L.; et al. Allosteric signaling through an mGlu2 and 5-HT2A heteromeric receptor complex and its potential contribution to schizophrenia. Sci. Signal. 2016, 9, ra5. [Google Scholar] [CrossRef]
- Lopez-Gimenez, J.F.; Canals, M.; Pediani, J.D.; Milligan, G. The alpha(1b)-adrenoceptor exists as a higher-order oligomer: Effective oligomerization is required for receptor maturation, surface delivery, and function. Mol. Pharmacol. 2007, 71, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Frade, J.M.; del Real, G.; Serrano, A.; Hernanz-Falcon, P.; Soriano, S.F.; Vila-Coro, A.J.; de Ana, A.M.; Lucas, P.; Prieto, I.; Martinez-A, C.; et al. Blocking HIV-1 infection via CCR5 and CXCR4 receptors by acting in trans on the CCR2 chemokine receptor. EMBO J. 2004, 23, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Sohy, D.; Parmentier, M.; Springael, J.Y. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J. Biol. Chem. 2007, 282, 30062–30069. [Google Scholar] [CrossRef] [PubMed]
- Sohy, D.; Yano, H.; de Nadai, P.; Urizar, E.; Guillabert, A.; Javitch, J.A.; Parmentier, M.; Springael, J.Y. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the Protean Effects of "Selective" Antagonists. J. Biol. Chem. 2009, 284, 31270–31279. [Google Scholar] [CrossRef] [PubMed]
- Grefen, C.; Blatt, M.R. A 2in1 cloning system enables ratiometric bimolecular fluorescence complementation (rBiFC). BioTechniques 2012, 53, 311–314. [Google Scholar] [CrossRef] [PubMed]
- White, C.W.; Vanyai, H.K.; See, H.B.; Johnstone, E.K.M.; Pfleger, K.D.G. Using nanoBRET and CRISPR/Cas9 to monitor proximity to a genome-edited protein in real-time. Sci. Rep. 2017, 7, 3187. [Google Scholar] [CrossRef] [PubMed]
- Zych, C.; Domling, A.; Ayyavoo, V. Development of a robust cell-based high-throughput screening assay to identify targets of HIV-1 viral protein R dimerization. Drug Des. Dev. Ther. 2013, 7, 403–412. [Google Scholar]
- Morell, M.; Espargaro, A.; Aviles, F.X.; Ventura, S. Detection of transient protein-protein interactions by bimolecular fluorescence complementation: The Abl-SH3 case. Proteomics 2007, 7, 1023–1036. [Google Scholar] [CrossRef]
- Auld, D.S.; Inglese, J. Interferences with Luciferase Reporter Enzymes. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Simeonov, A.; Davis, M.I. Interference with Fluorescence and Absorbance. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Caaveiro, J.M.M., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Wade, M.; Mendez, J.; Coussens, N.P.; Arkin, M.R.; Glicksman, M.A. Inhibition of Protein-Protein Interactions: Cell-Based Assays. In Assay Guidance Manual; Sittampalam, G.S., Coussens, N.P., Brimacombe, K., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Bejcek, B., Caaveiro, J.M.M., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2017. [Google Scholar]
- Wu, J.C.; Sundaresan, G.; Iyer, M.; Gambhir, S.S. Noninvasive optical imaging of firefly luciferase reporter gene expression in skeletal muscles of living mice. Mol. Ther. J. Am. Soc. Gene Ther. 2001, 4, 297–306. [Google Scholar] [CrossRef]
- Han, Y.; Wang, S.F.; Zhang, Z.P.; Ma, X.H.; Li, W.; Zhang, X.W.; Deng, J.Y.; Wei, H.P.; Li, Z.Y.; Zhang, X.E.; et al. In vivo imaging of protein-protein and RNA-protein interactions using novel far-red fluorescence complementation systems. Nucleic Acids Res. 2014, 42, e103. [Google Scholar] [CrossRef]
- Faron-Gorecka, A.; Szlachta, M.; Kolasa, M.; Solich, J.; Gorecki, A.; Kusmider, M.; Zurawek, D.; Dziedzicka-Wasylewska, M. Understanding GPCR dimerization. Methods Cell Biol. 2019, 149, 155–178. [Google Scholar] [PubMed]
- Maurice, P.; Guillaume, J.L.; Benleulmi-Chaachoua, A.; Daulat, A.M.; Kamal, M.; Jockers, R. GPCR-interacting proteins, major players of GPCR function. Adv. Pharmacol. (San Diego, Calif.) 2011, 62, 349–380. [Google Scholar]
- Milligan, G.; Ward, R.J.; Marsango, S. GPCR homo-oligomerization. Curr. Opin. Cell Biol. 2018, 57, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Gomes, I.; Ayoub, M.A.; Fujita, W.; Jaeger, W.C.; Pfleger, K.D.; Devi, L.A. G Protein-Coupled Receptor Heteromers. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 403–425. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reporter Protein | Source Species | Readout | Excitation Wavelength (nm) | Emission Wavelength (nm) | Substrate | Cofactor | Stability (h) | Maturation Time (t1/2) (min) | MW (kDa) |
|---|---|---|---|---|---|---|---|---|---|
| VENUS | Aequorea victoria | Fluorescence | 515 | 528 | - | N/A | - | 40 (in vitro) | 27 |
| GFP | Aequorea victoria | Fluorescence | 488 | 510 | - | N/A | - | 53 (in vitro) | 27 |
| mCherry | Discosoma | Fluorescence | 587 | 610 | - | N/A | - | 17 + 30 (S. cerevisiae) | 29 |
| Cerulean | Aequorea victoria | Fluorescence | 433 | 475 | - | N/A | - | nd | 27 |
| Tripartite-Split GFP | Aequorea victoria | Fluorescence | 488 | 530 | - | N/A | - | nd | 23 |
| EYFP | Aequorea victoria | Fluorescence | 514 | 527 | - | N/A | - | 23 (in vitro) | 26.4 |
| ECFP | Aequorea macrodactyla | Fluorescence | 405 | 485 | - | N/A | - | 49 (S. cerevisiae) | 26.8 |
| Citrine | Aequorea victoria | Fluorescence | 516 | 529 | - | N/A | - | nd | 27 |
| mRFP | Discosoma striata | Fluorescence | 584 | 607 | - | N/A | - | <60 | 25.9 |
| mKate | Discosoma striata | Fluorescence | 588 | 635 | - | N/A | - | 20 | 26 |
| DsRed monomer | Discosoma striata | Fluorescence | 558 | 583 | - | N/A | - | 600 | 28 |
| Renilla luciferase | Renilla reniformas | Luminescence | - | 480 | Coelenterazine | N/A | 4.5 h (cell) | - | 36 |
| Firefly Luciferase | Photinus pyralis | Luminescence | - | 550–570 | d-luciferin | ATP, O2 | 4.0 h (cell) | - | 62 |
| Gaussia Luciferase | Gaussia princeps | Luminescence | - | 485 | Coelenterazine | N/A | 60 h (cell media) | - | 20 |
| NanoBiT | Oplophorus gracilirostris | Luminescence | - | 460 | Furimazine | N/A | 6.0 h (cell) | - | 19 |
| β-lactamase | Bacillus licheniformis | Luminescence | - | 492 | Nitrocefin | N/A | nd | - | 29 |
| β-Galactosidase | Escherichia coli | Fluorescence | Reliant on the substrate | Reliant on the substrate | FDG, MUG a.o. | Mg2+ | 1.1 h (yeast cells) | - | 464 |
| Click Beetle luciferase | Pyrophorus plagiophthalamus | Luminescence | - | - | d-luciferin | Mg2+, ATP | - | - | 64 |
| GPCR Dimer | Oligomeric Type | PCA Type | Split Biosensor | Fragments | Negative Control | Cell-Type | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| CXCR4 − CXCR4/CC2 | Hetero-oligomer with CC2, homotetramer | BiLC and BiFC | RLucII, vYFP | RLucII: 1–330, 331–936 vYFP: 1–465, 466–720 | D2R | HEK293 | 2014 | [99] |
| A2a − D2R | heterotetramer | BiLC and BiFC | RLuc8, YFP | RLuc8: 1–229, 230–311 YFP: 1–155, 156–238 | A1R, D1R | HEK293 | 2015/2016 | [101,118] |
| D2SR − D2SR | Homo-oligomer | BiLC and BiFC | RLuc8, mVenus | RLuc8: 1–229, 230–311 mVenus: 1–155, 156–240 | CD8, TSHr | HEK293T | 2008 | [100] |
| β2AR − β2AR | homotetramer | BiLC and BiFC | GLuc, Venus | GLuc: 1–63, 64–185 | GLucN, VN, VC | HEK293 | 2008 | [77] |
| BiFC | |
|---|---|
| Advantages | Disadvantages |
| Straightforward technique | Need for tagged proteins (GPCRs) |
| High-throughput experiments | Autofluorescence |
| Imaging microscopy: localization of the interaction | Photobleaching |
| Study intact cells | Measuring dynamics: limited (maturation time) Not applicable for studying inhibition of interactions |
| BiLC | |
| Advantages | Disadvantages |
| Straightforward technique | Need for tagged proteins (GPCRs) |
| High-throughput experiments | Requires a substrate |
| Kinetic measurements | Detection of localization: limited |
| Study intact cells | |
| In vivo application | |
| GPCR Dimer | PCA Type | Linker | Ref |
|---|---|---|---|
| AT1 − AT2 | BiFC, Venus | GGGGSGGGG | [125] |
| CXCR4 − CXCR4 | BiLC, Rluc | (GGGS)2 | [99] |
| D2LR − D2LR | BiFC, Venus | LG | [100] |
| D2LR − D2LR A2a − D2R | BiLC, Rluc | ATGLDLELKASNSAVDGTAGPVAT | [112,117] |
| D2LR − D2LR | BiLC, NanoBiT | GNS-GSSGGGGSGGGGSSG | [112] |
| MOP − NPFF2 | BiFC, Venus | DGGSGGGS | [123] |
| GPCR Dimer | Oligomeric Type | PCA | Split Biosensor | Fragments | Negative Control | Cell-Type | Year | Ref. | In Vivo or Native Tissue Evidence | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| mGluR5− D2R | Heterodimer | BiFC | YFP | 1–155, 155–231 | GABAB2 | HEK | 2009 | [130] | Yes | [130] |
| D2R − D2R | Homodimer | BiFC | YFP | 1–155, 156–238 | D1R | HEK | 2015 | [118] | Yes | [131,132] |
| A2A − D2LR | Heterodimer | MBiFC | Venus/Cerulean | 1–172, 155–238 | D1 | CAD | 2008 | [59] | Yes | [130,133,134,135,136,137] |
| D2LR − CB1 | Heterodimer | MBiFC | Venus/Cerulean | 1–172, 155–238 | M4 | CAD | 2010 | [62] | Yes | [133] |
| D2LR − D2LR | Oligomer | MBiFC | Venus/Cerulean | 1–172, 155–238 | - | CAD | 2010 | [62] | - | - |
| D2SR − D2SR | Homodimer | BiFC | Venus | 1–155, 156–240 | CD8 | HEK293T | 2008 | [100] | Yes | [131,132] |
| AT1 − AT2 | Homo- and heterodimer | BiFC | Venus | 1–158,159–239 | ATIP | HEK293FT | 2011 | [125] | Yes | [138,139,140] |
| CXCR4 − CXCR4 | Homodimer | BiFC | vYFP | 1–465, 466–720 | D2R | HEK293 | 2014 | [99] | - | - |
| A2A − A2A | Homodimer | MBiFC | Venus/Cerulean | 1–172, 155–238 | - | CAD | 2008 | [59] | - | - |
| A2A − A2A | Homodimer | BiFC | YFP | 1–155, 155–238 | Non-fused A1 (competition) | HEK293T | 2018 | [65] | - | - |
| A2A − A1 | Heterodimer | BiFC | YFP | 1–155, 155–238 | - | HEK293T | 2018 | [65] | Yes | [141] |
| GHSR1a-OX1R | Heterodimer | BiFC | Venus | 1–172, 156–239 | - | HEK293T | 2018 | [64] | - | - |
| β2AR − β2AR | Oligomer | BiFC | − 15sfGFP | - | HeLa | 2019 | [67] | - | - | |
| A2A − A2A | Homodimer | BiFC | YFP | 1–155,156–239 | GABAB2 | CHO | 2018 | [66] | - | - |
| A2B − A2A | Heterodimer | BiFC | YFP | 1–155,156–239 | GABAB2 | CHO | 2018 | [66] | - | - |
| FFAR3 − FFAR3 | Homodimer | BiFC | Venus | 1–155 (I152L), 155–239 | P2RY1 | HEK293T | 2018 | [63] | - | - |
| FFAR2 − FFAR3 | Heterodimer | BiFC | Venus | 1–155 (I152L), 155–238 | P2RY1 | HEK293T | 2018 | [63] | - | - |
| mGluR2 − mGluR2 | Homodimer | BiFC | mCitrine | 1–172, 155–238 | - | HEK293T | 2016 | [142] | - | - |
| α1b − α1b | Homodimer | BiFC | eYFP | 1–172, 155–238 | - | HEK293T | 2007 | [143] | - | - |
| GPCR Dimer | Oligomeric Type | PCA Type | Split Biosensor | Fragments | Negative Control | Cell-Type | Year | Ref. | In Vivo or Native Tissue Evidence | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| CXCR4 − CXCR4/CC2 | Homodimer | BiLC | FLuc | NLuc-416 and CLuc-398 | β2-AR | HEK293T | 2009 | [98] | Yes | [144,145,146] |
| CXCR7 − CXCR7 | Homodimer | |||||||||
| CXCR4 − CXCR7 | Heterodimer | |||||||||
| A2a − D2R | heterodimer | BiLC | RLuc8 | 1–229, 230–311 | - | HEK293T | 2018 | [117] | Yes | [118,130,134,135,136,137] |
| A2a − A2a | homodimer | BiLC | RLuc8 | 1–229, 230–311 | A1 | HEK293 | 2016 | [101,118] | - | - |
| D2LR − D2LR | homodimer | NanoBiT | NanoLuc | 1–11, 12–167 | CB2 | HEK293T | 2018/2019 | [40,112] | Yes | [131,132] |
| Fluorescence | PCA (Excitation/Emission) | Fluorescent Marker (Excitation/Emission) |
|---|---|---|
| Venus (515/528) | CFP (433/475) | |
| mCherry (587/610) | CFP (433/475)/GFP (488/510)/YFP (514/527) | |
| GFP (488/510) | CFP (433/475) | |
| Cerulean (452/478) | mTagBFP (402/457) | |
| Luminescence | PCA | Fluorescence Marker |
| Rluc | CFP (433/475)/mCherry (587/610) * | |
| Fluc | CFP (433/475)/mCherry (587/610) * | |
| Nluc | CFP (433/475)/mCherry (587/610) * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wouters, E.; Vasudevan, L.; Crans, R.A.J.; Saini, D.K.; Stove, C.P. Luminescence- and Fluorescence-Based Complementation Assays to Screen for GPCR Oligomerization: Current State of the Art. Int. J. Mol. Sci. 2019, 20, 2958. https://doi.org/10.3390/ijms20122958
Wouters E, Vasudevan L, Crans RAJ, Saini DK, Stove CP. Luminescence- and Fluorescence-Based Complementation Assays to Screen for GPCR Oligomerization: Current State of the Art. International Journal of Molecular Sciences. 2019; 20(12):2958. https://doi.org/10.3390/ijms20122958
Chicago/Turabian StyleWouters, Elise, Lakshmi Vasudevan, René A. J. Crans, Deepak K. Saini, and Christophe P. Stove. 2019. "Luminescence- and Fluorescence-Based Complementation Assays to Screen for GPCR Oligomerization: Current State of the Art" International Journal of Molecular Sciences 20, no. 12: 2958. https://doi.org/10.3390/ijms20122958
APA StyleWouters, E., Vasudevan, L., Crans, R. A. J., Saini, D. K., & Stove, C. P. (2019). Luminescence- and Fluorescence-Based Complementation Assays to Screen for GPCR Oligomerization: Current State of the Art. International Journal of Molecular Sciences, 20(12), 2958. https://doi.org/10.3390/ijms20122958