Cellular Effects of Butyrate on Vascular Smooth Muscle Cells are Mediated through Disparate Actions on Dual Targets, Histone Deacetylase (HDAC) Activity and PI3K/Akt Signaling Network

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
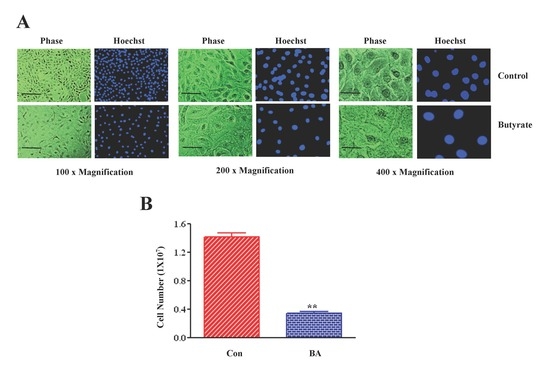
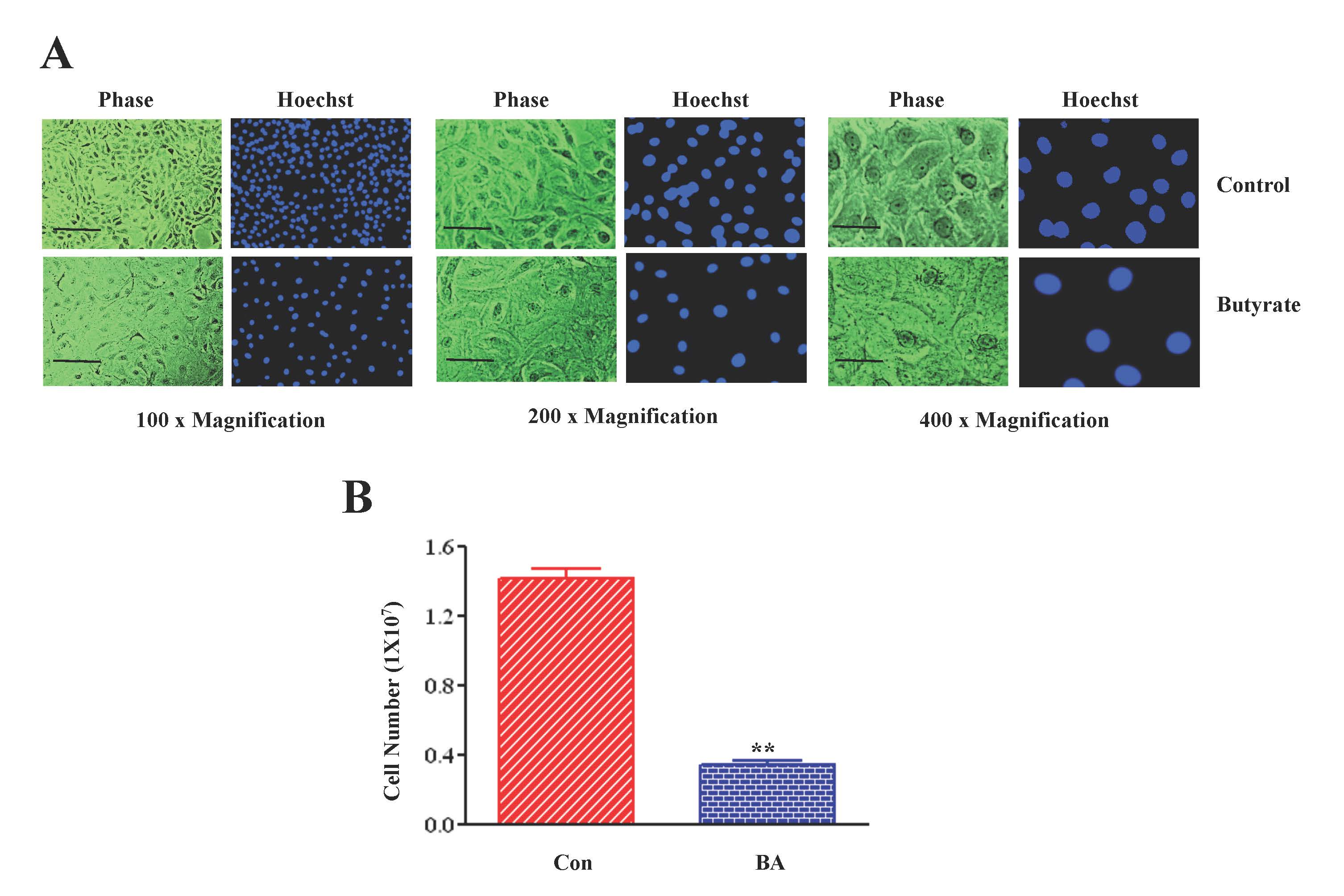
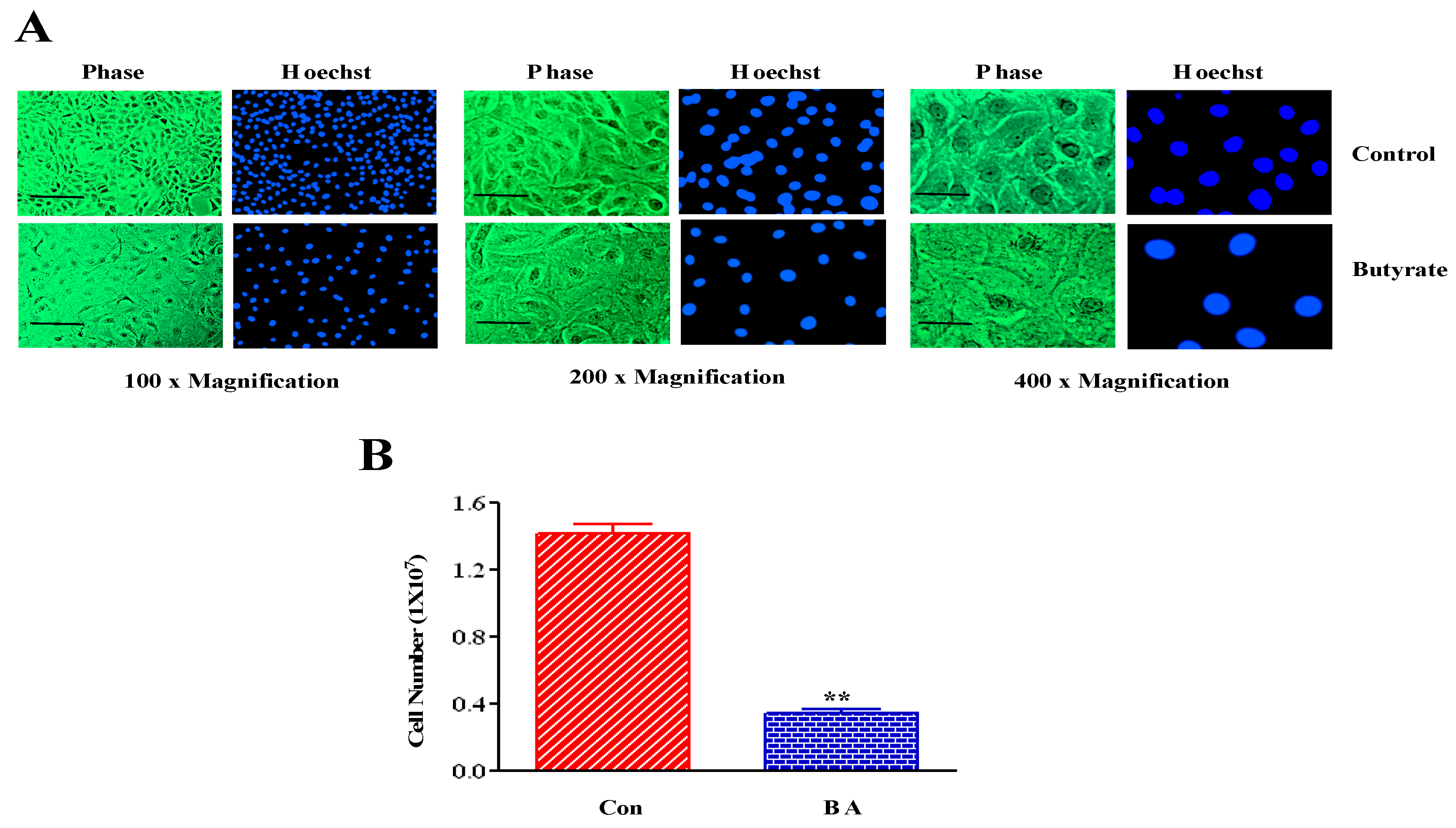
2.1. Butyrate Promotes VSMC Growth by Altering Cellular Size and Morphology, and Arresting VSMC Proliferation
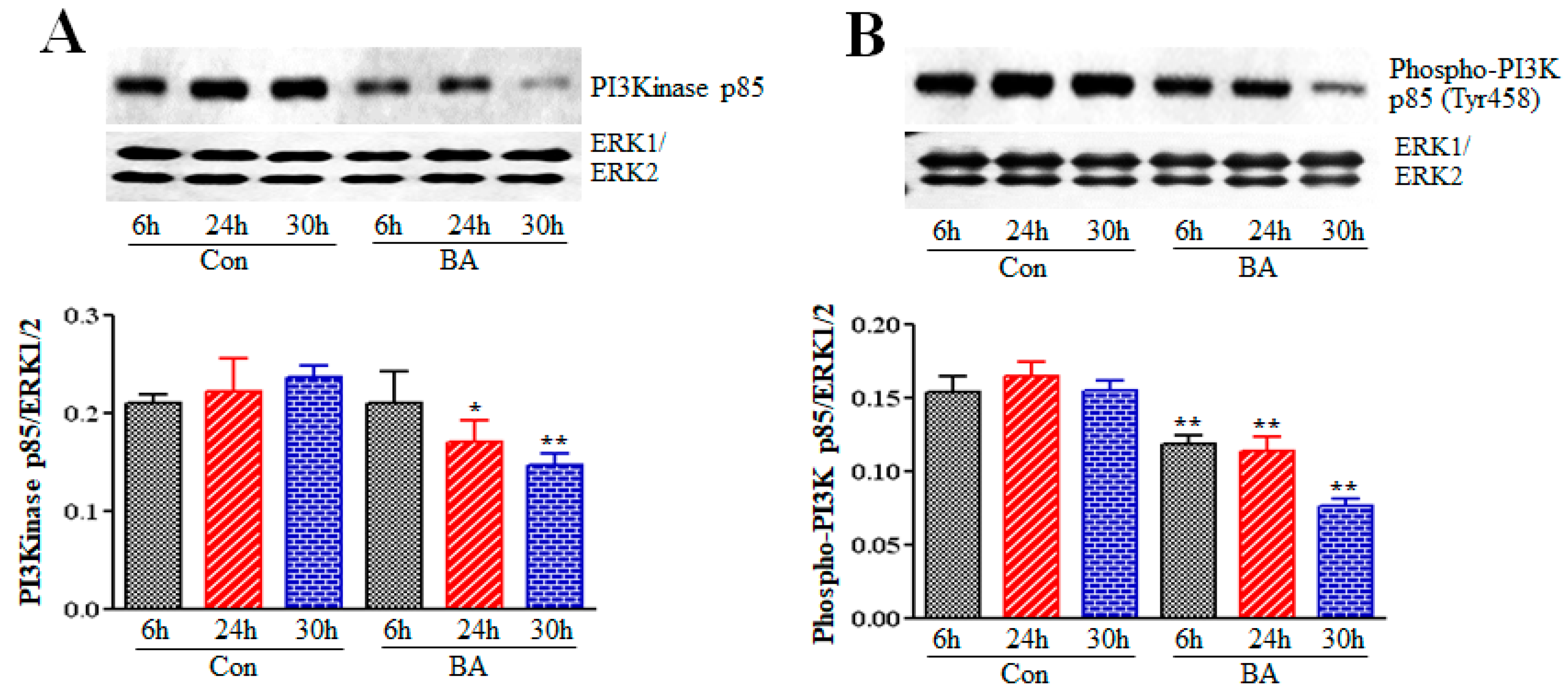
2.2. Effect of Butyrate on Phosphatidylinositol-3-Kinase (PI3K) in Proliferation Arrested VSMCs
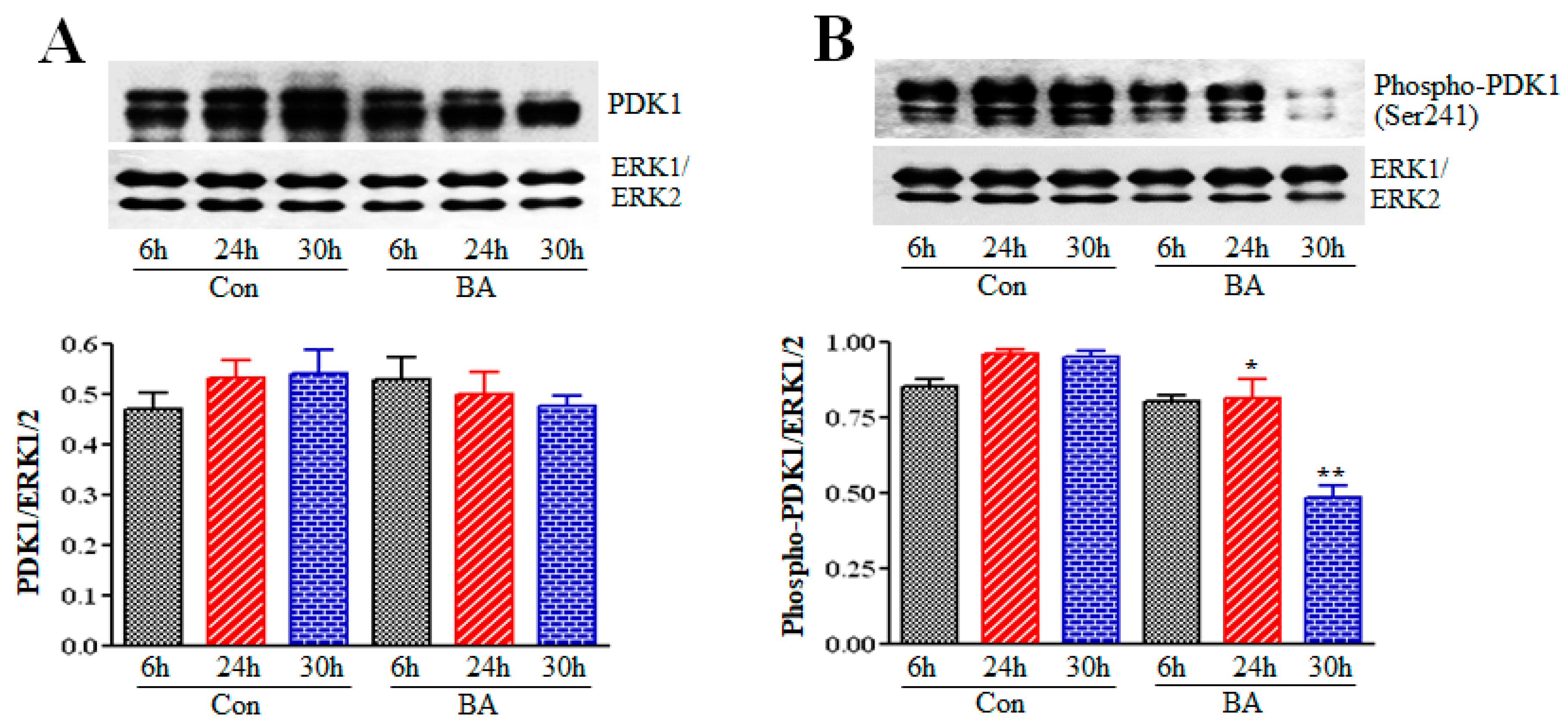
2.3. Influence of Butyrate Treatment of VSMCs on PDK1
2.4. Butyrate Treatment of VSMCs Impedes Activation of Akt
2.5. Role of Downstream Targets of Akt in Butyrate-Induced Cellular Effects of VSMCs
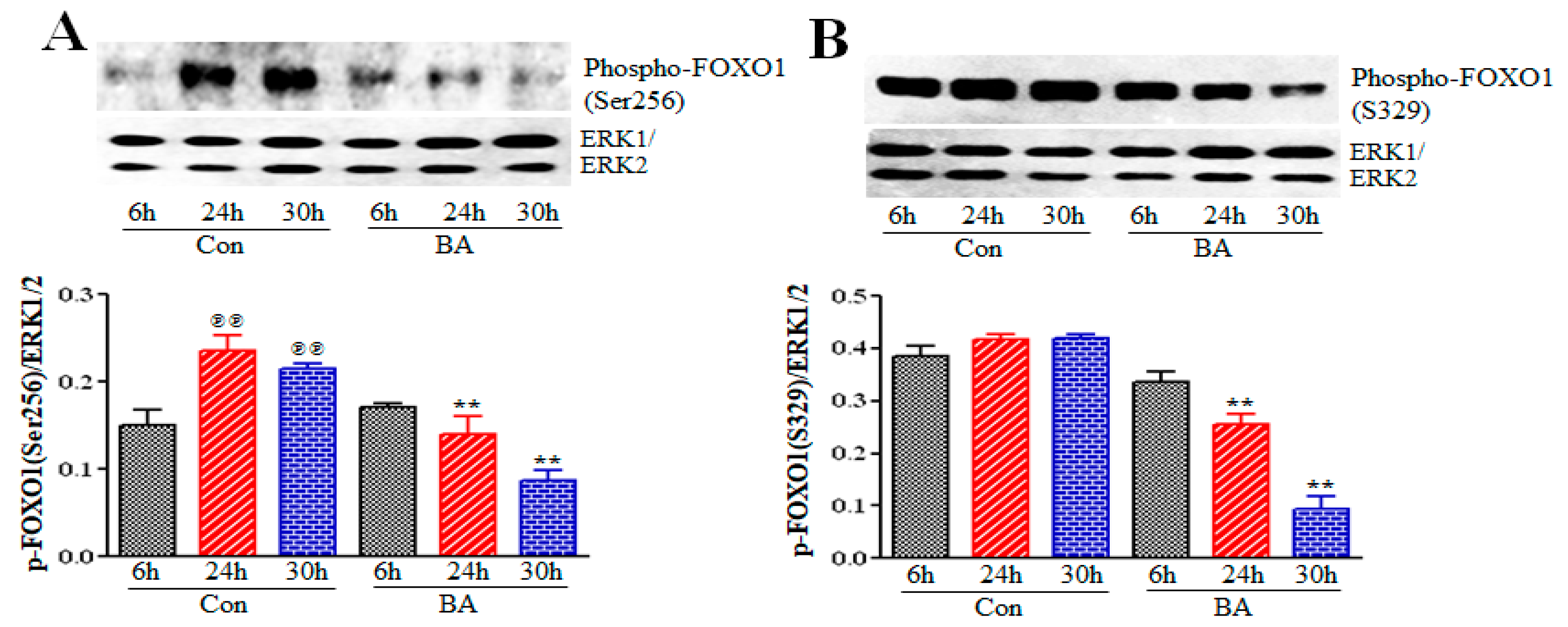
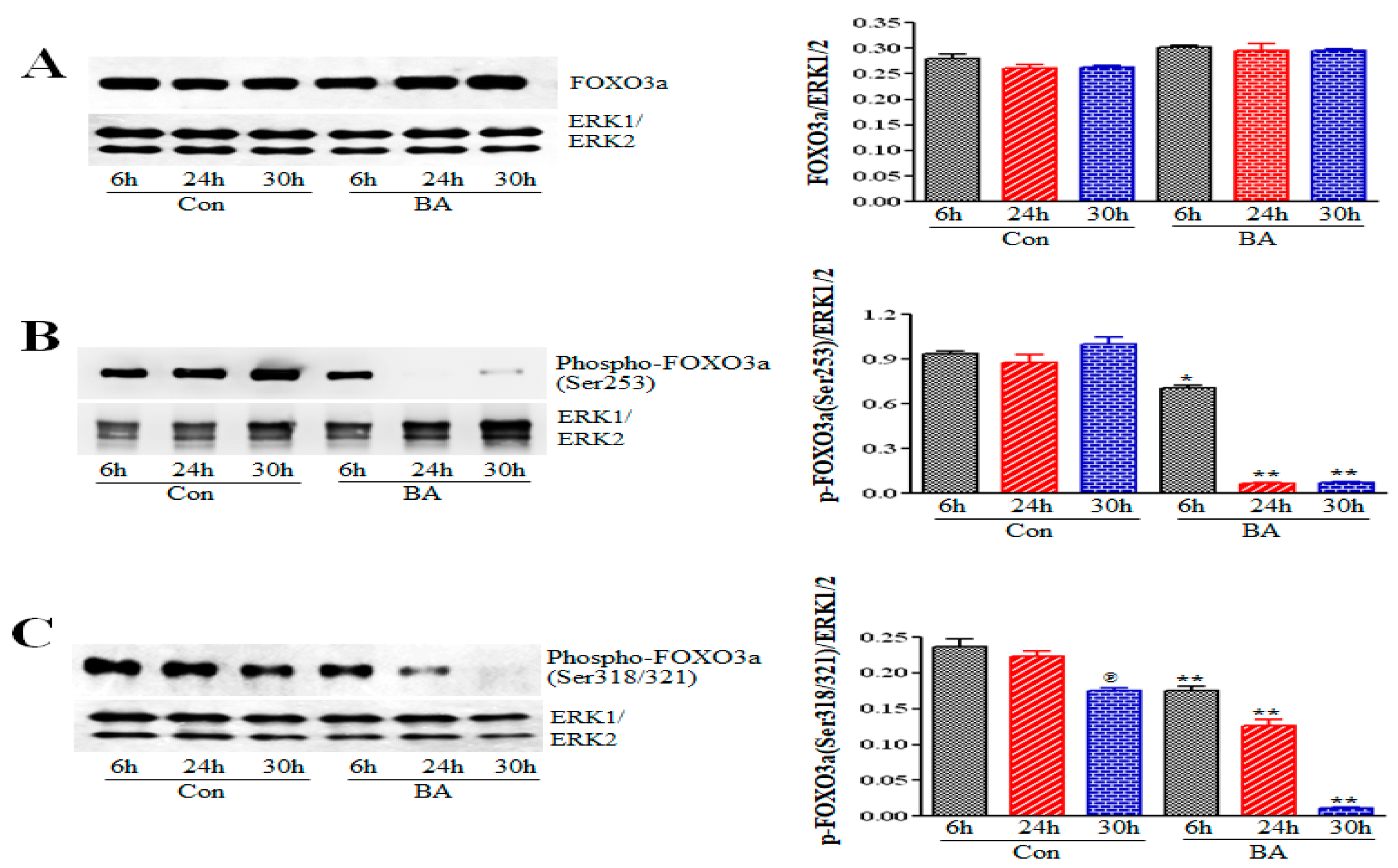
2.5.1. Impact of Butyrate-Inhibited Akt Activation on FOXO1 and FOXO3 Transcription Factors
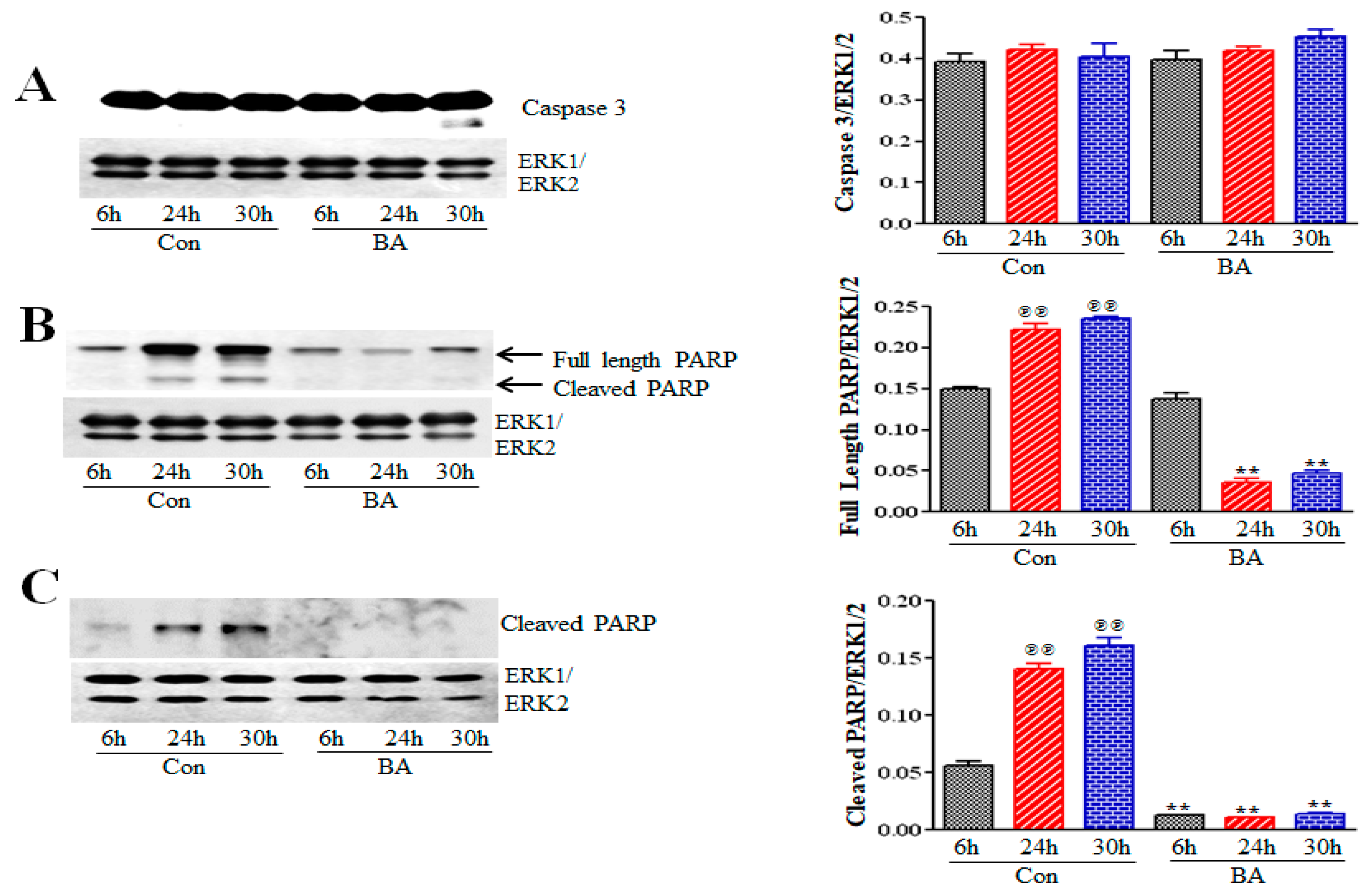
2.5.2. Butyrate Effect on Caspase 3 and Poly (ADP-Ribose) Polymerase (PARP) in VSMCs
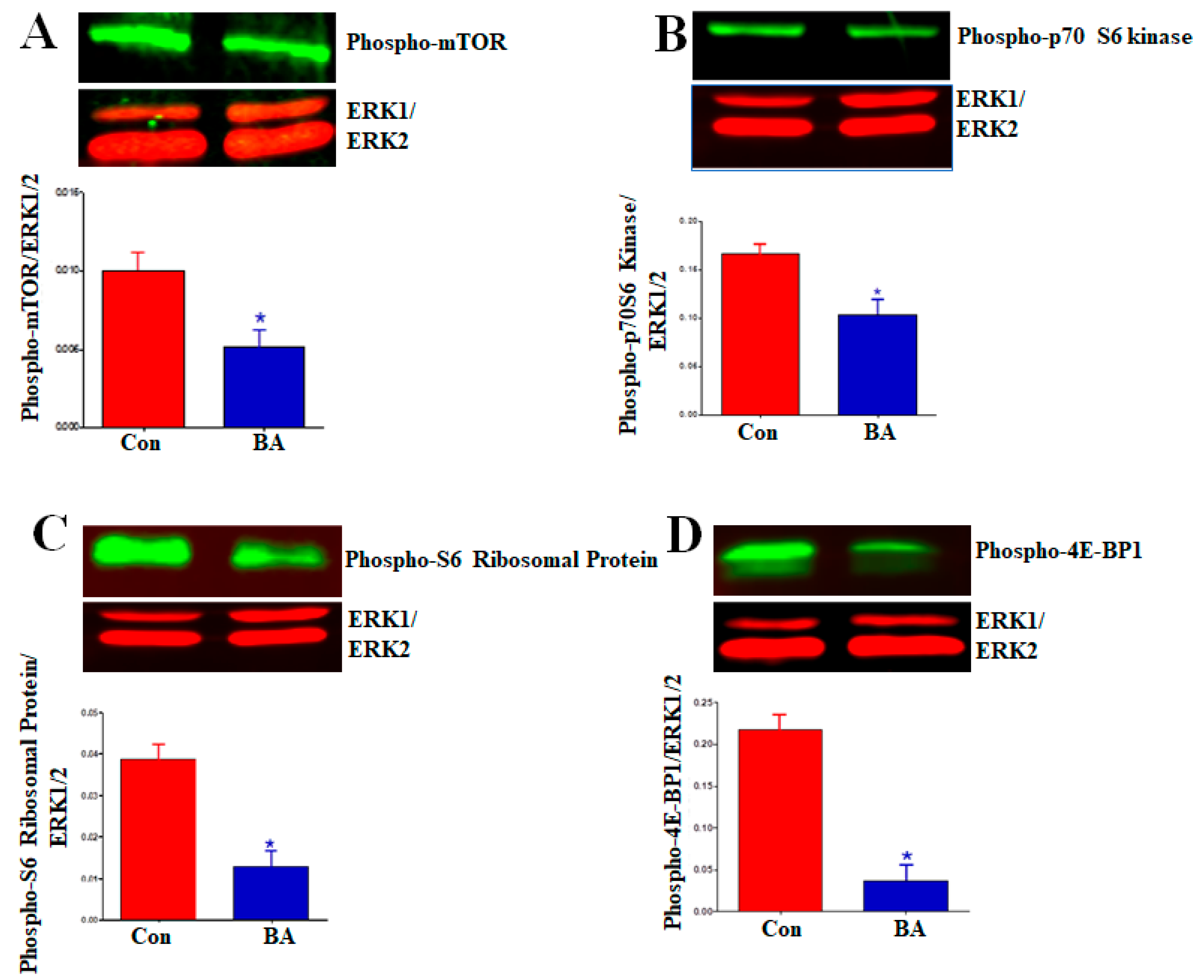
2.5.3. Impact of Butyrate-Inhibited Akt Activation on Mammalian Target of Rapamycin (mTOR)
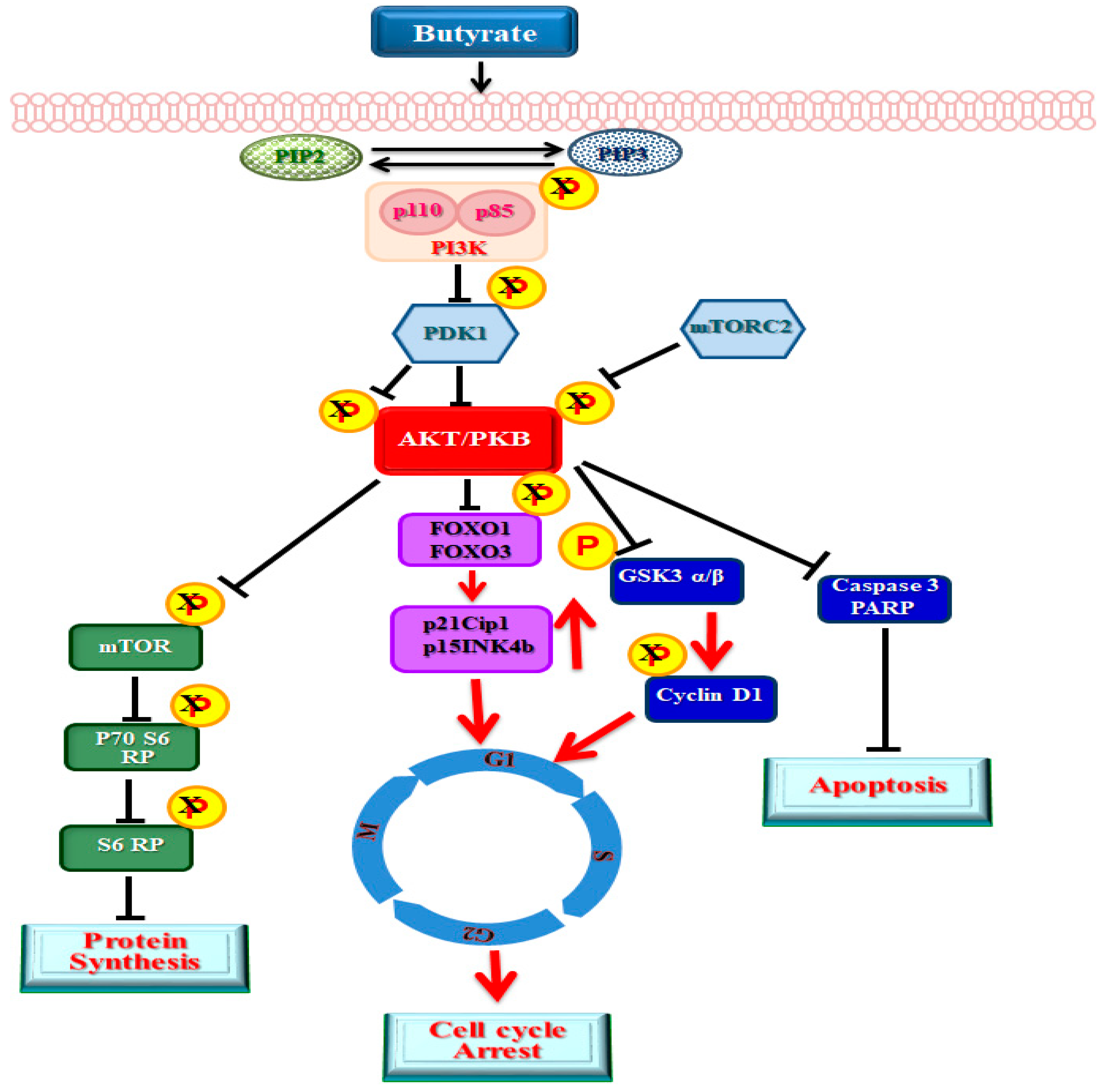
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment of VSMCs
4.2. Butyrate-Induced Cellular Effects on VSMCs
4.3. Western Blot Analysis
4.4. Data Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Go, A.S.; Muzaffarabad, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics–2013 update: A report from the American Heart Association. Circulation 2013, 127, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Pons, D.; de Vries, F.R.; van den Elsen, P.J.; Heijmans, B.T.; Quax, P.H.; Jukema, J.W. Epigenetic histone acetylation modifiers in vascular remodeling: New targets for therapy in cardiovascular disease. Eur. Heart J. 2009, 30, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Ranganna, K.; Yatsu, F.M.; Mathew, O.P. Insights into the pathogenesis and intervention of atherosclerosis. Vasc. Dis. Prev. 2006, 3, 375–390. [Google Scholar] [CrossRef]
- Weinhold, B. Epigenetics: The science of change. Envion. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- McKay, J.A.; Mathers, J.C. Diet induced epigenetic changes and their implications for health. Acta Physiol. Oxf. 2011, 202, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Phillips, T. The role of methylation in gene expression. Nat. Educ. 2008, 1, 116. [Google Scholar]
- Mattick, J.S.; Amaral, P.P.; Dinger, M.E.; Mercer, T.R.; Mehler, M.F. RNA regulation of epigenetic processes. Bioessays 2009, 31, 51–59. [Google Scholar] [CrossRef]
- Bergmann, A.; Lane, M.E. Hidden targets of microRNAs for growth control. Trends Biochem. Sci. 2003, 28, 461–463. [Google Scholar] [CrossRef]
- Simmons, D. Epigenetic influence and disease. Nat. Educ. 2008, 1, 1–69. [Google Scholar]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, M.; Akbarian, S. Epigenetic mechanisms in neurological diseases. Nat. Med. 2012, 18, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Ballestar, E. An introduction to epigenetics. In Epigenetic Contribution in Autoimmune Disease; Ballestar, E.L., Ed.; Bioscience/Springer Science + Business Media: Berlin/Heidelberg, Germany, 2011; pp. 1–11. ISBN 978-1-4419-8215-5. [Google Scholar]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Shirodkar, A.V.; Marsden, P.A. Epigenetics in cardiovascular disease. Curr. Opin. Cardiol. 2011, 26, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, A.; Ghosh, S. Environmental exposures, epigenetics and cardiovascular disease. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Udali, S.; Guarini, P.; Moruzzi, S.; Choi, S.-W.; Friso, S. Cardiovascular epigenetics: From DNA methylation to microRNA. Mol. Asp. Med. 2013, 34, 883–901. [Google Scholar] [CrossRef]
- Verdone, L.; Caserta, M.; Mauro, E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef]
- Ververis, K.; Hiong, A.; Karagiannis, T.C.; Licciardi, P.V. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Biologics 2013, 7, 47–60. [Google Scholar]
- Ranganna, K.; Yatsu, F.M.; Mathew, O.P. Emerging epigenetic therapy for vascular proliferative diseases. In Atherogenesis; Parthasarathy, S., Ed.; InTech: London, UK, 2011; ISBN 978-953-307-992-9. [Google Scholar]
- Ranganna, K.; Yatsu, F.M.; Hayes, B.E.; Milton, S.G.; Jayakumar, A. Butyrate inhibits proliferation-induced proliferating cell nuclear antigen expression (PCNA) in rat vascular smooth muscle cells. Mol. Cell Biochem. 2000, 205, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Mathew, O.P.; Ranganna, K.; Yatsu, F.M. Butyrate, an HDAC inhibitor, stimulates interplay between different posttranslational modifications of histone H3 and differently alters G1-specific cell cycle proteins in vascular smooth muscle cells. Biomed. Pharmacother. 2010, 64, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [PubMed]
- Feinburg, A.P. Epigenetics at the epicenter of modern medicine. JAMA 2008, 299, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. Histone deacetylase inhibitors: A chemical genetics approach to understanding cellular functions. Biochim. Biophys. Acta 2010, 1799, 717–725. [Google Scholar] [CrossRef]
- Li, C.J.; Elsasser, T.H. Butyrate-induced apoptosis and cell cycle arrest in bovine kidney epithelial cells: Involvement of caspase and proteasome pathways. J. Anim. Sci. 2005, 83, 89–97. [Google Scholar] [CrossRef]
- Chen, J.; Ghazawi, F.M.; Bakkar, W.; Li, Q. Valproic acid and butyrate induce apoptosis in human cancer cells through inhibition of gene expression of Akt/protein kinase B. BMC Mol. Cancer 2006, 5, 71. [Google Scholar]
- Lim, S.-J.; Choi, M.-K.; Kim, M.J.; Lee, C.-H. Effect of butyrate on the heregulin/ErbB-mediated proliferation of human colorectal cancer cells. Mol. Med. Rep. 2009, 2, 497–502. [Google Scholar] [CrossRef][Green Version]
- Li, Q.; Ding, C.; Meng, T.; Lu, W.; Liu, W.; Hao, H. Butyrate suppresses motility of colorectal cancer cells via deactivating Akt/ERK signaling in histone deacetylase dependent manner. J. Pharm. Sci. 2017, 135, 148–155. [Google Scholar] [CrossRef]
- Park, J.K.; Cho, C.H.; Ramachandran, S.; Shin, S.J.; Kwon, S.H.; Kwon, S.Y.; Cha, S.D. Augmentation of sodium butyrate-induced apoptosis by phosphatidylinositol 3-kinase inhibition in the human cervical cancer cell-line. Cancer Res. Treat. 2006, 38, 112–117. [Google Scholar] [CrossRef]
- Bai, Z.; Zhang, Z.; Ye, Y.; Wang, S. Sodium butyrate induces differentiation of gastric cancer cells to intestinal cells via PTEN/phosphoinositide 3-kinase pathway. Cell Biol. Int. 2010, 34, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Ranganna, K.; Yatsu, F.M.; Hayes, B.E. Butyrate, a small pleiotropic molecule with multiple cellular and molecular actions: Its role as an anti-atherogenic agent. Recent Res. Dev. Mol. Cell Biochem. 2005, 2, 123–151. [Google Scholar]
- Ranganna, K.; Mathew, O.P.; Milton, S.G. Regulation of cellular processes by epigenetic mechanisms of butyrate. In Butyrate: Food Sources, Functions and Health Benefits; Li, C.-J., Ed.; Biochemistry Research Trends Series; Bovine Functional Genomics Laboratory, ARS, USDA: Beltsville, MD, USA; Nova Science Publishers: Hauppauge, NY, USA, 2014; pp. 219–232. ISBN 978-1-63117-657-9. [Google Scholar]
- Milton, S.G.; Mathew, O.P.; Yatsu, F.M.; Ranganna, K. Differential cellular and molecular effects of butyrate and trichostatin A on vascular smooth muscle cells. Pharmaceuticals 2012, 5, 925–943. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, S.; Galletti, M.; Zambelli, F.; Valente, S.; Ponti, F.; Tassinari, R.; Pasquinelli, G.; Galie, N.; Ventura, C. Sodium butyrate inhibits platelet-derived growth factor-induced proliferation and migration in pulmonary artery smooth muscle cells through Akt inhibition. FEBS J. 2013, 280, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.C.; Leonel, A.J.; Teixeira, L.G.; Silva, A.R.; Silva, J.F.; Pelaez, J.M.; Capettini, L.S.; Lemos, V.S.; Santos, R.A.; Alvarez-Leite, J.I. Butyrate impairs atherogenesis by reducing plaque inflammation and vulnerability and decreasing NF-kB activation. Nutr. Metab. Cardivasc. Dis. 2014, 24, 606–613. [Google Scholar] [CrossRef]
- Ho, K.J.; Xiong, L.; Hubert, N.J.; Nadimpalli, A.; Wun, K.; Chang, E.B.; Kibbe, M.R. Vancomycin treatment and butyrate supplementation modulate gut microbe composition and severity of neointimal hyperplasia after arterial injury. Physiol. Rep. 2015, 3, e12627. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Ge, L.; Akyhani, N.; Liau, G. Sodium butyrate is a potent modulator of smooth muscle cell proliferation and gene expression. Cell Prolif. 1996, 29, 231–241. [Google Scholar] [CrossRef]
- Mathew, O.P.; Ranganna, K.; Milton, S.G. Involvement of antioxidant effect and anti-inflammatory response in butyrate inhibited vascular smooth muscle cell proliferation. Pharmaceuticals 2014, 7, 1008–1027. [Google Scholar] [CrossRef]
- Spange, S.; Wagner, T.; Heinzel, T.; Kramer, O.H. Acetylation of non-histone proteins modulates cellular signaling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Ocker, M. Deacetylase inhibitors-focus on non-histone targets and effects. World J. Biol. Chem. 2010, 1, 55–61. [Google Scholar] [CrossRef]
- Morello, F.; Perino, A.; Hirsch, E. Phosphoinositide 3-kinase signaling in the vascular system. Cardivasc. Res. 2009, 82, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathna, P.; Su, Y. The critical role of Akt in cardiovascular function. Vascul. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signaling: The path to discovery and understanding. Nat. Rev. Mol. Cell Biol. 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. Akt/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Toker, A. 3′-Phosphoinositide-dependent kinase-1 (PDK1) in PI3-kinase signaling. Front. Biosci. 2002, 7, d886–d902. [Google Scholar] [CrossRef] [PubMed]
- Allard, D.; Figg, N.; Bennett, M.R.; Littlewood, T.D. Akt regulates the survival of vascular smooth muscle cells via inhibition of FOXO3a and GSK3. J. Biol. Chem. 2008, 283, 19739–19747. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.K.; Srivastava, R.K.; Shankar, S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J. Mol. Signal 2010, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.M.; Steelman, S.L.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef]
- Nunes, R.O.; Schmidt, M.; Dueck, G.; Baarsma, H.; Halayko, A.J.; Kerstjens, H.A.; Meurs, H.; Gosens, R. GSK-3/β-catenin signaling axis in airway smooth muscle: Role in mitogeneic signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L1110–L1118. [Google Scholar] [CrossRef]
- Ranganna, K.; Mathew, O.P.; Yatsu, F.M.; Yousefipour, Z.; Hayes, B.E.; Milton, S.G. Involvement of glutathione/glutathione S-transferase antioxidant system in butyrate-inhibited vascular smooth muscle cell proliferation. FEBS J. 2007, 274, 5962–5978. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.J.; Abraham, R.T.; Yu, K. Mammailan target of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin. Oncol. 2009, 36, S3–S17. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.; Siegel, N.; Valli, A.; Fuchs, C.; Hengstschlager, M. mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids 2010, 38, 223–228. [Google Scholar] [CrossRef]
- Schwartz, S.M.; deBlois, D.; O’Brien, E.R. The intima: Soil for atherosclerosis and restenosis. Circ. Res. 1995, 77, 445–465. [Google Scholar] [CrossRef]
- Alfonso, F.; Byrne, R.A.; Rivero, F.; Kastrati, A. Current treatment of in-stent restenosis. J. Am. Coll. Cardiol. 2014, 163, 2659–2673. [Google Scholar] [CrossRef] [PubMed]
- Shawky, N.M.; Segar, L. Sulforaphane inhibits platelet-derived growth factor-induced vascular smooth muscle cell proliferation by targeting mTOR/p70S6kinase signaling independent of Nrf2 activation. Pharmacol. Res. 2017, 119, 251–264. [Google Scholar] [CrossRef]
- Clarke, M.C.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Littlewood, T.D.; Bennett, M.R. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat. Med. 2006, 12, 1075–1080. [Google Scholar] [CrossRef]
- Li, H.; Zhang, T.-B.; Jia, D.-H.; Sun, W.-Q.; Wang, C.-L.; Gu, A.-Z.; Yang, X.-M. Genipin inhibits the growth of human bladder cancer cells via inactivation of PI3K/Akt signaling. Oncol. Lett. 2018, 15, 2619–2624. [Google Scholar] [CrossRef]
- Guan, F.; Yang, X.; Li, J.; Dong, W.; Zhang, X.; Liu, N.; Gao, S.; Wang, J.; Zhang, L.; Lu, D. New molecular mechanism underlying myc-mediated cytochrome P450 2E1 upregulation in apoptosis and energy metabolism in the myocardium. J. Am. Heart Assoc. 2019, 8, e009871. [Google Scholar] [CrossRef]
- Liu, K.; Shi, Y.; Guo, X.H.; Ouyang, Y.B.; Wang, S.S.; Liu, D.J.; Wang, A.N.; Li, N.; Chen, D.X. Phosphorylated AKT inhibits the apoptosis induced by DRAM-mediated mitophagy in hepatocellular carcinoma by preventing the translocation of DRAM to mitochondria. Cell Death Dis. 2014, 5, e1078. [Google Scholar] [CrossRef]
- Jung, F.; Haendeler, J.; Goebel, C.; Zeiher, A.M.; Dimmeler, S. Growth factor-induced phosphoinositide 3-OH kinase/Akt phosphorylation in smooth muscle cells: Induction of cell proliferation and inhibition of cell death. Cardiovasc. Res. 2000, 48, 148–157. [Google Scholar] [CrossRef]
- Nim, T.H.; Luo, L.; White, J.K.; Clement, M.-V.; Tucker-Kellogg, L. Non-canonical Activation of Akt in Serum-Stimulated Fibroblasts, Revealed by Comparative Modeling of Pathway Dynamics. PLoS Comput. Biol. 2015, 11, e1004505. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes Cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FOXO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Daitoku, H.; Sakamaki, J.-I.; Fukamizu, A. Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochim. Biophys. Acta 2011, 1831, 1954–1960. [Google Scholar] [CrossRef]
- Barkauskaite, E.; Jankevicius, G.; Ahel, I. Structures and mechanisms of enzymes employed in the synthesis and degradation of PARP-dependent protein ADP-ribosylation. Mol. Cell 2015, 58, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Marek Banasik, M.; Stedeford, T.; Strosznajder, R.P. Natural Inhibitors of Poly (ADP-ribose). Polymerase-1. Mol. Neurobiol. 2012, 46, 55–63. [Google Scholar] [CrossRef]
- Dominguez-Calderon, A.; Avila-Flores, A.; Ponce, A.; Lopez-Bayghen, E.; Calderon-Salinas, J.-V.; Reyes, J.L.; Chávez-Munguía, B.; Segovia, J.; Angulo, C.; Ramírez, L.; et al. ZO-2 silencing induces renal hypertrophy through a cell cycle mechanism and the activation of YAP and the mTOR pathway. MBoC 2016, 27, 1581–1595. [Google Scholar] [CrossRef]
- Lloyd, A.C. The regulation of cell size. Cell 2013, 154, 1194–1205. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.A. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Preisig, P.A. TGF-beta1-mediated hypertrophy involves inhibiting pRB- phosphorylation by blocking activation of cyclin E kinase. Am. J. Physiol. 1999, 277, F186–F194. [Google Scholar] [PubMed]
- Franch, H.A.; Shay, J.W.; Alpern, R.J.; Preisig, P.A. Involvement of pRB family in TGF-beta-dependent epithelial cell hypertrophy. J. Cell Biol. 1995, 129, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Monkawa, T.; Hiromura, K.; Wolf, G.; Shanklland, S.J. The hypertrophic effect of transforming growth factor-beta is reduced in the absence of cyclin-dependent kinase inhibitors p21 and p27. J. Am. Soc. Nephrol. 2002, 13, 1172–1178. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wolf, G.; Jablonski, K.; Schroder, R.; Reinking, R.; Shankland, S.J.; Stahl, R.A. Angiotensin II-induced hypertrophy of proximal tubular cells requires p27Kip1. Kidney Int. 2003, 64, 71–81. [Google Scholar] [CrossRef]
- Fu, M.; Wang, C.; Li, Z.; Sakamaki, T.; Pestell, R.G. Cyclin D1: Normal and abnormal functions. Endocrinology 2004, 145, 5439–5447. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathew, O.P.; Ranganna, K.; Mathew, J.; Zhu, M.; Yousefipour, Z.; Selvam, C.; Milton, S.G. Cellular Effects of Butyrate on Vascular Smooth Muscle Cells are Mediated through Disparate Actions on Dual Targets, Histone Deacetylase (HDAC) Activity and PI3K/Akt Signaling Network. Int. J. Mol. Sci. 2019, 20, 2902. https://doi.org/10.3390/ijms20122902
Mathew OP, Ranganna K, Mathew J, Zhu M, Yousefipour Z, Selvam C, Milton SG. Cellular Effects of Butyrate on Vascular Smooth Muscle Cells are Mediated through Disparate Actions on Dual Targets, Histone Deacetylase (HDAC) Activity and PI3K/Akt Signaling Network. International Journal of Molecular Sciences. 2019; 20(12):2902. https://doi.org/10.3390/ijms20122902
Chicago/Turabian StyleMathew, Omana P., Kasturi Ranganna, Joseph Mathew, Meiling Zhu, Zivar Yousefipour, Chelliah Selvam, and Shirlette G. Milton. 2019. "Cellular Effects of Butyrate on Vascular Smooth Muscle Cells are Mediated through Disparate Actions on Dual Targets, Histone Deacetylase (HDAC) Activity and PI3K/Akt Signaling Network" International Journal of Molecular Sciences 20, no. 12: 2902. https://doi.org/10.3390/ijms20122902
APA StyleMathew, O. P., Ranganna, K., Mathew, J., Zhu, M., Yousefipour, Z., Selvam, C., & Milton, S. G. (2019). Cellular Effects of Butyrate on Vascular Smooth Muscle Cells are Mediated through Disparate Actions on Dual Targets, Histone Deacetylase (HDAC) Activity and PI3K/Akt Signaling Network. International Journal of Molecular Sciences, 20(12), 2902. https://doi.org/10.3390/ijms20122902