Roles of E-cadherin and Noncoding RNAs in the Epithelial–mesenchymal Transition and Progression in Gastric Cancer

Abstract
1. Introduction
2. E-cadherin and Its Biological Significance
3. Role of E-cadherin in Carcinogenesis: The Epithelial–Mesenchymal Transition (EMT)
4. Signaling Pathways Regulated by E-cadherin
5. Mechanisms of E-cadherin Inactivation in the Tumors
6. Role of MiRNAs in Regulation of E-cadherin Expression in Gastric Cancer
MiRNAs in Diagnostic and Therapy of Gastric Cancer
7. Long Noncoding RNAs Involved in the EMT and Regulation of E-cadherin Expression in Gastric Cancer
7.1. Oncogenic ncRNAs in Gastric Cancer
7.2. Tumour Suppressor ncRNAs in Gastric Cancer
7.3. LncRNAs in Diagnostic and Therapy of Gastric Cancer
8. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Przeglad Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, F. Stomach cancer. World Cancer Rep. 2014, 383–391. [Google Scholar]
- Jang, B.-G.; Kim, W.H. Molecular pathology of gastric carcinoma. Pathobiol. J. Immunopathol. Mol. Cell. Biol. 2011, 78, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Janjigian, Y.Y.; Kelsen, D.P. Genomic dysregulation in gastric tumors. J. Surg. Oncol. 2013, 107, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Röcken, C. Molecular classification of gastric cancer. Expert Rev. Mol. Diagn. 2017, 17, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [CrossRef]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Taft, R.J.; Pheasant, M.; Mattick, J.S. The relationship between non-protein-coding DNA and eukaryotic complexity. BioEssays News Rev. Mol. Cell. Dev. Biol. 2007, 29, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Bure, I.V.; Kuznetsova, E.B.; Zaletaev, D.V. Long Noncoding RNAs and Their Role in Oncogenesis. Mol. Biol. 2018, 52, 907–920. [Google Scholar] [CrossRef]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Hyafil, F.; Babinet, C.; Jacob, F. Cell–cell interactions in early embryogenesis: A molecular approach to the role of calcium. Cell 1981, 26, 447–454. [Google Scholar] [CrossRef]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-cadherin-integrin crosstalk in cancer invasion and metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Ratheesh, A.; Yap, A.S. A bigger picture: Classical cadherins and the dynamic actin cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 673–679. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial–mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Beetham, H.; Black, M.A.; Priya, R.; Telford, B.J.; Guest, J.; Wiggins, G.A.R.; Godwin, T.D.; Yap, A.S.; Guilford, P.J. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial–mesenchymal transition. BMC Cancer 2014, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Peeters, J.K.; Smid, M.; Timmermans, M.; Verhoog, L.C.; Westenend, P.J.; Heine, A.A.J.; Chan, A.; Sieuwerts, A.M.; Wiemer, E.A.C.; et al. Loss of E-cadherin is not a necessity for epithelial to mesenchymal transition in human breast cancer. Breast Cancer Res. Treat. 2013, 138, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Villarejo, A.; Cortés-Cabrera, A.; Molina-Ortíz, P.; Portillo, F.; Cano, A. Differential role of Snail1 and Snail2 zinc fingers in E-cadherin repression and epithelial to mesenchymal transition. J. Biol. Chem. 2014, 289, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, C.; Gao, W.; Chen, T.; Qian, T.; Hu, J.; Tan, Y. FOXA2 attenuates the epithelial to mesenchymal transition by regulating the transcription of E-cadherin and ZEB2 in human breast cancer. Cancer Lett. 2015, 361, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhang, L.; Zhu, Y.; Ke, X.; Wang, Q.; Min, H. Regulation of Proliferation and Epithelial-to-Mesenchymal Transition (EMT) of Gastric Cancer by ZEB1 via Modulating Wnt5a and Related Mechanisms. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, H.; Basilicata, M.F.; Shehwana, H.; Kosowan, T.; Schreck, I.; Braeutigam, C.; Konu, O.; Brabletz, T.; Stemmler, M.P. Enhancer cooperativity as a novel mechanism underlying the transcriptional regulation of E-cadherin during mesenchymal to epithelial transition. Biochim. Biophys. Acta 2015, 1849, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.M.; Fang, C.M.; Chuah, L.-H.; Leong, C.O.; Ngai, S.C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit. Rev. Oncol. Hematol. 2018, 121, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef]
- Pan, Y.; Bi, F.; Liu, N.; Xue, Y.; Yao, X.; Zheng, Y.; Fan, D. Expression of seven main Rho family members in gastric carcinoma. Biochem. Biophys. Res. Commun. 2004, 315, 686–691. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Yong, X.; Tang, B.; Li, B.-S.; Xie, R.; Hu, C.-J.; Luo, G.; Qin, Y.; Dong, H.; Yang, S.-M. Helicobacter pylori virulence factor CagA promotes tumorigenesis of gastric cancer via multiple signaling pathways. Cell Commun. Signal. CCS 2015, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Suriano, G.; Oliveira, M.J.; Huntsman, D.; Mateus, A.R.; Ferreira, P.; Casares, F.; Oliveira, C.; Carneiro, F.; Machado, J.C.; Mareel, M.; et al. E-cadherin germline missense mutations and cell phenotype: Evidence for the independence of cell invasion on the motile capabilities of the cells. Hum. Mol. Genet. 2003, 12, 3007–3016. [Google Scholar] [CrossRef] [PubMed]
- Bremm, A.; Walch, A.; Fuchs, M.; Mages, J.; Duyster, J.; Keller, G.; Hermannstädter, C.; Becker, K.-F.; Rauser, S.; Langer, R.; et al. Enhanced activation of epidermal growth factor receptor caused by tumor-derived E-cadherin mutations. Cancer Res. 2008, 68, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Mateus, A.R.; Seruca, R.; Machado, J.C.; Keller, G.; Oliveira, M.J.; Suriano, G.; Luber, B. EGFR regulates RhoA-GTP dependent cell motility in E-cadherin mutant cells. Hum. Mol. Genet. 2007, 16, 1639–1647. [Google Scholar] [CrossRef] [PubMed]
- Soto, E.; Yanagisawa, M.; Marlow, L.A.; Copland, J.A.; Perez, E.A.; Anastasiadis, P.Z. p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression. J. Cell Biol. 2008, 183, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Cowell, C.F.; Yan, I.K.; Eiseler, T.; Leightner, A.C.; Döppler, H.; Storz, P. Loss of cell–cell contacts induces NF-kappaB via RhoA-mediated activation of protein kinase D1. J. Cell. Biochem. 2009, 106, 714–728. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Sokolova, O.; Naumann, M. NF-κB Signaling in Gastric Cancer. Toxins 2017, 9, 119. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Kuphal, S.; Poser, I.; Jobin, C.; Hellerbrand, C.; Bosserhoff, A.K. Loss of E-cadherin leads to upregulation of NFkappaB activity in malignant melanoma. Oncogene 2004, 23, 8509–8519. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Shin, J.-H.; Kee, S.-H. E-cadherin expression increases cell proliferation by regulating energy metabolism through nuclear factor-κB in AGS cells. Cancer Sci. 2017, 108, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chu, K.-M. E-cadherin and gastric cancer: Cause, consequence, and applications. BioMed Res. Int. 2014, 2014, 637308. [Google Scholar] [CrossRef] [PubMed]
- van der Post, R.S.; Vogelaar, I.P.; Carneiro, F.; Guilford, P.; Huntsman, D.; Hoogerbrugge, N.; Caldas, C.; Schreiber, K.E.C.; Hardwick, R.H.; Ausems, M.G.E.M.; et al. Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J. Med. Genet. 2015, 52, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Melo, S.; Carneiro, P.; Moreira, A.M.; Fernandes, M.S.; Ribeiro, A.S.; Guilford, P.; Paredes, J.; Seruca, R. Clinical spectrum and pleiotropic nature of CDH1 germline mutations. J. Med. Genet. 2019, 56, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Sivakumaran, S.; Agakov, F.; Theodoratou, E.; Prendergast, J.G.; Zgaga, L.; Manolio, T.; Rudan, I.; McKeigue, P.; Wilson, J.F.; Campbell, H. Abundant pleiotropy in human complex diseases and traits. Am. J. Hum. Genet. 2011, 89, 607–618. [Google Scholar] [CrossRef]
- Simões-Correia, J.; Silva, D.I.; Melo, S.; Figueiredo, J.; Caldeira, J.; Pinto, M.T.; Girão, H.; Pereira, P.; Seruca, R. DNAJB4 molecular chaperone distinguishes WT from mutant E-cadherin, determining their fate in vitro and in vivo. Hum. Mol. Genet. 2014, 23, 2094–2105. [Google Scholar] [CrossRef]
- Virani, S.; Virani, S.; Colacino, J.A.; Kim, J.H.; Rozek, L.S. Cancer epigenetics: A brief review. ILAR J. 2012, 53, 359–369. [Google Scholar] [CrossRef]
- Qu, Y.; Dang, S.; Hou, P. Gene methylation in gastric cancer. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 424, 53–65. [Google Scholar] [CrossRef]
- Park, J.; Jang, K.L. Hepatitis C virus represses E-cadherin expression via DNA methylation to induce epithelial to mesenchymal transition in human hepatocytes. Biochem. Biophys. Res. Commun. 2014, 446, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Seto, Y. CpG island methylator phenotype, Helicobacter pylori, Epstein-Barr virus, and microsatellite instability and prognosis in gastric cancer: A systematic review and meta-analysis. PLoS ONE 2014, 9, e86097. [Google Scholar] [CrossRef] [PubMed]
- Fukayama, M.; Ushiku, T. Epstein-Barr virus-associated gastric carcinoma. Pathol. Res. Pract. 2011, 207, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Toyota, M.; Suzuki, H.; Nojima, M.; Yamamoto, E.; Kamimae, S.; Watanabe, Y.; Kai, M.; Akashi, H.; Maruyama, R.; et al. DNA methylation of interferon regulatory factors in gastric cancer and noncancerous gastric mucosae. Cancer Sci. 2010, 101, 1708–1716. [Google Scholar] [CrossRef] [PubMed]
- Nemtsova, M.V.; Strelnikov, V.V.; Tanas, A.S.; Bykov, I.I.; Zaletaev, D.V.; Rudenko, V.V.; Glukhov, A.I.; Kchorobrich, T.V.; Li, Y.; Tarasov, V.V.; et al. Implication of Gastric Cancer Molecular Genetic Markers in Surgical Practice. Curr. Genomics 2017, 18, 408–415. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Polk, D.B.; Peek, R.M. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Calcagno, D.Q.; Gigek, C.O.; Chen, E.S.; Burbano, R.R.; Smith Mde, A. DNA and histone methylation in gastric carcinogenesis. World J. Gastroenterol. 2013, 19, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.; Stadler, S.C. Role of epigenetic mechanisms in epithelial-to-mesenchymal transition of breast cancer cells. Transl. Res. J. Lab. Clin. Med. 2015, 165, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.-I.; Evers, B.M.; Zhou, B.P. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef]
- Fukagawa, A.; Ishii, H.; Miyazawa, K.; Saitoh, M. δEF1 associates with DNMT1 and maintains DNA methylation of the E-cadherin promoter in breast cancer cells. Cancer Med. 2015, 4, 125–135. [Google Scholar] [CrossRef]
- Naidu, S.; Magee, P.; Garofalo, M. MiRNA-based therapeutic intervention of cancer. J. Hematol. Oncol. 2015, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Geng, Y.; Feng, R.; Zhu, Q.; Miao, B.; Cao, J.; Fei, S. The Human RNA Surveillance Factor UPF1 Modulates Gastric Cancer Progression by Targeting Long Non-Coding RNA MALAT1. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 2194–2206. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhang, Y.; She, Q.; Li, X.; Peng, L.; Wang, X.; Liu, S.; Shen, X.; Zhang, W.; Dong, Y.; et al. Long Noncoding RNA H19/miR-675 Axis Promotes Gastric Cancer via FADD/Caspase 8/Caspase 3 Signaling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 2364–2376. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Tian, J.; Shi, W.; Xia, H.; Zhu, Y. LncRNA SNHG6 is Associated with Poor Prognosis of Gastric Cancer and Promotes Cell Proliferation and EMT through Epigenetically Silencing p27 and Sponging miR-101-3p. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Ye, X.; Du, Q.; Zhu, B.; Zhai, Q.; Li, X.-X. The Long Non-Coding RNA CRNDE Promotes Colorectal Carcinoma Progression by Competitively Binding miR-217 with TCF7L2 and Enhancing the Wnt/β-Catenin Signaling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 41, 2489–2502. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.-S.; Gao, W.; Chan, J.Y.-W. Interactions between E-cadherin and microRNA deregulation in head and neck cancers: The potential interplay. BioMed Res. Int. 2014, 2014, 126038. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M. An overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Bure, I.; Haller, F.; Zaletaev, D.V. Coding and Non-coding: Molecular Portrait of GIST and its Clinical Implication. Curr. Mol. Med. 2018, 18, 252–259. [Google Scholar] [CrossRef]
- Ma, D.-N.; Chai, Z.-T.; Zhu, X.-D.; Zhang, N.; Zhan, D.-H.; Ye, B.-G.; Wang, C.-H.; Qin, C.-D.; Zhao, Y.-M.; Zhu, W.-P.; et al. MicroRNA-26a suppresses epithelial–mesenchymal transition in human hepatocellular carcinoma by repressing enhancer of zeste homolog 2. J. Hematol. Oncol. J. Hematol. Oncol. 2016, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Kurata, A.; Yamada, M.; Ohno, S.-I.; Inoue, S.; Hashimoto, H.; Fujita, K.; Takanashi, M.; Kuroda, M. Expression level of microRNA-200c is associated with cell morphology in vitro and histological differentiation through regulation of ZEB1/2 and E-cadherin in gastric carcinoma. Oncol. Rep. 2018, 39, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J.; van Grieken, N.C.; Pereira, P.M.; Sousa, S.; Tijssen, M.; Buffart, T.E.; Diosdado, B.; Grabsch, H.; Santos, M.A.S.; Meijer, G.; et al. Lack of microRNA-101 causes E-cadherin functional deregulation through EZH2 up-regulation in intestinal gastric cancer. J. Pathol. 2012, 228, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, J.; Zhu, Y.; Dai, Y.; Zeng, T.; Liu, L.; Li, J.; Wang, H.; Qin, Y.; Zeng, M.; et al. MicroRNA-9 promotes tumor metastasis via repressing E-cadherin in esophageal squamous cell carcinoma. Oncotarget 2014, 5, 11669–11680. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kumar, S.M.; Lu, H.; Liu, A.; Yang, R.; Pushparajan, A.; Guo, W.; Xu, X. MicroRNA-9 up-regulates E-cadherin through inhibition of NF-κB1-Snail1 pathway in melanoma. J. Pathol. 2012, 226, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.M.; Ferreira, R.M.; Pinto-Ribeiro, I.; Sougleri, I.S.; Oliveira, M.J.; Carreto, L.; Santos, M.A.; Sgouras, D.N.; Carneiro, F.; Leite, M.; et al. Helicobacter pylori Activates Matrix Metalloproteinase 10 in Gastric Epithelial Cells via EGFR and ERK-mediated Pathways. J. Infect. Dis. 2016, 213, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, X.; Du, J.; Yin, Y.; Li, Y. Involvement of microRNAs-MMPs-E-cadherin in the migration and invasion of gastric cancer cells infected with Helicobacter pylori. Exp. Cell Res. 2018, 367, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Naito, Y.; Oue, N.; Sentani, K.; Uraoka, N.; Zarni Oo, H.; Yanagihara, K.; Aoyagi, K.; Sasaki, H.; Yasui, W. MicroRNA-148a is downregulated in gastric cancer, targets MMP7, and indicates tumor invasiveness and poor prognosis. Cancer Sci. 2014, 105, 236–243. [Google Scholar] [CrossRef]
- Wang, S.-H.; Li, X.; Zhou, L.-S.; Cao, Z.-W.; Shi, C.; Zhou, C.-Z.; Wen, Y.-G.; Shen, Y.; Li, J.-K. microRNA-148a suppresses human gastric cancer cell metastasis by reversing epithelial-to-mesenchymal transition. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2013, 34, 3705–3712. [Google Scholar] [CrossRef]
- Cui, H.; Wang, L.; Gong, P.; Zhao, C.; Zhang, S.; Zhang, K.; Zhou, R.; Zhao, Z.; Fan, H. Deregulation between miR-29b/c and DNMT3A is associated with epigenetic silencing of the CDH1 gene, affecting cell migration and invasion in gastric cancer. PLoS ONE 2015, 10, e0123926. [Google Scholar] [CrossRef]
- Li, L.-Q.; Pan, D.; Chen, Q.; Zhang, S.-W.; Xie, D.-Y.; Zheng, X.-L.; Chen, H. Sensitization of Gastric Cancer Cells to 5-FU by MicroRNA-204 Through Targeting the TGFBR2-Mediated Epithelial to Mesenchymal Transition. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wu, J.; Huang, W.; Peng, J.; Ye, J.; Yang, L.; Yuan, Y.; Chen, C.; Zhang, C.; Cai, S.; et al. Long Non-Coding RNA RP11-789C1.1 Suppresses Epithelial to Mesenchymal Transition in Gastric Cancer Through the RP11-789C1.1/MiR-5003/E-Cadherin Axis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 2432–2444. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, Y.-Q. MiR-217 is involved in the carcinogenesis of gastric cancer by down-regulating CDH1 expression. Kaohsiung J. Med. Sci. 2018, 34, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.F.; Carvalho, J.; Pinto-Ribeiro, I.; Almeida, C.; Wengel, J.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Targeting miR-9 in gastric cancer cells using locked nucleic acid oligonucleotides. BMC Mol. Biol. 2018, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, Y.; Muramatsu, T.; Uetake, H.; Kozaki, K.; Inazawa, J. miR-544a induces epithelial–mesenchymal transition through the activation of WNT signaling pathway in gastric cancer. Carcinogenesis 2015, 36, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-P.; Sun, X.-H.; Cao, X.-L.; Jiang, W.-W.; Wang, X.-X.; Zhang, Y.-F.; Wang, J.-L. MicroRNA-217 suppressed epithelial-to-mesenchymal transition in gastric cancer metastasis through targeting PTPN14. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1759–1767. [Google Scholar] [PubMed]
- Zhao, X.; He, L.; Li, T.; Lu, Y.; Miao, Y.; Liang, S.; Guo, H.; Bai, M.; Xie, H.; Luo, G.; et al. SRF expedites metastasis and modulates the epithelial to mesenchymal transition by regulating miR-199a-5p expression in human gastric cancer. Cell Death Differ. 2014, 21, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liang, Y.; Ma, M.-H.; Wu, K.-Z.; Zhang, C.-D.; Dai, D.-Q. Downregulation of microRNA-376a in Gastric Cancer and Association with Poor Prognosis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 51, 2010–2018. [Google Scholar] [CrossRef]
- Cao, Q.; Liu, F.; Ji, K.; Liu, N.; He, Y.; Zhang, W.; Wang, L. MicroRNA-381 inhibits the metastasis of gastric cancer by targeting TMEM16A expression. J. Exp. Clin. Cancer Res. CR 2017, 36, 29. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Y.; Li, N.; Deng, W.Y.; Ma, Y.J.; Han, X.L.; Zhang, Z.Y.; Xie, J.L.; Luo, S.X. miRNA-96-5p inhibits the proliferation and migration of gastric cancer cells by targeting FoxQ1. Zhonghua Zhong Liu Za Zhi 2019, 41, 193–199. [Google Scholar]
- Wang, M.; Zhang, R.; Zhang, S.; Xu, R.; Yang, Q. MicroRNA-574-3p regulates epithelial mesenchymal transition and cisplatin resistance via targeting ZEB1 in human gastric carcinoma cells. Gene 2019, 700, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Shi, L.; Yang, C.; Ge, Y.; Lin, L.; Fan, H.; He, Y.; Zhang, D.; Miao, Y.; Yang, L. miR-1254 inhibits cell proliferation, migration, and invasion by down-regulating Smurf1 in gastric cancer. Cell Death Dis. 2019, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xu, M.; Guo, Y.; Ye, L.; Long, L.; Wang, H.; Tan, P.; Xu, M. MicroRNA-588 regulates invasion, migration and epithelial–mesenchymal transition via targeting EIF5A2 pathway in gastric cancer. Cancer Manag. Res. 2018, 10, 5187–5197. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tian, S.; Chen, Y.; Ji, M.; Qu, Y.; Hou, P. miR-218 inhibits gastric tumorigenesis through regulating Bmi-1/Akt signaling pathway. Pathol. Res. Pract. 2019, 215, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fu, Y.; Liu, G.; Ye, Y.; Zhang, X. miR-218 Inhibits Proliferation, Migration, and EMT of Gastric Cancer Cells by Targeting WASF3. Oncol. Res. 2017, 25, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sun, T.; Yu, Y.; Gao, Y.; Wang, X.; Chen, Z. MicroRNA-370 inhibits the proliferation, invasion and EMT of gastric cancer cells by directly targeting PAQR4. J. Pharmacol. Sci. 2018, 138, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.-S.; Li, D.-F.; Tang, Y.-P.; Chen, Y.-Z.; Deng, W.-B.; Chen, J.; Zhou, W.-W.; Liao, A.-J. Inhibition of epithelial-mesenchymal transition in gastric cancer cells by miR-711-mediated downregulation of CD44 expression. Oncol. Rep. 2018, 40, 2844–2853. [Google Scholar] [CrossRef]
- Xu, J.; Wang, F.; Wang, X.; He, Z.; Zhu, X. miRNA-543 promotes cell migration and invasion by targeting SPOP in gastric cancer. OncoTargets Ther. 2018, 11, 5075–5082. [Google Scholar] [CrossRef]
- Tian, L.; Zhao, Z.; Xie, L.; Zhu, J. MiR-361-5p inhibits the mobility of gastric cancer cells through suppressing epithelial–mesenchymal transition via the Wnt/β-catenin pathway. Gene 2018, 675, 102–109. [Google Scholar] [CrossRef]
- Tian, L.; Zhao, Z.; Xie, L.; Zhu, J. MiR-361-5p suppresses chemoresistance of gastric cancer cells by targeting FOXM1 via the PI3K/Akt/mTOR pathway. Oncotarget 2018, 9, 4886–4896. [Google Scholar] [CrossRef]
- He, Y.; Ge, Y.; Jiang, M.; Zhou, J.; Luo, D.; Fan, H.; Shi, L.; Lin, L.; Yang, L. MiR-592 Promotes Gastric Cancer Proliferation, Migration, and Invasion Through the PI3K/AKT and MAPK/ERK Signaling Pathways by Targeting Spry2. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 1465–1481. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-H.; Lin, C.; Liu, C.-C.; Jiang, W.-W.; Huang, M.-Z.; Liu, X.; Guo, W.-J. MiR-616-3p promotes angiogenesis and EMT in gastric cancer via the PTEN/AKT/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 501, 1068–1073. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Jian, M.; Qi, H.; Mao, W.-Z. MicroRNA 495 Inhibits Proliferation and Metastasis and Promotes Apoptosis by Targeting Twist1 in Gastric Cancer Cells. Oncol. Res. 2019, 27, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.-J.; Deng, J.; Liu, Y.-W.; Wan, L.-Y.; Feng, M.; Chen, J.; Xiong, J.-P. MiR-1271 Inhibits Cell Proliferation, Invasion and EMT in Gastric Cancer by Targeting FOXQ1. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 1382–1394. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Wang, L.-N.; Li, W.; Zuo, Q.-F.; Li, M.-M.; Zou, Q.-M.; Xiao, B. Downregulation of miR-491-5p promotes gastric cancer metastasis by regulating SNAIL and FGFR4. Cancer Sci. 2018, 109, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Wu, Z.; Lin, L.; Zhou, M.; Wang, L.; Ma, H.; Xia, J.; Bin, J.; Liao, Y.; Liao, W. MiR-338-3p inhibits epithelial–mesenchymal transition in gastric cancer cells by targeting ZEB2 and MACC1/Met/Akt signaling. Oncotarget 2015, 6, 15222–15234. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-L.; Gao, H.-L.; Lv, X.-K.; Hei, Y.-R.; Li, P.-Z.; Zhang, J.-X.; Lu, N. MicroRNA-124 inhibits cell invasion and epithelial–mesenchymal transition by directly repressing Snail2 in gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3389–3396. [Google Scholar] [PubMed]
- Xu, M.; Qin, S.; Cao, F.; Ding, S.; Li, M. MicroRNA-379 inhibits metastasis and epithelial–mesenchymal transition via targeting FAK/AKT signaling in gastric cancer. Int. J. Oncol. 2017, 51, 867–876. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, M.; Xia, R.; Zhang, E.; Liu, X.; Zhang, Z.; Xu, T.; De, W.; Liu, B.; Wang, Z. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015, 6, e1802. [Google Scholar] [CrossRef]
- Song, Y.; Wang, R.; Li, L.-W.; Liu, X.; Wang, Y.-F.; Wang, Q.-X.; Zhang, Q. Long non-coding RNA HOTAIR mediates the switching of histone H3 lysine 27 acetylation to methylation to promote epithelial-to-mesenchymal transition in gastric cancer. Int. J. Oncol. 2019, 54, 77–86. [Google Scholar] [CrossRef]
- Jia, J.; Zhan, D.; Li, J.; Li, Z.; Li, H.; Qian, J. The contrary functions of lncRNA HOTAIR/miR-17-5p/PTEN axis and Shenqifuzheng injection on chemosensitivity of gastric cancer cells. J. Cell. Mol. Med. 2019, 23, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.-Y.; Yu, Q.-M.; Du, Y.-A.; Yang, L.-T.; Dong, R.-Z.; Huang, L.; Yu, P.-F.; Cheng, X.-D. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial–mesenchymal transition in gastric cancer. Int. J. Biol. Sci. 2013, 9, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Y.; Chen, Z.-H.; Peng, J.-J.; Wu, J.-L.; Yuan, Y.-J.; Zhai, E.-T.; Cai, S.-R.; He, Y.-L.; Song, W. Up-regulation of long non-coding RNA XLOC_010235 regulates epithelial-to-mesenchymal transition to promote metastasis by associating with Snail1 in gastric cancer. Sci. Rep. 2017, 7, 2461. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Liang, W.; Fu, M.; Huang, Z.-H.; Li, X.; Zhang, W.; Zhang, P.; Qian, H.; Jiang, P.-C.; Xu, W.-R.; et al. Exosomes-mediated transfer of long noncoding RNA ZFAS1 promotes gastric cancer progression. J. Cancer Res. Clin. Oncol. 2017, 143, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, F.; Chen, H.; Tan, Q.; Qiu, S.; Chen, S.; Jing, W.; Yu, M.; Liang, C.; Ye, S.; et al. Increased expression of long-noncoding RNA ZFAS1 is associated with epithelial–mesenchymal transition of gastric cancer. Aging 2016, 8, 2023–2038. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, L.; Wang, K.; Yu, H.; Wang, Y.; Liu, J.; Guo, Y.; Zhang, H. The role of MALAT-1 in the invasion and metastasis of gastric cancer. Scand. J. Gastroenterol. 2017, 52, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Lee, J.H.; Ivan, C.; Ling, H.; Zhang, X.; Park, C.H.; Calin, G.A.; Lee, S.K. MALAT1 promoted invasiveness of gastric adenocarcinoma. BMC Cancer 2017, 17, 46. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Chen, J.; He, B.; Li, Q.; Li, Y.; Gao, Y. A FOXM1 related long non-coding RNA contributes to gastric cancer cell migration. Mol. Cell. Biochem. 2015, 406, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Huang, Z.; Zang, X.; Pan, L.; Liang, W.; Chen, J.; Qian, H.; Xu, W.; Jiang, P.; Zhang, X. Long noncoding RNA LINC00978 promotes cancer growth and acts as a diagnostic biomarker in gastric cancer. Cell Prolif. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.-K.; Gong, Y.; Chen, X.-H.; Ye, F.; Yin, Z.-M.; Gong, Q.-N.; Huang, J.-S. TGFβ1-Induced LncRNA UCA1 Upregulation Promotes Gastric Cancer Invasion and Migration. DNA Cell Biol. 2017, 36, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; He, X.; Yin, D.; Han, L.; Qiu, M.; Xu, T.; Xia, R.; Xu, L.; Yin, R.; De, W. Increased expression of long noncoding RNA TUG1 predicts a poor prognosis of gastric cancer and regulates cell proliferation by epigenetically silencing of p57. Cell Death Dis. 2016, 7, e2109. [Google Scholar] [CrossRef]
- Sun, J.; Ding, C.; Yang, Z.; Liu, T.; Zhang, X.; Zhao, C.; Wang, J. The long non-coding RNA TUG1 indicates a poor prognosis for colorectal cancer and promotes metastasis by affecting epithelial–mesenchymal transition. J. Transl. Med. 2016, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, W.; Yang, P.; Zhou, J.; Wang, K.; Tao, Q. Knockdown of linc00152 inhibits the progression of gastric cancer by regulating microRNA-193b-3p/ETS1 axis. Cancer Biol. Ther. 2019, 20, 461–473. [Google Scholar] [CrossRef]
- Chen, D.-L.; Ju, H.-Q.; Lu, Y.-X.; Chen, L.-Z.; Zeng, Z.-L.; Zhang, D.-S.; Luo, H.-Y.; Wang, F.; Qiu, M.-Z.; Wang, D.-S.; et al. Long non-coding RNA XIST regulates gastric cancer progression by acting as a molecular sponge of miR-101 to modulate EZH2 expression. J. Exp. Clin. Cancer Res. CR 2016, 35, 142. [Google Scholar] [CrossRef]
- Saito, T.; Kurashige, J.; Nambara, S.; Komatsu, H.; Hirata, H.; Ueda, M.; Sakimura, S.; Uchi, R.; Takano, Y.; Shinden, Y.; et al. A Long Non-coding RNA Activated by Transforming Growth Factor-β is an Independent Prognostic Marker of Gastric Cancer. Ann. Surg. Oncol. 2015, 22 (Suppl. 3), S915–S922. [Google Scholar] [CrossRef]
- Guo, W.; Huang, J.; Lei, P.; Guo, L.; Li, X. LncRNA SNHG1 promoted HGC-27 cell growth and migration via the miR-140/ADAM10 axis. Int. J. Biol. Macromol. 2019, 122, 817–823. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Zhao, M.; Huang, S.; Zhang, Q.; Lin, H.; Wang, W.; Li, K.; Li, Z.; Huang, W.; et al. Long noncoding RNA SNHG6 regulates p21 expression via activation of the JNK pathway and regulation of EZH2 in gastric cancer cells. Life Sci. 2018, 208, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, H.; Zhu, L.; Hao, B.; Zhang, W.; Hua, J.; Gu, H.; Jin, W.; Zhang, G. Helicobacter pylori infection related long noncoding RNA (lncRNA) AF147447 inhibits gastric cancer proliferation and invasion by targeting MUC2 and up-regulating miR-34c. Oncotarget 2016, 7, 82770–82782. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Guo, H.; Zhou, B.; Feng, J.; Li, Y.; Han, T.; Liu, L.; Li, L.; Zhang, S.; Liu, Y.; et al. Long non-coding RNA SNHG5 suppresses gastric cancer progression by trapping MTA2 in the cytosol. Oncogene 2016, 35, 5770–5780. [Google Scholar] [CrossRef]
- Yu, Y.; Li, L.; Zheng, Z.; Chen, S.; Chen, E.; Hu, Y. Long non-coding RNA linc00261 suppresses gastric cancer progression via promoting Slug degradation. J. Cell. Mol. Med. 2017, 21, 955–967. [Google Scholar] [CrossRef]
- Qiao, C.-F.; Zhang, Y.; Jin, L.; Du, X.-G.; Qiao, Z.-J. High expression of lncRNA AFAP1-AS1 promotes cell proliferation and invasion by inducing epithelial-to-mesenchymal transition in gastric cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 393–400. [Google Scholar]
- Guo, J.-Q.; Li, S.-J.; Guo, G.-X. Long Noncoding RNA AFAP1-AS1 Promotes Cell Proliferation and Apoptosis of Gastric Cancer Cells via PTEN/p-AKT Pathway. Dig. Dis. Sci. 2017, 62, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Xiang, S.; Ma, J.; Hui, P.; Wang, T.; Meng, W.; Shi, M.; Wang, Y. Long non-coding RNA CASC15 regulates gastric cancer cell proliferation, migration and epithelial mesenchymal transition by targeting CDKN1A and ZEB1. Mol. Oncol. 2018, 12, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.-L.; Cai, J.; Chen, Z.-Q.; Zhang, Y.; Liang, L.-H.; Wang, J.-F.; Wang, J.-G.; Wu, J.; Mao, J.-D. The utility of long non-coding RNA ZEB1-AS1 as a prognostic biomarker in human solid tumors: A meta-analysis. Clin. Chim. Acta Int. J. Clin. Chem. 2018, 485, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-L.; Zhang, L.-F.; Guo, X.-H.; Zhang, D.-Z.; Yang, F.; Fan, Y.-Y. Downregulation of miR-335-5p by Long Noncoding RNA ZEB1-AS1 in Gastric Cancer Promotes Tumor Proliferation and Invasion. DNA Cell Biol. 2018, 37, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.-W.; Kong, Y.; Sun, X. Long noncoding RNA NEAT1 is an unfavorable prognostic factor and regulates migration and invasion in gastric cancer. J. Cancer Res. Clin. Oncol. 2016, 142, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-Y.; Wang, C.; Liu, G.; Zhou, X. Long noncoding RNA NEAT1-modulated miR-506 regulates gastric cancer development through targeting STAT3. J. Cell. Biochem. 2019, 120, 4827–4836. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Liu, Y.; Peng, J.; Liang, H.; Chen, H.; Chen, J.; He, W.; Xu, J.; Cai, S.; He, Y. Identification of differentially expressed signatures of long non-coding RNAs associated with different metastatic potentials in gastric cancer. J. Gastroenterol. 2016, 51, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Xie, X.; Xiao, Y.-F.; Tang, B.; Hu, C.-J.; Wang, S.-M.; Wu, Y.-Y.; Dong, H.; Li, B.-S.; Yang, S.-M. Long noncoding RNA LINC00675 enhances phosphorylation of vimentin on Ser83 to suppress gastric cancer progression. Cancer Lett. 2018, 412, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Ding, C.; Rodrìguez, R.A.; Del Mar Requena Mullor, M. The SOX2OT/miR-194-5p axis regulates cell proliferation and mobility of gastric cancer through suppressing epithelial–mesenchymal transition. Oncol. Lett. 2018, 16, 6361–6368. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-Z.; Cheng, T.-T.; He, Q.-J.; Lei, Z.-Y.; Chi, J.; Tang, Z.; Liao, Q.-X.; Zhang, H.; Zeng, L.-S.; Cui, S.-Z. LINC01133 as ceRNA inhibits gastric cancer progression by sponging miR-106a-3p to regulate APC expression and the Wnt/β-catenin pathway. Mol. Cancer 2018, 17, 126. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Meng, L.; Yuan, D.; Li, K.; Zhang, Y.; Dang, C.; Zhu, K. MEG3/miR-21 axis affects cell mobility by suppressing epithelial-mesenchymal transition in gastric cancer. Oncol. Rep. 2018, 40, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yan, Y.; Cao, S.; Chen, Y. Long non-coding RNA SNHG14 contributes to gastric cancer development through targeting miR-145/SOX9 axis. J. Cell. Biochem. 2018, 119, 6905–6913. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-T.; Xing, A.-Y.; Chen, X.; Ma, R.-R.; Wang, Y.-W.; Shi, D.-B.; Zhang, H.; Li, P.; Chen, H.-F.; Li, Y.-H.; et al. MicroRNA-27b, microRNA-101 and microRNA-128 inhibit angiogenesis by down-regulating vascular endothelial growth factor C expression in gastric cancers. Oncotarget 2015, 6, 37458–37470. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhou, B.; Shi, L.; Wang, H.; Chu, Q.; Xu, F.; Li, Y.; Chen, R.; Shen, C.; Schinckel, A.P. lncRNA AK017368 promotes proliferation and suppresses differentiation of myoblasts in skeletal muscle development by attenuating the function of miR-30c. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, T.; Li, M.; Cao, N.; Han, J. The Functional SOCS3 RS115785973 Variant Regulated by MiR-4308 Promotes Gastric Cancer Development in Chinese Population. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 38, 1796–1802. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Verma, S.; Pathak, R.U.; Mishra, R.K. Long Noncoding RNAs in Mammalian Development and Diseases. Adv. Exp. Med. Biol. 2017, 1008, 155–198. [Google Scholar] [PubMed]
- Sun, M.; Kraus, W.L. From discovery to function: The expanding roles of long noncoding RNAs in physiology and disease. Endocr. Rev. 2015, 36, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Heery, R.; Finn, S.P.; Cuffe, S.; Gray, S.G. Long Non-Coding RNAs: Key Regulators of Epithelial–mesenchymal Transition, Tumour Drug Resistance and Cancer Stem Cells. Cancers 2017, 9, 38. [Google Scholar] [CrossRef]
- Deng, G.; Sui, G. Noncoding RNA in oncogenesis: A new era of identifying key players. Int. J. Mol. Sci. 2013, 14, 18319–18349. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, H.; Shen, L.; Wang, J.; Pu, W.; Chen, Z.; Shen, X.; Fu, J.; Zhuang, Z. Rs56288038 (C/G) in 3′UTR of IRF-1 Regulated by MiR-502-5p Promotes Gastric Cancer Development. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 40, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zhao, Z.-Y.; Wu, R.; Zhang, Y.; Zhang, Z.-Y. Prognostic value of long noncoding RNAs in gastric cancer: A meta-analysis. OncoTargets Ther. 2018, 11, 4877–4891. [Google Scholar] [CrossRef] [PubMed]
- Hurt, J.A.; Robertson, A.D.; Burge, C.B. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013, 23, 1636–1650. [Google Scholar] [CrossRef] [PubMed]
- Ramón Y Cajal, S.; Segura, M.F.; Hümmer, S. Interplay Between ncRNAs and Cellular Communication: A Proposal for Understanding Cell-Specific Signaling Pathways. Front. Genet. 2019, 10, 281. [Google Scholar] [CrossRef]
- Bolha, L.; Ravnik-Glavač, M.; Glavač, D. Long Noncoding RNAs as Biomarkers in Cancer. Dis. Markers 2017, 2017, 7243968. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, D.; Dinger, M.E.; Mazar, J.; Crawford, J.; Smith, M.A.; Mattick, J.S.; Perera, R.J. The melanoma-upregulated long noncoding RNA SPRY4-IT1 modulates apoptosis and invasion. Cancer Res. 2011, 71, 3852–3862. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Nie, F.; Sun, M.; Xia, R.; Liu, Y.; Zhou, P.; De, W.; Liu, X. Decreased long noncoding RNA SPRY4-IT1 contributing to gastric cancer cell metastasis partly via affecting epithelial–mesenchymal transition. J. Transl. Med. 2015, 13, 250. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Ding, Q.; Yu, W.; Gao, M.; Wang, Y. Long noncoding RNA SPRY4-IT1 promotes malignant development of colorectal cancer by targeting epithelial–mesenchymal transition. OncoTargets Ther. 2016, 9, 5417–5425. [Google Scholar]
- Peng, W.; Wu, G.; Fan, H.; Wu, J.; Feng, J. Long noncoding RNA SPRY4-IT1 predicts poor patient prognosis and promotes tumorigenesis in gastric cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 6751–6758. [Google Scholar] [CrossRef]
- Wang, M.; Dong, X.; Feng, Y.; Sun, H.; Shan, N.; Lu, T. Prognostic role of the long non-coding RNA, SPRY4 Intronic Transcript 1, in patients with cancer: A meta-analysis. Oncotarget 2017, 8, 33713–33724. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Chen, J.; Gao, S.; Huang, J. LEIGC long non-coding RNA acts as a tumor suppressor in gastric carcinoma by inhibiting the epithelial-to-mesenchymal transition. BMC Cancer 2014, 14, 932. [Google Scholar] [CrossRef] [PubMed]


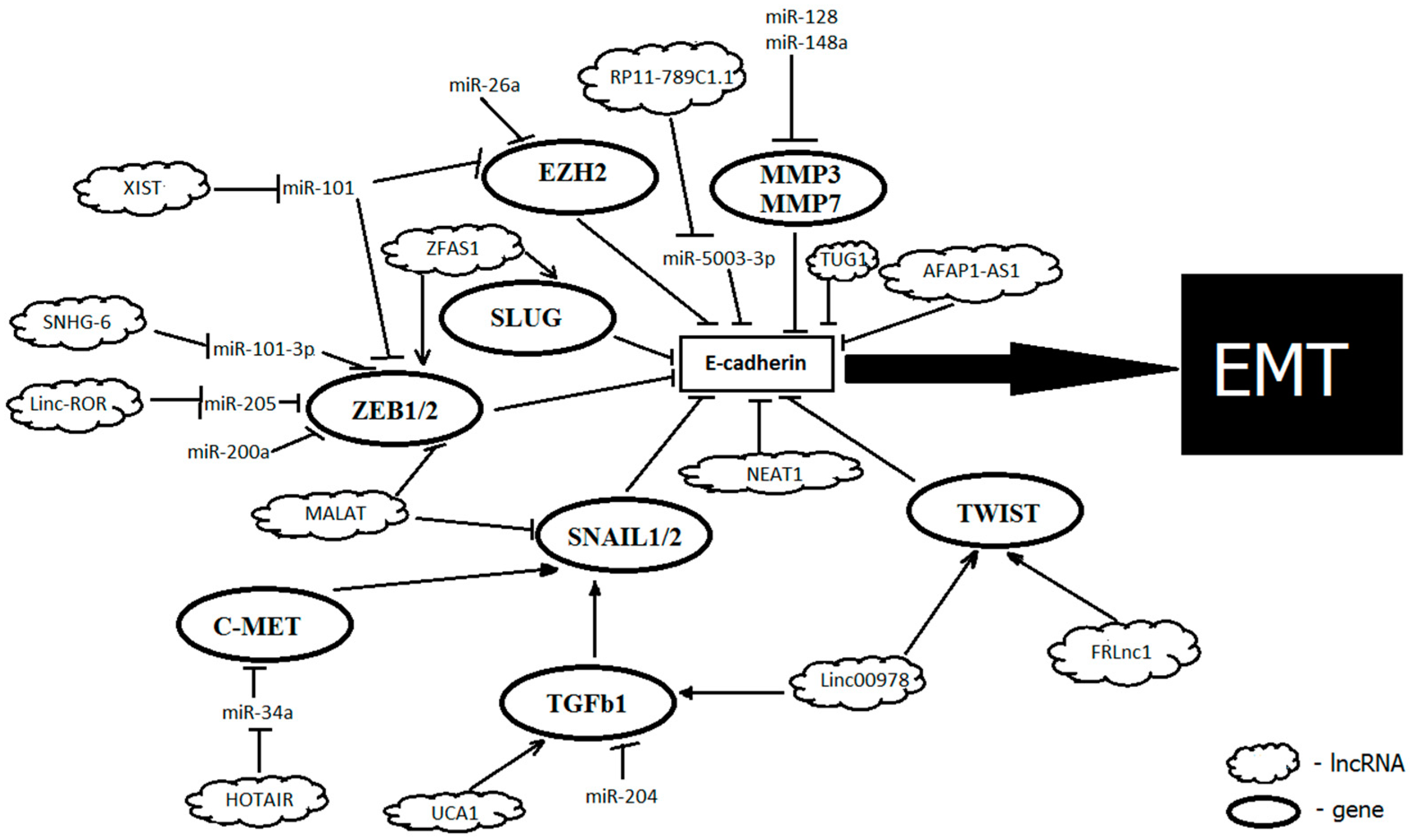{kind=link}
| NcRNA | Status in GC | Targets | Mechanism of Action | Functional Role | Reference |
|---|---|---|---|---|---|
| microRNAs: | |||||
| miR-5003-3p | Up | CDH1 | directly targets the 3′UTR of CDH1 at sites A and B | promotes migration, invasion and EMT | [82] |
| miR-200c | Down | ZEB1 | targets ZEB1 and thus increase E-cadherin expression | suppresses invasion and EMT | [72] |
| miR-101 | Down | EZH2 | targets EZH2 and thus increase E-cadherin expression | suppresses EMT | [73] |
| miR-148a | Down | SMAD2 | binds the 3′UTR of the SMAD4 and suppresses TGFβ-induced EMT | suppresses EMT, cell invasion and migration; low expression associated with advanced clinical stage and poor prognosis in GC | [79] |
| miR-29b/c | Down | DNMT3A | targets DNMT3A, thus modulating methylation of CDH1 promoter | suppresses EMT; significantly correlates with the degree of differentiation and invasion of the GC cells | [80] |
| miR-204 | Down | TGFBR2 | targets TGFBR2, regulating TGF-β | suppresses EMT, proliferation, invasion and migration | [81] |
| miR-217 | Up/Down | CDH1, PTPN14 | directly targets the 3′UTR of CDH1; directly targets the 3′UTR of PTPN14 | promotes cells proliferation; suppresses EMT, low expression is correlated with metastasis | [83,86] |
| miRNA-9 | Up/Down | CDH1, RAB34, NFKB1, CDX-2 | targets 3′UTR of CDH1; being downregulated, target RAB34 and NFKB1, regulating E-cadherin indirectly | triggering cell motility and invasiveness, regulates EMT | [84] |
| miR-544a | Up | CDH1 | directly targets CDH1 and AXIN2, regulate WNT signaling pathway | promotes EMT, cell motility and invasion; potential therapeutic target for metastatic GC | [85] |
| miR-199a-5p | Up | CDH1 | directly targets the 3′UTR of CDH1 | promotes EMT, cell invasion and metastasis; potential therapeutic targets and biomarkers for GC progression | [87] |
| miR-376a | Down | n/a | n/a | associated with advanced GC and poor prognosis | [88] |
| miR-381 | Down | TMEM16A | targets TMEM16A, thus regulating the TGF-β signaling pathway and EMT | suppresses EMT, decreases cell proliferation, migration and invasion | [89] |
| miRNA-96-5p | Down | FoxQ1 | binds to the 3′UTR of FoxQ1, decreasing the protein level of FoxQ1; upregulates the expression of E-cadherin and downregulates the expression of vimentin | suppresses the proliferation, migration and EMT | [90] |
| miR-574-3p | Down | ZEB1 | bounds 3′-UTR of ZEB1, thus upregulating E-cadherin expression, and concomitantly downregulating the expression of vimentin. | inhibits cancer cell migration, invasion, EMT; modulates cisplatin sensitivity in vitro and in vivo | [91] |
| miR-1254 | Down | SMURF1 | downregulating SMURF1 and thus inhibits EMT and decreases the PI3K/AKT signaling pathway | inhibits proliferation, migration, invasion, and EMT | [92] |
| miR-588 | Down | EIF5A2 | directly binds to 3′-UTR of EIF5A2 | suppresses cell invasion, migration, and progression of EMT | [93] |
| miR-218 | Down | BMI1, WASF3 | inhibits the expression of BMI1 and its downstream targets p-Akt473 and MMPs; directly inhibits expression of WASF3 | inhibits EMT, proliferation, invasion, and migration | [94,95] |
| miR-370 | Down | PAQR4 | directly inhibits expression of PAQR4 | inhibits the proliferation, invasion, and EMT | [96] |
| miR-711 | Down | CD44 | targets CD44 and thus downregulates vimentin expression and upregulates E-cadherin expression | inhibits the invasion, migration, and EMT | [97] |
| miR-543 | Up | SPOP | directly inhibits expression of SPOP | promotes EMT, cell migration, and invasion | [98] |
| miR-361-5p | Down | FOXM1 | suppresses the expression of MMP-3, MMP-9 and VEGF, increases expression of E-cadherin; acting through Wnt/β-catenin pathway; targets FOXM1, acting through the PI3K/Akt/mTOR pathway | inhibits EMT, cell proliferation, and mobility; low expression is correlated with larger tumor size and advanced TNM stage. | [99,100] |
| miR-592 | Up | SPRY2 | targets SPRY2 and acting through PI3K/AKT and MAPK/ERK signaling pathways | promotes proliferation, migration, and invasion, induces the EMT | [101] |
| miR-616-3p | Up | PTEN | directly inhibits expression of PTEN | promotes EMT, angiogenesis and metastasis; high expression is correlated with poor prognosis | [102] |
| miR-495 | Down | TWIST1 | directly inhibits expression of TWIST1 | decreases cell viability and migration, increases apoptosis and inhibits the EMT | [103] |
| miR-1271 | Down | FOXQ1 | directly suppressing FOXQ1 expression | suppressed cell proliferation, invasion, and EMT; correlated with tumor size, tumor stage, lymph node metastasis, and TNM stage | [104] |
| miR-491-5p | Down | SNAIL | directly inhibits SNAIL expression; indirectly inhibits FGFR4, also decreasing the SNAIL level | suppresses EMT and tumor metastasis | [105] |
| miR-338-3p | Down | ZEB2 and MACC1 | targets ZEB2 and MACC1/Met/Akt signaling, thus upregulating the E-cadherin and downregulating the N-cadherin, fibronectin, and vimentin | inhibits EMT, migration, and invasion | [106] |
| miR-124 | Down | SNAIL2 | represses the SNAIL2 expression | inhibits EMT, cell proliferation, and invasion; lower expression is associated with tumor size, lymphatic metastasis, and TNM stage | [107] |
| miR-379 | Down | FAK | directly binds to 3′-UTR of FAK, resulting in suppression of AKT signaling | inhibited cell migration, invasion and EMT; low expression is associated with poor prognosis, lymph node metastasis, and advanced TNM stage | [108] |
| Long non-coding RNAs: | |||||
| HOTAIR | Up | PCR2, miR-34a, c-MET, SNAIL1, CDH1, miR-152 | switching the acetylation of histone H3 lysine 27 to the methylation of the E-cadherin promoter, inducing its transcriptional inhibition; inactivates miR-34a, which activates the HGF/c-MET/SNAIL pathway and thus indirectly inhibits E-cadherin; targets miR-17-5p and thus regulates expression of PTEN | promotes EMT, facilitates viability, proliferation, and metastasis; higher expression correlates with lymphatic metastasis and TNM stage | [109,110,111,112] |
| XLOC_010235 | Up | SNAIL1 | inactivates SNAIL1, thereby upregulating E-cadherin expression | promotes EMT; high expression correlates with metastasis and TNM stage | [113] |
| ZFAS1 | Up | ZEB1 | activates the EMT inducer ZEB1 | promotes EMT | [114,115] |
| MALAT1 | Up | SNAIL, N-cadherin, ZEB1 | targets SNAIL, N-cadherin, and ZEB1, thus decreasing E-cadherin expression | promotes EMT, invasion, angiogenesis, and metastasis | [116,117] |
| FRLnc1 | Up | TWIST, TGFβ-1 | activates the TGFβ-1 and TWIST | promotes EMT, invasion, and migration of cells | [118] |
| LINC00978 | Up | TGFβ/SMAD, TWIST, SLUG | activates the TGF-β/SMAD regulatory pathway, thus decreasing E-cadherin expression | promotes EMT, invasion, and migration of cells, decreases apoptosis | [119] |
| UCA1 | Up | TGFβ | targets TGFβ, decreases the levels of vimentin and SNAIL, thus regulating levels of E-cadherin and ZO-1 | promotes EMT, associated with invasion and metastasis | [120] |
| TUG1 | Up | CDH1 | interacts with PRC2, epigenetically repressing cyclin-dependent kinase inhibitors (P15, P16, P21, and P57); downregulation of E-cadherin | promotes EMT, cell proliferation, and metastases, predicts a poor prognosis | [121,122] |
| Linc00152 | Up | miR-193b-3p | directly inhibits expression of miR-193b-3p, leading additionally to ETS1 upregulation | promotes EMT, proliferation, migration, and invasion | [123] |
| XIST | Up | miR-101 | acts as a sponge for miR-101, and modulates EZH2 expression | promotes EMT, cell proliferation, and invasion | [124] |
| lncRNA-ATB | Up | miR-200 | acts through the TGF-β/miR-200/ZEB regulatory axis, thus decreasing E-cadherin expression | promotes EMT | [125] |
| SNHG1 | Up | miR-140 | acting as a sponge, repress miR-140 expression and thereby elevated its down-stream target ADAM10 | promotes EMT, proliferation, and invasion; linked with poor prognosis in cancer patients. | [126] |
| SNHG6 | Up | miR-101-3p | acts as sponge for miR-101-3p, thereby upregulating ZEB1 at the post-transcriptional level and regulating E-cadherin; epigenetically inactivates P27 through EZH2-dependent histone H3 methylation in the promoter of the P27; activates the JNK pathway and upregulate P21 | promotes EMT, invasion, migration, and metastasis | [65,127] |
| AF147447 | Down | MUC2, miR-34c | acts as sponge for miR-34c, thus regulating MUC2, EGFR, and CD44 expression | suppresses EMT, cell invasion, and proliferation | [128] |
| SNHG5 | Down | MTA2 | provides a cytoplasmic trap for MTA2, directly binding to it and preventing its transfer from the cytoplasm into the nucleus | suppresses EMT, cell invasion, proliferation, and metastases | [129] |
| Linc00261 | Down | SLUG | promotes SLUG degradation | suppresses EMT, cell invasion, and proliferation | [130] |
| AFAP1-AS1 | Up | CDH1 | upregulates E-cadherin and downregulates N-cadherin and vimentin | promotes EMT, invasion, and proliferation, associated with invasion in lymph nodes, distant metastasis, advanced TNM stages, and poor prognosis. | [131,132] |
| CASC15 | Up | CDH1, miR-33a-5p, EZH2 | targets CHD1; interacts with EZH2 and WDR5, recruits them to the CDKN1A promoter region, and thus modulates CDKN1A expression in the nucleus; acts as a sponge for miR-33a-5p and activates ZEB1 in the cytoplasm | promotes EMT, invasion, and proliferation, associated with poor prognosis | [133] |
| ZEB1-AS1 | Up | miR-335-5p | downregulates miR-335-5p expression by acting as a molecular sponge | promotes EMT, invasion, and proliferation, correlates with lymph node metastasis, TNM stage, and poor overall survival of patients | [134,135] |
| NEAT1 | Up | miR-506, CDH1 | acts through the NEAT1/miR-506/STAT3 regulatory axis; targets CHD1 | promotes EMT, invasion, and migration, correlates with more advanced stages, metastasis, and a low overall survival in patients | [136,137] |
| RP11-789C1.1 | Down | miR-5003-3p | acts as sponge for miR-5003-3p | promotes migration, invasion, and EMT; correlates with metastases | [82,138] |
| LINC00675 | Down | vimentin | regulates vimentin expression | suppresses proliferation, migration, invasion, and EMT | [139] |
| SOX2OT | Up | miR-194-5p | act as sponge for miR-194-5p | promotes EMT, cell proliferation, invasion, and migration | [140] |
| LINC01133 | Down | miR-106a-3p | act as sponge for miR-106a-3p, which specifically targets the APC, and thus inactivates the Wnt/β-catenin pathway | inhibits proliferation, migration, EMT and metastasis | [141] |
| MEG3 | Down | miR-21 | act as sponge for miR-21; downregulating the expression of MMP-3, MMP-9, and VEGF; increases the expression of E-cadherin and downregulates the expression of N-cadherin, Snail, and β-catenin | suppresses EMT and cell mobility | [142] |
| SNHG14 | Up | miR-145 | negatively regulates miR-145 and thus affects its direct target; involved in PI3K/AKT/mTOR pathway | promotes EMT, cell viability, migration, invasion, and inhibits apoptosis | [143] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bure, I.V.; Nemtsova, M.V.; Zaletaev, D.V. Roles of E-cadherin and Noncoding RNAs in the Epithelial–mesenchymal Transition and Progression in Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 2870. https://doi.org/10.3390/ijms20122870
Bure IV, Nemtsova MV, Zaletaev DV. Roles of E-cadherin and Noncoding RNAs in the Epithelial–mesenchymal Transition and Progression in Gastric Cancer. International Journal of Molecular Sciences. 2019; 20(12):2870. https://doi.org/10.3390/ijms20122870
Chicago/Turabian StyleBure, Irina V., Marina V. Nemtsova, and Dmitry V. Zaletaev. 2019. "Roles of E-cadherin and Noncoding RNAs in the Epithelial–mesenchymal Transition and Progression in Gastric Cancer" International Journal of Molecular Sciences 20, no. 12: 2870. https://doi.org/10.3390/ijms20122870
APA StyleBure, I. V., Nemtsova, M. V., & Zaletaev, D. V. (2019). Roles of E-cadherin and Noncoding RNAs in the Epithelial–mesenchymal Transition and Progression in Gastric Cancer. International Journal of Molecular Sciences, 20(12), 2870. https://doi.org/10.3390/ijms20122870