Sphingomyelin Synthase 2 Promotes Endothelial Dysfunction by Inducing Endoplasmic Reticulum Stress

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
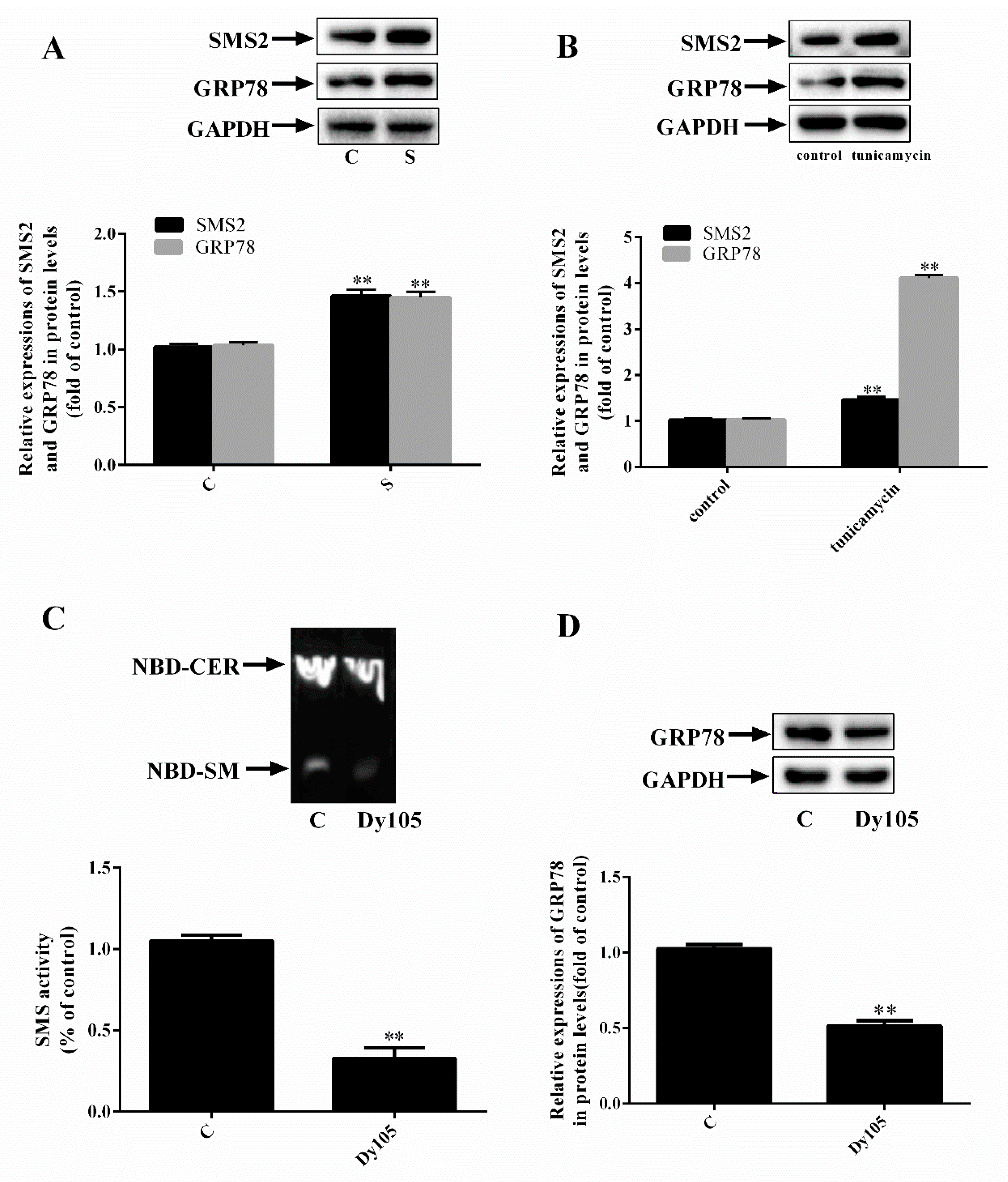
2.1. SMS2 Can Activate ER Stress
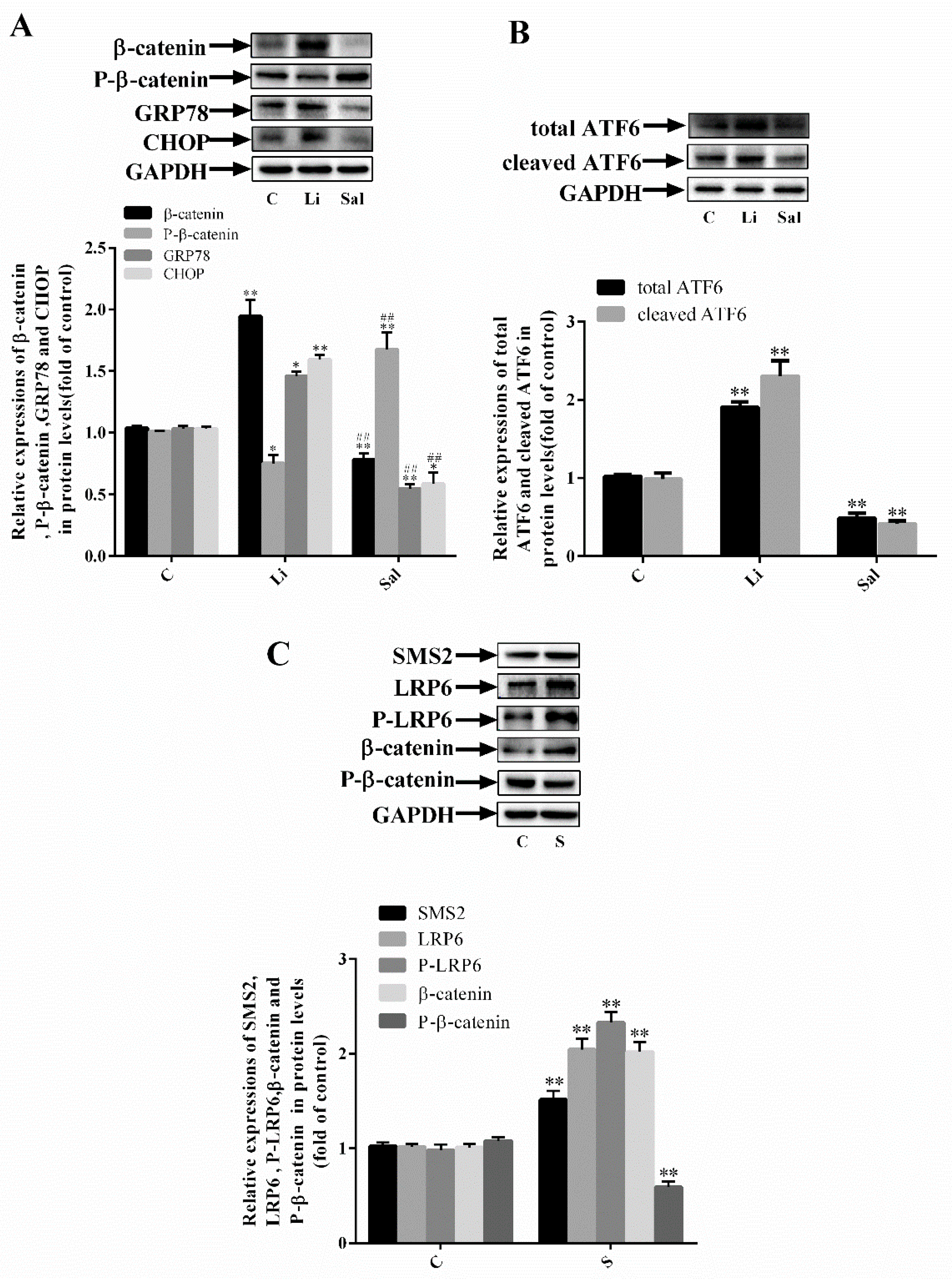
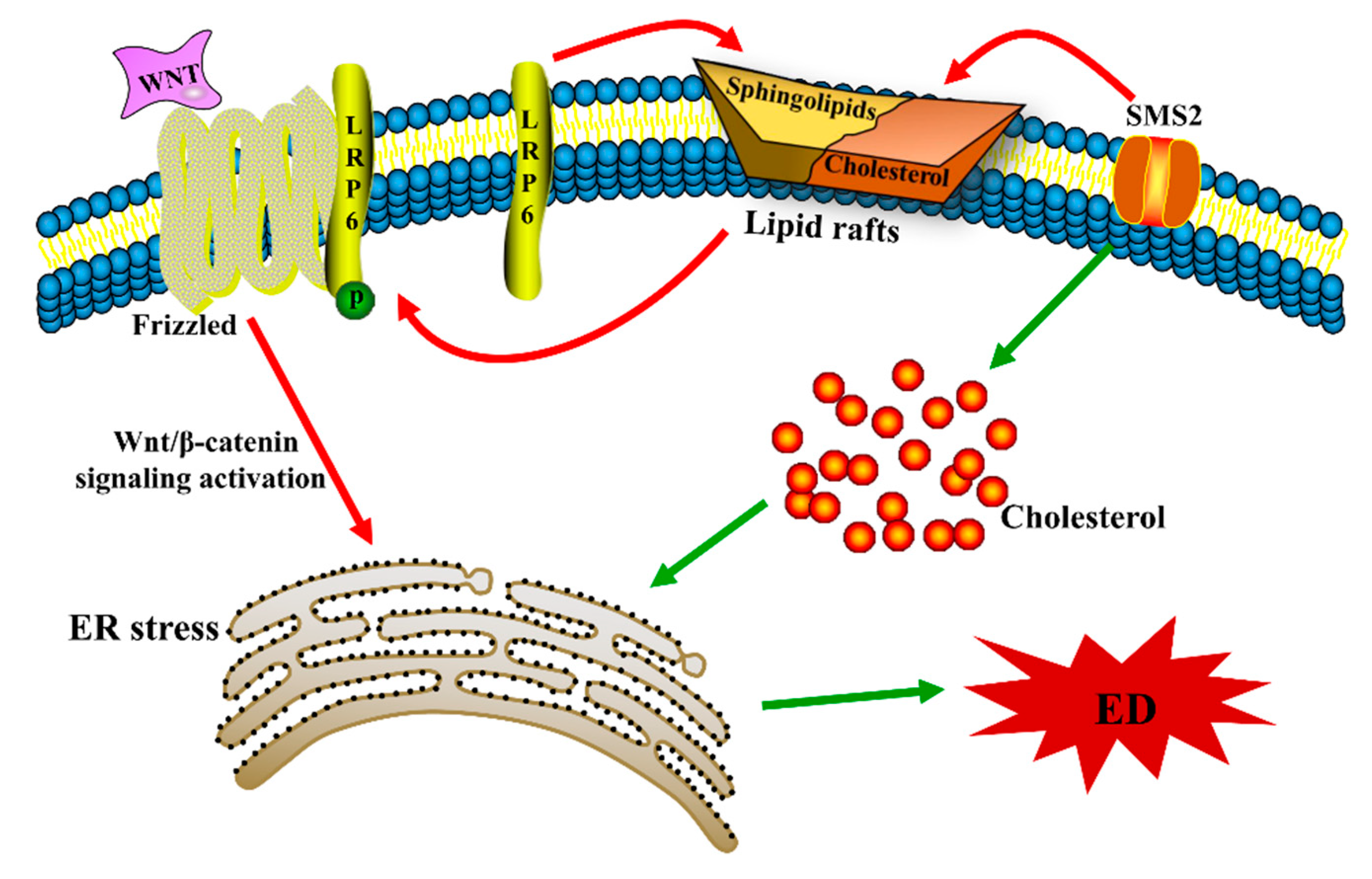
2.2. SMS2 Can Trigger ER Stress by Provoking the Wnt/β-Catenin Pathway
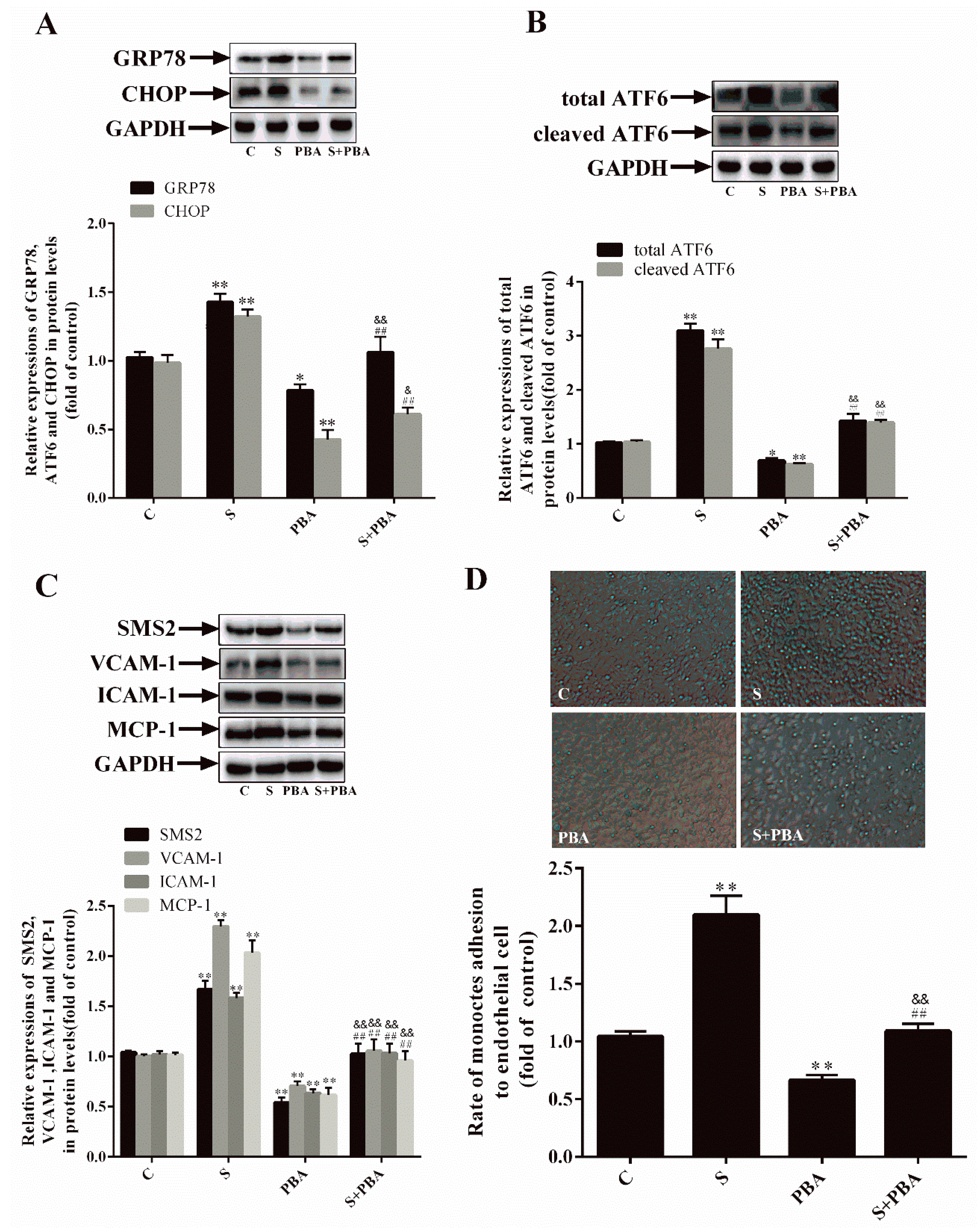
2.3. Inhibition of ER Stress Can Decrease SMS2-Induced ED
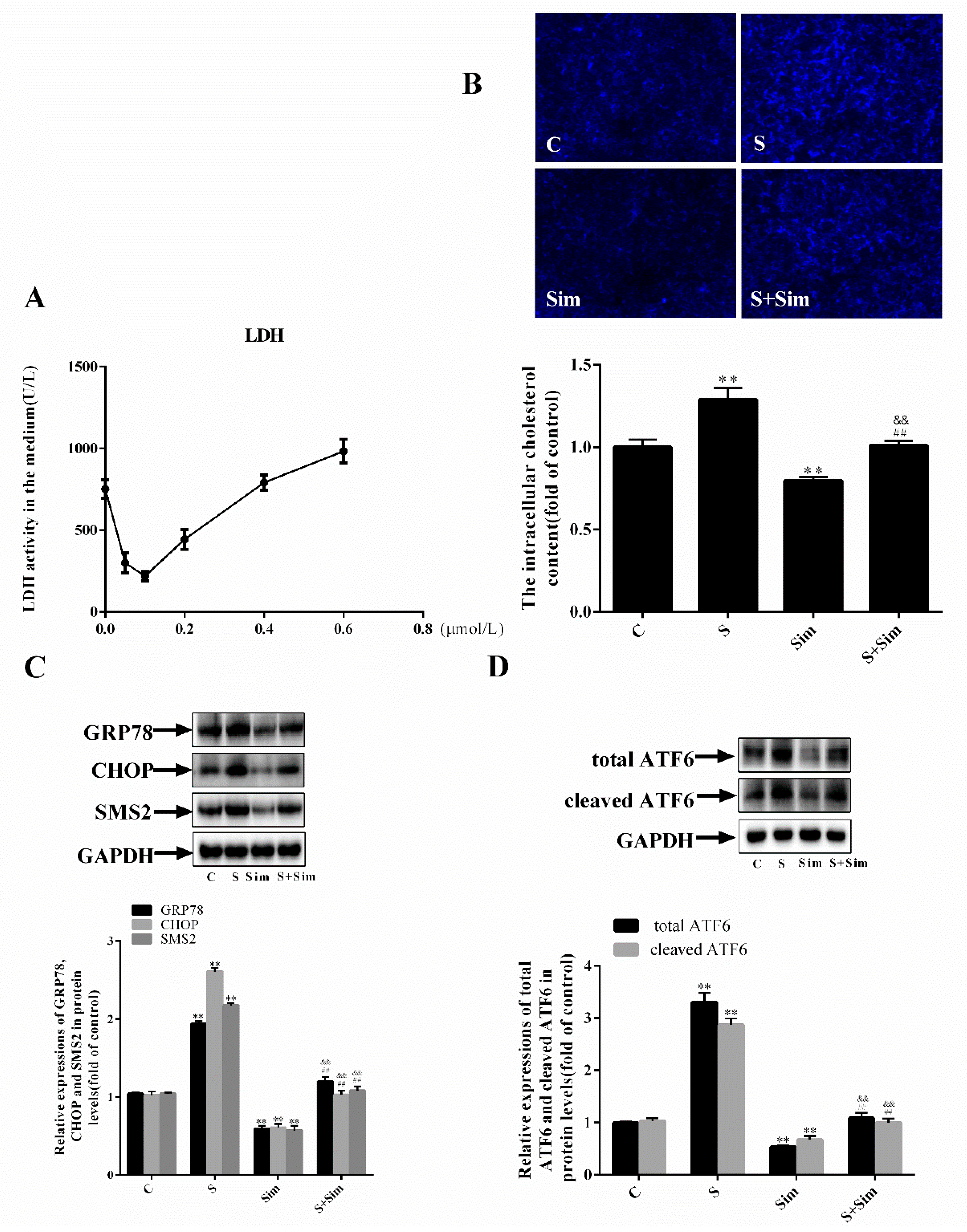
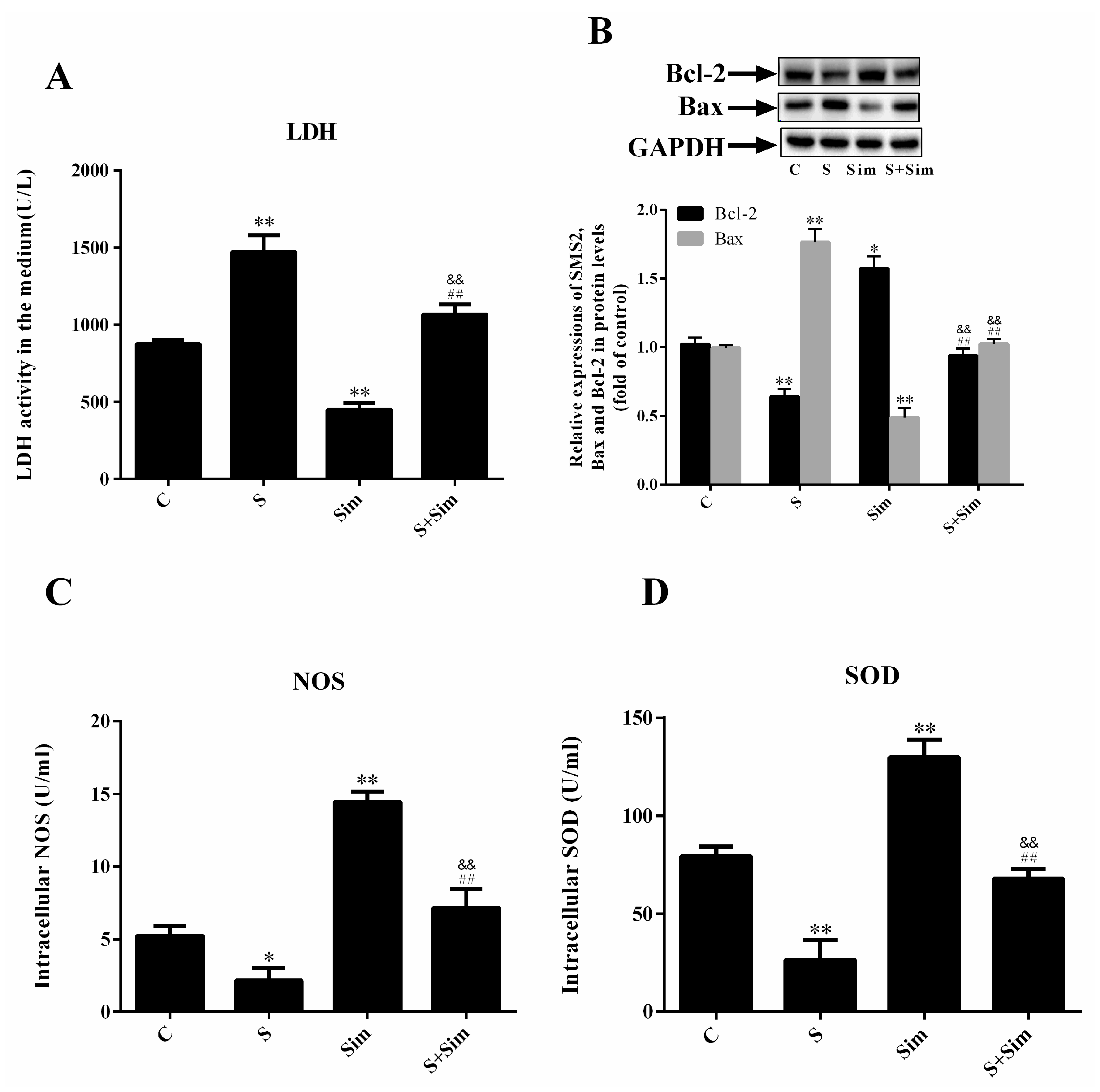
2.4. Simvastatin Can Attenuate the ER Stress Induced by SMS2
2.5. Simvastatin Can Attenuate the Injury Induced by SMS2
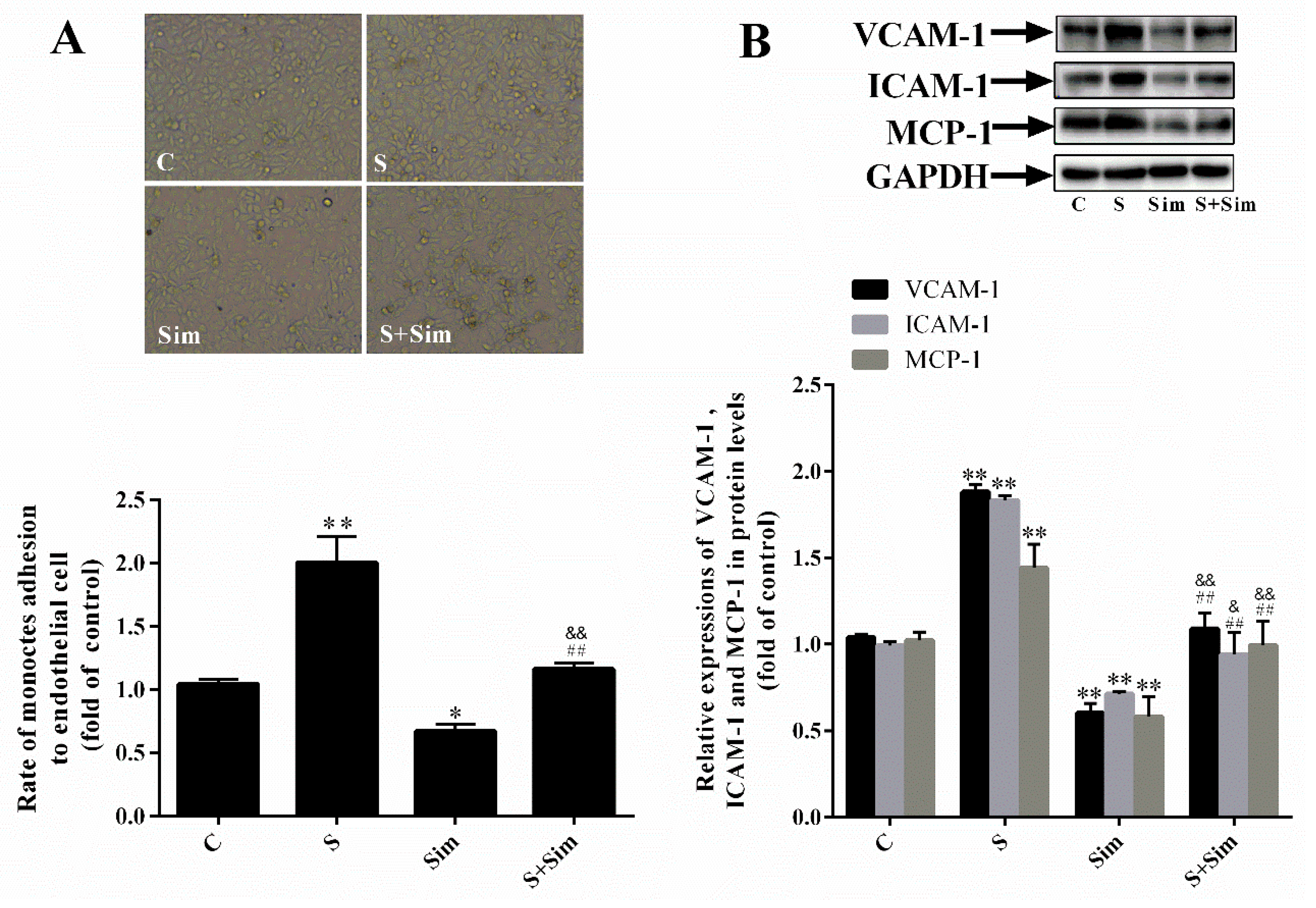
2.6. Simvastatin Can Attenuate the Adhesion Capacity Induced by SMS2
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Transfection with an SMS2 Overexpression Plasmid
4.3. Measurement of the Degree of Oxidative Stress
4.4. Filipin Staining
4.5. Cell Adhesion Assay
4.6. Western Blot Analysis
4.7. LiCl or Salinomycin Treatment of HUVECs
4.8. Sphingomyelin Synthase Enzyme Activity Assay
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ross, R. Atherosclerosis—An Inflammatory Disease—NEJM. N. Engl. J. Med. 1999, 340, 115. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- El Assar, M.; Angulo, J.; Rodríguez-Maas, L. Oxidative stress and vascular inflammation in aging. Free Radic. Biol. Med. 2013, 65, 380–401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Pan, H.; Xu, Y.; Wang, X.; Qiu, Z.; Jiang, L. Allicin decreases lipopolysaccharide-induced oxidative stress and infammation in human umbilical vein endothelial cells through suppression of mitochondrial dysfunction and activation of Nrf2. Cell Physiol. Biochem. 2017, 41, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Del Río, L.A. ROS and RNS in plant physiology: An overview. J. Exp. Bot. 2015, 66, 2827–2837. [Google Scholar] [CrossRef] [PubMed]
- Adada, M.; Luberto, C.; Canals, D. Inhibitors of the sphingomyelin cycle: Sphingomyelin synthases and sphingomyelinases. Chem. Phys. Lipids 2016, 197, 45–59. [Google Scholar] [CrossRef]
- Fessler, M.B.; Parks, J.S. Intracellular Lipid Flux and Membrane Microdomains as Organizing Principles in Inflammatory Cell Signaling. J. Immunol. 2011, 187, 1529–1535. [Google Scholar] [CrossRef]
- Yamaoka, S.; Miyaji, M.; Kitano, T.; Umehara, H.; Okazaki, T. Expression Cloning of a Human cDNA Restoring Sphingomyelin Synthesis and Cell growth in Sphingomyelin Synthase-defective Lymphoid Cells. J. Biol. Chem. 2004, 279, 18688–18693. [Google Scholar] [CrossRef]
- Jiang, X.C.; Liu, J. Sphingolipid metabolism and atherosclerosis. Handb. Exp. Pharmacol. 2013, 216, 133. [Google Scholar]
- Zilversmit, D.B.; Mccandless, E.L.; Jordan, P.H.; Henly, W.S.; Ackerman, R.F. The Synthesis of Phospholipids in Human Atheromatous Lesions. Circulation 1961, 23, 370. [Google Scholar] [CrossRef]
- Park, T.S.; Panek, R.L.; Mueller, S.B.; Hanselman, J.C.; Rosebury, W.S.; Robertson, A.W.; Kindt, E.K.; Homan, R.; Karathanasis, S.K.; Rekhter, M.D. Inhibition of Sphingomyelin Synthesis Reduces Atherogenesis in Apolipoprotein E-Knockout Mice. Circulation 2004, 110, 3465–3471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hua, L.; Hou, H.; Du, X.; He, Z.; Liu, M.; Hu, X.; Yan, N. Sphingomyelin synthase 2 promotes H2O2-induced endothelial dysfunction by activating the Wnt/β-catenin signaling pathway. Int. J. Mol. Med. 2018, 42, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- De Jaime-Soguero, A.; Abreu de Oliveira, W.A.; Lluis, F. The pleiotropic effects of the canonical Wnt pathway in early development and pluripotency. Genes 2018, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Matthijs, B.W.; Hermans, K.C. Wnt signaling in atherosclerosis. Eur. J. Pharmacol. 2015, 763, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Otterdal, K.; Lekva, T.; Halvorsen, B.; Gabrielsen, A.; Sandberg, W.J.; Paulsson-Berne, G.; Pedersen, T.M.; Folkersen, L.; Gullestad, L.; et al. Dickkopf-1 Enhances Inflammatory Interaction Between Platelets and Endothelial Cells and Shows Increased Expression in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1228–1234. [Google Scholar] [CrossRef]
- Bhatt, P.M.; Lewis, C.J.; House, D.L. Increased Wnt5a mRNA Expression in Advanced Atherosclerotic Lesions, and Oxidized LDL Treated Human Monocyte-Derived Macrophages. Open Circ. Vasc. J. 2012, 5, 1–7. [Google Scholar] [CrossRef]
- Malgor, R.; Bhatt, P.M.; Connolly, B.A.; Jacoby, D.L.; Feldmann, K.J.; Silver, M.J.; Nakazawa, M.; McCall, K.D.; Goetz, D.J. Wnt5a, TLR2 and TLR4 are elevated in advanced human atherosclerotic lesions. Inflamm. Res. 2014, 63, 277–285. [Google Scholar] [CrossRef][Green Version]
- Kim, J.; Kim, J.; Kim, D.W.; Ha, Y.; Ihm, M.H.; Kim, H.; Song, K.; Lee, I. Wnt5a Induces Endothelial Inflammation via β-Catenin-Independent Signaling. J. Immunol. 2010, 185, 1274–1282. [Google Scholar] [CrossRef]
- Vikram, A.; Kim, Y.R.; Kumar, S.; Naqvi, A.; Hoffman, T.A.; Kumar, A.; Miller, F.J., Jr.; Kim, C.S.; Irani, K. Canonical Wnt Signaling Induces Vascular endothelial dysfunction via p66Shc-Regulated Reactive Oxygen Species Significance. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2301–2309. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Sozen, E.; Karademir, B.; Ozer, N.K. Basic mechanisms in endoplasmic reticulum stress and relation to cardiovascular diseases. Free Radic. Biol. Med. 2015, 78, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Patel, S.; McAlpine, C.S.; Werstuck, G.H. The Role of Endoplasmic Reticulum Stress-Glycogen Synthase Kinase-3 Signaling in Atherogenesis. Int. J. Mol. Sci. 2018, 19, 1607. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Chiba, T.; Kokame, K.; Miyata, T.; Kaneko, E.; Shimokado, K. A deficiency of Herp, an endoplasmic reticulum stress protein, suppresses atherosclerosis in ApoE knockout mice by attenuating inflammatory responses. PLoS ONE 2013, 8, e75249. [Google Scholar] [CrossRef] [PubMed]
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Kumar Puli, L.; Färber, K.; Harkany, T.; Schulte, G. WNT signaling in activated microglia is proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Amodio, G.; Moltedo, O.; Faraonio, R.; Remondelli, P. Targeting the Endoplasmic Reticulum Unfolded Protein Response to Counteract the Oxidative Stress-Induced endothelial dysfunction. Oxid. Med. Cell. Longev. 2018, 2018, 4946289. [Google Scholar] [CrossRef] [PubMed]
- Battson, M.L.; Lee, D.M.; Gentile, C.L. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H355–H367. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Watanabe, K.; Itoh, M.; Huan, C.-R.; Tong, X.-P.; Nakamura, T.; Miki, M.; Iwao, H.; Nakajima, A.; Kawanami, T.S.T.; et al. CD4+ T-cell dysfunctions through the imp aired lipid rafts ameliorate concanavalin A-induced hepatitis in sphingomyelin synthas1 knockout mice. Int. Immunol. 2012, 24, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Li, Z.; Hailemariam, T.; Mukherjee, S.; Maxfield, F.R.; Wu, M.P.; Jiang, X.C. SMS overexpression and knockdown: Impact on cellular sphingomyelin and diacylglycerol metabolism, and cell apoptosis. J. Lipid Res. 2008, 49, 376–385. [Google Scholar] [CrossRef]
- Li, Y.; Guan, J.; Wang, W.; Hou, C.; Zhou, L.; Ma, J.; Cheng, Y.; Jiao, S.; Zhou, Z. TRAF3-interacting JNK-activating modulator promotes inflammation by stimulating translocation of Toll-like receptor 4 to lipid rafts. J. Biol. Chem. 2018, 294, 2744–2756. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Komekado, H.; Kikuchi, A. Caveolin is necessary for Wnt-3a-dependent internalization of lrp6 and accumulation of b-catenin. Dev. Cell 2006, 11, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Bedel, A.; Nègre-Salvayre, A.; Heeneman, S.; Grazide, M.H.; Thiers, J.C.; Salvayre, R.; Maupas-Schwalm, F. E-cadherin/β-catenin/T-cell factor pathway is involved in smooth muscle cell proliferation elicited by oxidized low-density lipoprotein. Circ. Res. 2008, 103, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Roth, B.; Bu, G. Tyrosine-based signal mediates LRP6 receptor endocytosis and desensitization of Wnt/β-catenin pathway signaling. J. Biol. Chem. 2014, 289, 27562–27570. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Yao, S.; Tian, H.; Jiao, P.; Yang, N.; Zhu, P.; Qin, S. Pigment epithelium-derived factor alleviates endothelial injury by inhibiting Wnt/β-catenin pathway. Lipids Health Dis. 2017, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, K.; Keasey, M.P.; Razskazovskiy, V.; Visavadiya, N.P.; Jia, C.; Hagg, T. Reduced FAK-STAT3 signaling contributes to ER stress-induced mitochondrial dysfunction and death in endothelial cells. Cell. Signal. 2017, 36, 154–162. [Google Scholar] [CrossRef]
- SchrDer, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wu, S.; Muhammad, S.; Ren, Q.; Sun, C. miR-103/107 promote ER stress-mediated apoptosis via targeting the Wnt3a/β-catenin/ATF6 pathway in preadipocytes. Lipid Res. 2018, 59, 843–853. [Google Scholar] [CrossRef]
- Cao, L.; Lei, H.; Chang, M.Z.; Liu, Z.Q.; Bie, X.H. Down-regulation of 14-3-3 β exerts anti-cancer effects through inducing ER stress in human glioma U87 cells: Involvement of CHOP-Wnt pathway. Biochem. Biophys. Res. Commun. 2015, 462, 389–395. [Google Scholar] [CrossRef]
- Jia, X.; Chen, Y.; Zhao, X.; Lv, C.; Yan, J. Oncolytic vaccinia virus inhibits human epatocellular carcinoma MHCC97-H cell proliferation via endoplasmic reticulum stress, autophagy and Wnt pathways. J. Gene Med. 2016, 18, 211–219. [Google Scholar] [CrossRef]
- Chaube, R.; Kallakunta, V.M.; Espey, M.G.; McLarty, R.; Faccenda, A.; Ananvoranich, S.; Mutus, B. Endoplasmic reticulum stress-mediated inhibition of NSMase2 elevates plasma membrane cholesterol and attenuates NO production in endothelial cells. Biochim. Biophys. Acta 2012, 1821, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Duan, W.; Nizigiyimana, P.; Gao, L.; Liao, Z.; Xu, B.; Liu, L.; Lei, M. α-Mangostin attenuates diabetic nephropathy in association with suppression of acid sphingomyelianse and endoplasmic reticulum stress. Biochem. Biophys. Res. Commun. 2018, 496, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Sozen, E.; Ozer, N.K. Impact of high cholesterol and endoplasmic reticulum stress on metabolic diseases: An updated mini-review. Redox. Biol. 2017, 12, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Röhrl, C.; Stangl, H. Cholesterol metabolism-physiological regulation and pathophysiological deregulation by the endoplasmic reticulum. Wien. Med. Wochenschr. 2018, 168, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Mou, D.; Yang, H.; Qu, C.; Chen, J.; Zhang, C. Pharmacological Activation of Peroxisome Proliferator-Activated Receptor Increases Sphingomyelin Synthase Activity in THP-1 Macrophage-Derived Foam Cell. Inflammation 2016, 39, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Lesimple, A.; Denis, M.; Vincent, J.; Larsen, A.; Mamer, O.; Krimbou, L.; Genest, J.; Marcil, M. Increased sphingomyelin content impairs HDL biogenesis and maturation in human Niemann-Pick disease type B. J. Lipid Res. 2006, 47, 622–632. [Google Scholar] [CrossRef]
- Yan, N.; Ding, T.; Dong, J.; Li, Y.; Wu, M. Sphingomyelin synthase overexpression increases cholesterol accumulation and decreases cholesterol secretion in liver cells. Lipids Health Dis. 2011, 10, 46. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, L.; Wu, N.; Zhao, R.; He, X.; Liu, Q.; Li, X.; He, Z.; Yu, L.; Yan, N. Sphingomyelin Synthase 2 Promotes Endothelial Dysfunction by Inducing Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2019, 20, 2861. https://doi.org/10.3390/ijms20122861
Hua L, Wu N, Zhao R, He X, Liu Q, Li X, He Z, Yu L, Yan N. Sphingomyelin Synthase 2 Promotes Endothelial Dysfunction by Inducing Endoplasmic Reticulum Stress. International Journal of Molecular Sciences. 2019; 20(12):2861. https://doi.org/10.3390/ijms20122861
Chicago/Turabian StyleHua, Lingyue, Na Wu, Ruilin Zhao, Xuanhong He, Qian Liu, Xiatian Li, Zhiqiang He, Lehan Yu, and Nianlong Yan. 2019. "Sphingomyelin Synthase 2 Promotes Endothelial Dysfunction by Inducing Endoplasmic Reticulum Stress" International Journal of Molecular Sciences 20, no. 12: 2861. https://doi.org/10.3390/ijms20122861
APA StyleHua, L., Wu, N., Zhao, R., He, X., Liu, Q., Li, X., He, Z., Yu, L., & Yan, N. (2019). Sphingomyelin Synthase 2 Promotes Endothelial Dysfunction by Inducing Endoplasmic Reticulum Stress. International Journal of Molecular Sciences, 20(12), 2861. https://doi.org/10.3390/ijms20122861