1. Introduction
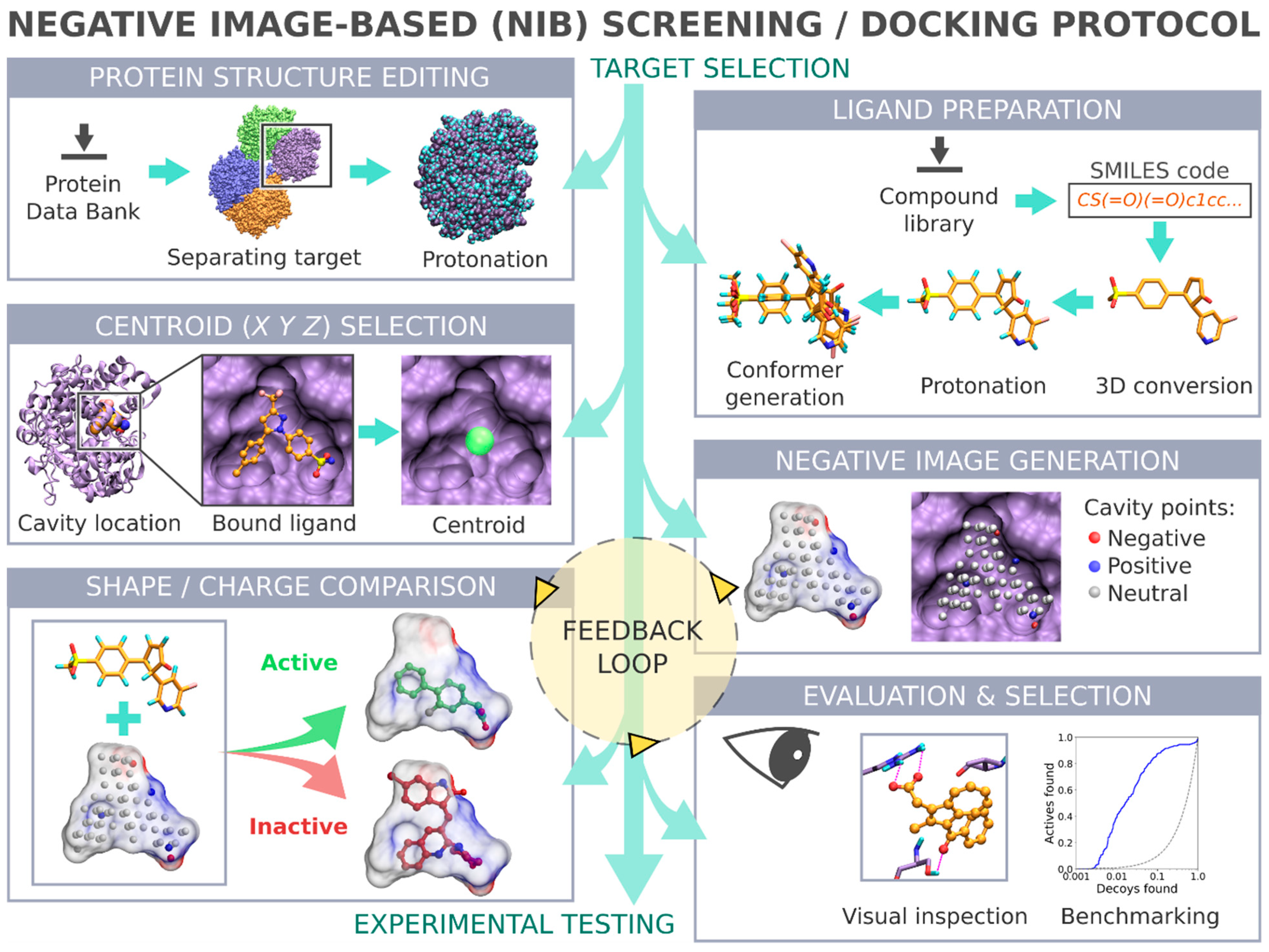
Negative image-based (NIB) screening (
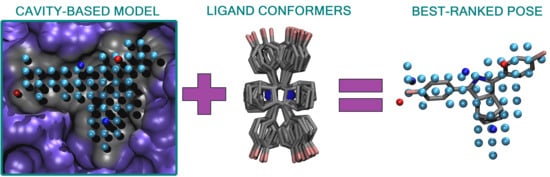
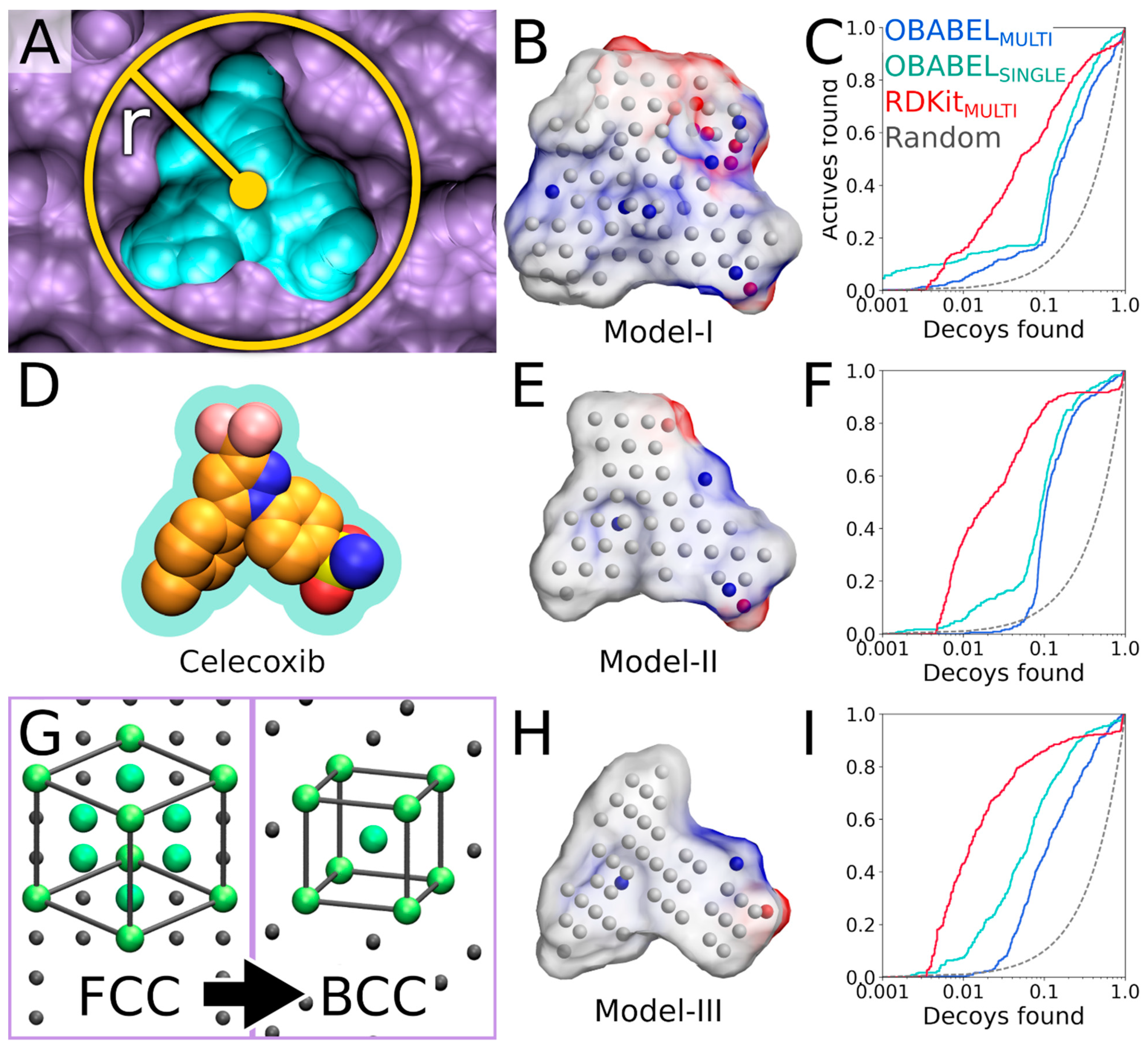
Figure 1) is a rigid molecular docking methodology that combines the key strengths of both the structure- and ligand-based computer-aided drug discovery approaches [
1]. The NIB relies primarily on the 3D coordinates of the target protein’s structure, especially its ligand-binding cavity (
Figure 1), and the geometry optimization (or rigid docking) is performed similarly to the traditional ligand-based screening.
In the NIB screening, a negative image is built based on the target protein’s ligand-binding cavity shape and electrostatics (
Figure 1). The NIB model ideally encompasses those key shape features of the target’s cavity required for the potent ligand binding. The NIB model generation, which is done using the cavity detection software PANTHER [
3], takes into account explicit water molecules, cofactors and ions, user-defined restrictions, and alternative residue protonation. A NIB model can be built based solely on protein 3D structure information (
Figure 1) and, thus, without prior knowledge on target-specific active and inactive ligands. The resulting NIB model functions as a template or pseudo-ligand directly in the shape/electrostatics similarity comparison against ligand 3D conformers included in the screening compound libraries. The ligand preparation and similarity comparison against the model (
Figure 1) is done using established ligand-based screening tools [
4,
7].
Whereas standard flexible docking relies on estimating the favorability of ligand-receptor complexes by summing up the weak interactions, such as hydrogen bonding and the hydrophobic effect, the NIB focuses squarely on the shape/electrostatics similarity of the molecular recognition process. Despite the apparent simplicity of this shape-centric approach, the benchmarking has shown thatthe NIB produces high enrichment as indicated by the area under curve (AUC) values and early enrichment factors with various targets [
1,
3,
8]. The methodology is especially suitable for the targets with well-defined cavities such as nuclear receptors, but, in practice, even a sub-cavity or a shallow groove can be used to build an effective negative image. As such, the NIB has been used to assist the structure-activity relationship analysis of the 3-phenylcoumarin analog series with the 17-hydroxysteroid dehydrogenase 1 [
9], monoamine oxidase B [
10], and UDP-glucuronosyltransferase 1A10 [
11], as well as to facilitate the discovery of novel estrogen receptor α ligands [
12] and retinoic acid-related orphan receptor γ(t) inverse agonists [
13].
Applying 3D similarity- or shape-based methods in the virtual screening schemes increases the diversity of the discovered compounds [
14]. With the NIB, the docked ligand and protein can overlap somewhat, and, while this can weaken the compound’s ranking, no ligands are skipped entirely due to the clashes as can happen with the flexible docking algorithms. The upside of tolerating the overlaps is that those novel scaffolds or functional moieties producing a good partial match with the target’s cavity are readily put forward. This is advantageous because docking can put forth not only new compounds but also functional fragments to be incorporated into novel drug constructs via organic synthesis [
15]. Moreover, Molecular Mechanics/Generalized Born and Surface Area (MM/GBSA) calculations, for example, can be performed to optimize the rigid docking poses inside the target protein’s cavity for improving the NIB enrichment [
8].
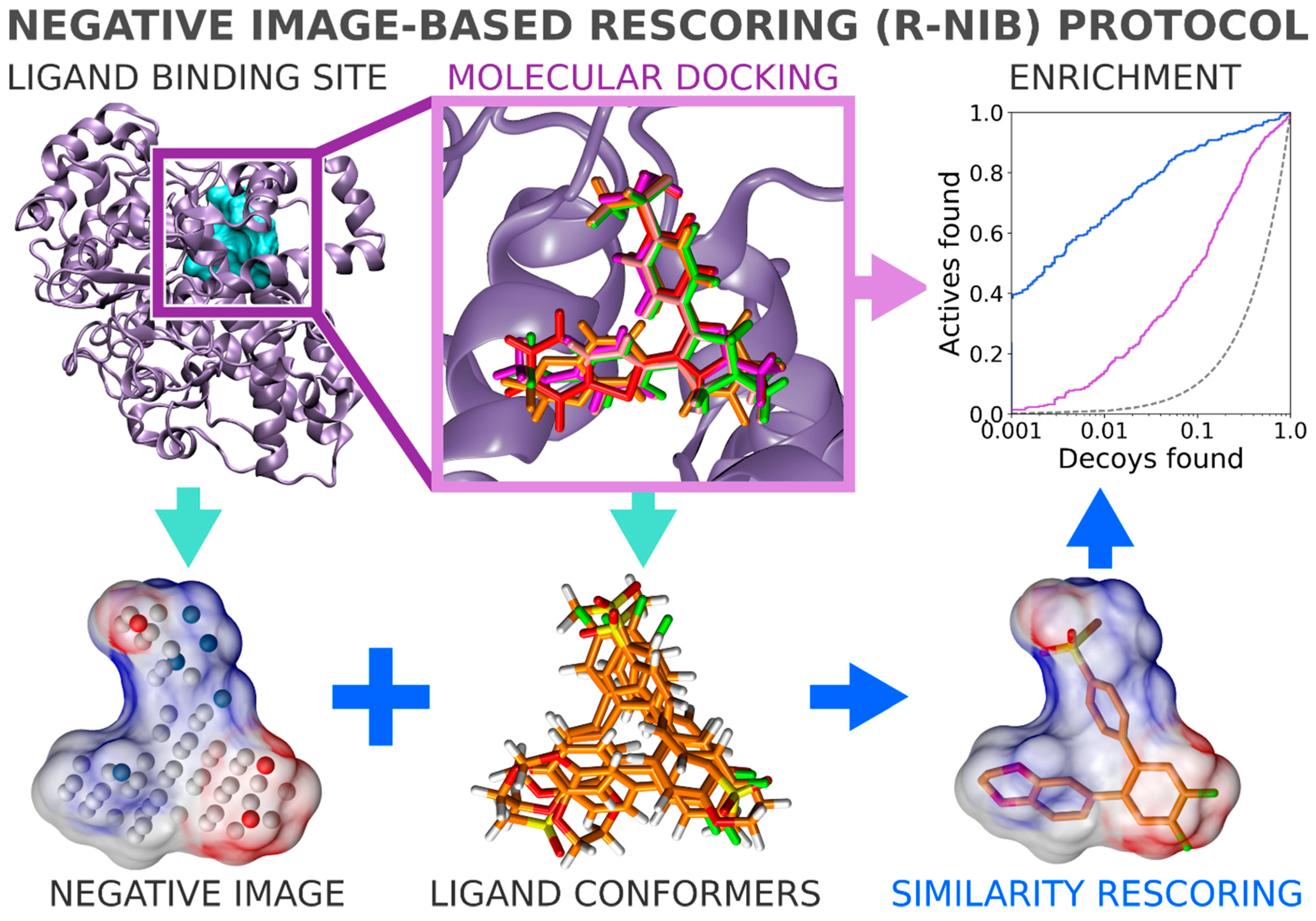
In general, flexible docking is better positioned to sample the possible ligand poses than the rigid docking approach. Therefore, the NIB methodology was recently repurposed for rescoring existing molecular docking solutions [
16]. The NIB rescoring (R-NiB;
Figure 2) of explicit docking poses was shown to improve the docking performance markedly, especially the very early enrichment, with several targets. This includes cyclooxygenase-2 (COX-2; enzyme commission number 1.14.99.1;
Figure 1 and
Figure 2), which catalyzes the conversion of arachidonic acid to prostaglandin endoperoxide H2 and was used as a NIB screening and docking rescoring example in this study. In short, the NIB is not only a powerful docking technique (
Figure 1), but it is also a docking rescoring (
Figure 2) methodology that has the potential for wide-scale application.
The study provides simple step-by-step instructions on how to perform rigid docking (
Figure 1) or docking rescoring (
Figure 2) using the NIB methodology with non-commercial software. The in-depth examination of the settings together with discussion on the notable exceptions is outlined using practical COX-2 screening examples (
Figure 1 and
Figure 2). Furthermore, several popular ligand 3D conformer generation algorithms are tested with the COX-2 test sets and compared to outline the optimal scheme for the rigid docking with the NIB methodology.
2. Results
The negative image-based (NIB;
Figure 1) screening [
1,
3,
8] and the negative image-based rescoring (R-NiB;
Figure 2) [
16] protocols are presented below as stepwise workflows.
The practical aspects of the NIB and R-NiB methodologies are discussed below using a virtual screening or benchmarking example, i.e., the screening is performed using the directory of useful decoys (DUD) test set [
18,
19] and a celecoxib-bound cyclooxygenase-2 (COX-2) protein 3D structure (
Figure 1 and
Figure 2; Protein Data Bank (PDB): 3LN1 [
2]). Note that the NIB protocol (commands #1–23) is executed in the BASH command line interface (or terminal) in the UNIX/LINUX environment. Furthermore, three alternative conformer generators (
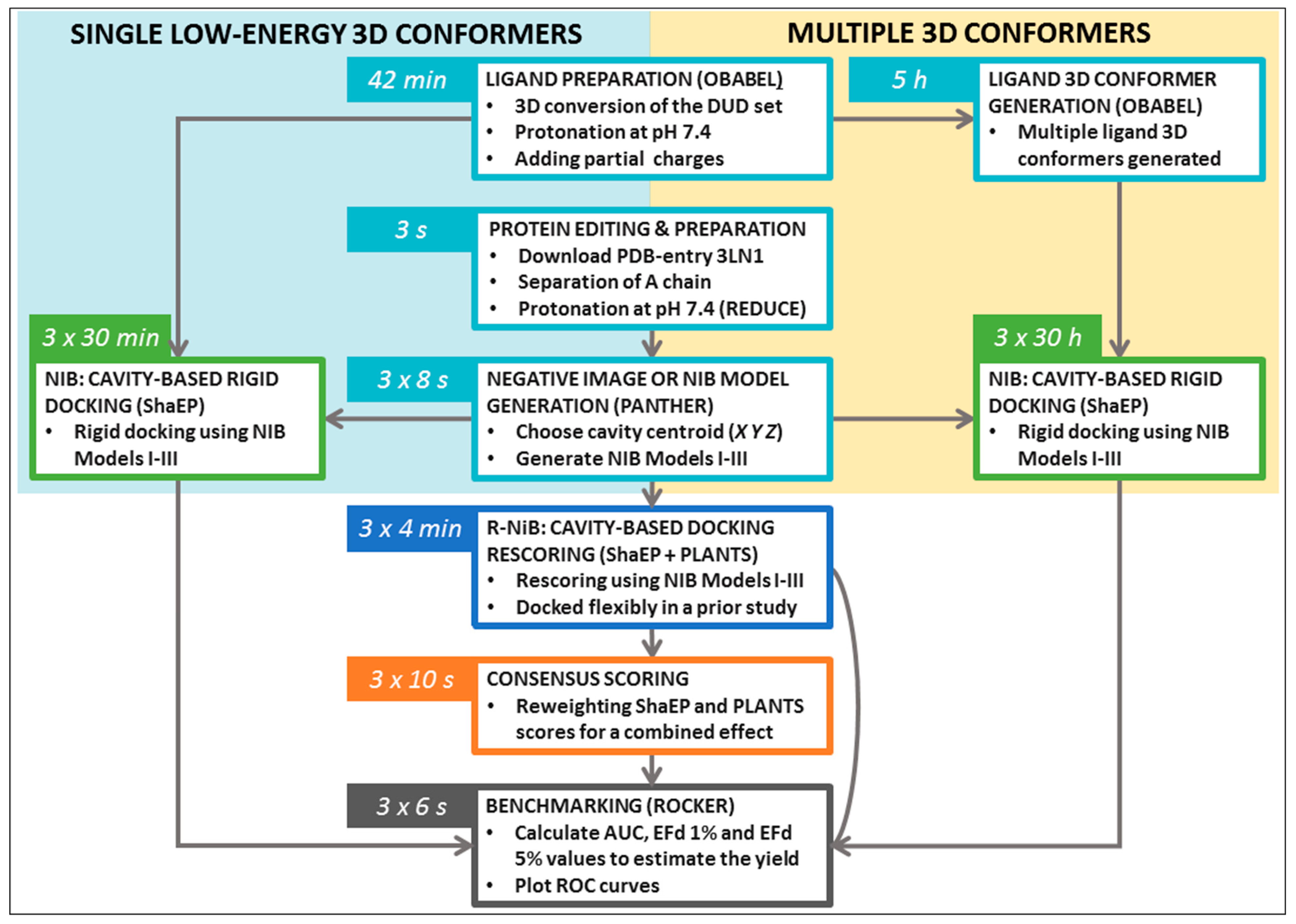
Table 1) were tested for the NIB in addition to OBABEL, which is used in the benchmarking example. Finally, the R-NiB is performed using the flexible docking poses generated by PLANTS to improve the enrichment. The rescoring relies either solely on the ShaEP-based complementarity or similarity scoring (commands #24–35) or the combined and re-weighted PLANTS- and ShaEP-based consensus scoring (commands #36–41).
The terminal commands and further practical information are given in the
Supplementary Material (README.txt, commands.txt) to assist the execution of trial runs of the protocols. The NIB protocol testing using the single low-energy conformers (
Table 2) takes ~10-fold less time than with the multiple conformers (
Table 3); furthermore, the R-NiB testing (
Table 4) is substantially faster than the rigid docking, because the flexible docking poses are provided premade, and no geometry optimization is performed with ShaEP (
Figure 3). Though the specific commands are not given to avoid repetition, the protocols were also tested using the DUD-E test set and the PDB-entry 1CX2 [
20].
2.1. Ligand Preparation: 3D Conversion, Protonation, and Partial Charges
In the NIB screening (
Figure 1), the rigidly docked ligand 3D conformers are generated ab initio with a separate software (
Table 1); however, depending on the target protein and the ligand sets one can acquire high enrichment using only a few or even a single low-energy conformer. Before performing the cavity-based rigid docking with a single conformer or multiple conformers, the 3D coordinates (simplified molecular-input line-entry system (SMILES)-to-MOL2), partial charges and ionization/protonation states of the small-molecules need to be generated (
Figure 1). This is achieved using, for example, LIGPREP in MAESTRO or MARVIN, but non-commercial software such as RDKit or OBABEL [
21] can also be used. It is crucial that the pH is set to match the conditions of the activity assay (e.g., pH 7.4) during the protonation.
The DUD [
19] ligands for COX-2 were converted from the SMILES format into the MOL2 format using OBABEL [
21] (command #1). A single 3D conformer was generated for each ligand included in the set. Next, the protonation of the ligands was set to match pH 7.4 (command #2), and the partial charges were inserted using the Merck Molecular Force Field 94 (MMFF94) [
22] (command #3) with OBABEL [
21]. For comparison, the ligands were also prepped using LIGPREP in MAESTRO, MARVIN, and RDKit (
Table 1). With COX-2, the NIB screening produces high enrichment directly using these single low-energy 3D conformers, and, for this reason, one can choose to skip the 3D conformer generation step to save time when going through the protocol (
Figure 3).
2.2. Ligand Preparation: 3D Conformer Generation
Ultra-fast speed and computational efficiency are hallmarks of both the NIB screening (
Figure 1) and the ligand-based screening [
1,
3,
8]. This is largely because the different ligand 3D conformers are not sampled on the fly against the protein 3D structure during the rigid docking, as is done in the flexible molecular docking. Instead, several low-energy conformers are generated for each ligand prior to the eventual similarity screening and geometry optimization with the cavity-based NIB model (
Figure 1). The ligand 3D conformer generation can be done using either non-commercial or commercial software tools with varying results (
Table 1). The conformer generation, as well as the eventual cavity-based rigid docking using the multiple conformers, is a lot more time consuming than performing the NIB screening with single low-energy conformers (
Figure 3). Alas, one should not expect that single conformers would work in all screening experiments, although this is the case with the COX-2 benchmarking.
The protonated ligand 3D coordinates were used as an input to generate multiple conformers using the --confab option in OBABEL [
21] (command #4). By default, an extensive number of conformers is generated, and, to avoid this, the output was limited with two basic options: The maximum number of conformers (-conf; from 1,000,000 to 100,000) and the root mean square deviation cutoff (--rcutoff; from 0.1 to 1.0). For comparison, the ligand 3D conformer generation was also done using other conformer generators (
Table 1).
2.3. Selecting the Target Protein 3D Structure
The success of the NIB screening is dependent on the input protein 3D structure, especially its ligand-binding cavity conformation, used as a template for the negative image generation (
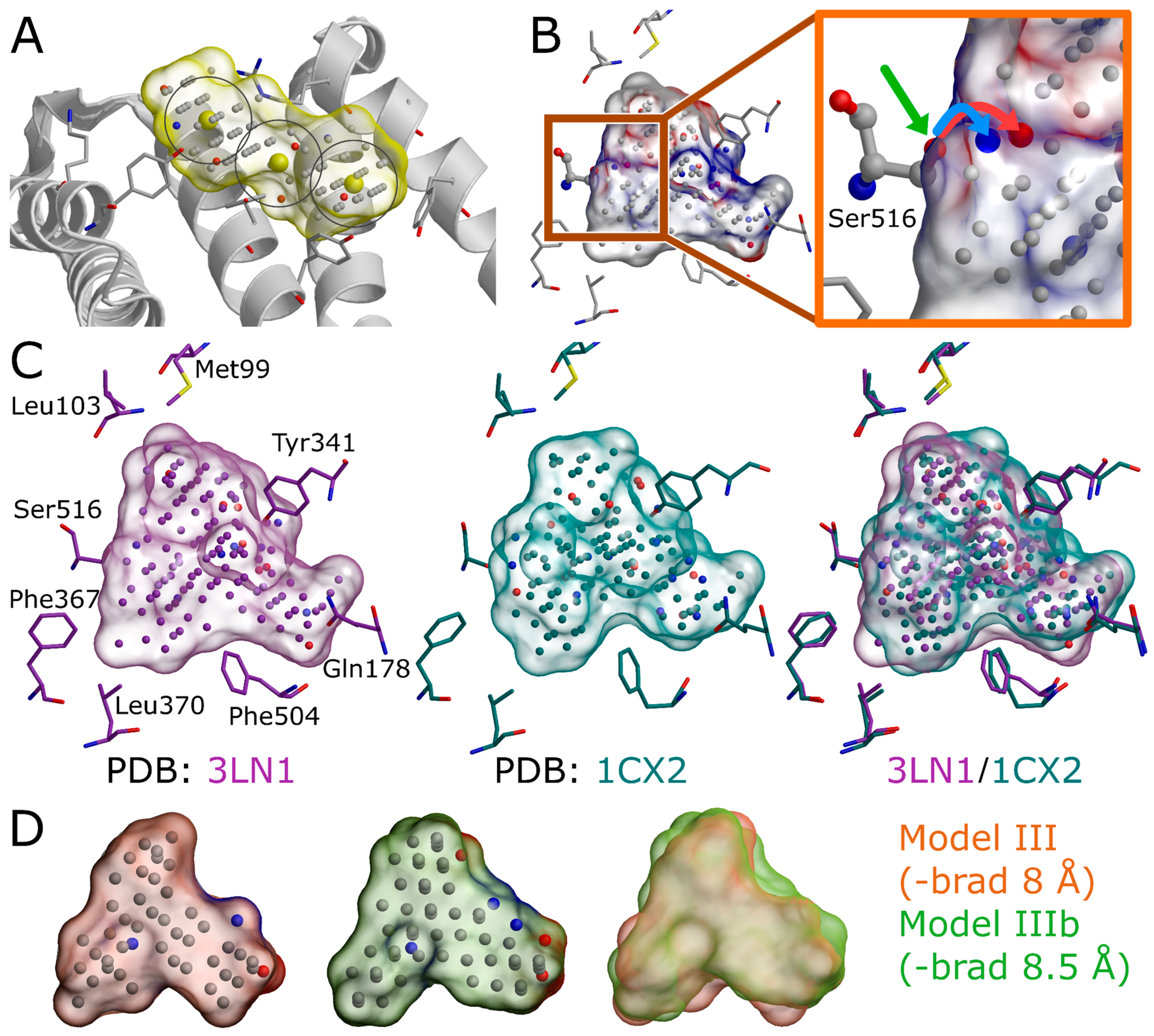
Figure 1). The input structure selection follows the basic criteria that apply to standard molecular docking as well: The resolution should be sufficiently high, and the protein conformation should be able to accommodate the binding ligand. In principle, the PDB-entry does not have to house any known active compounds prior to the model generation, but a bound ligand can affect the cavity geometry via induced-fit effects. If included, the bound ligand(s) can assist in the NIB model generation by providing centroid coordinates (
Figure 1A and
Figure 4A), and they can assist in limiting the model scope to the known binding area. In some cases, using multiple protein structures in the model generation originating, for example, from molecular dynamics (MD) simulation trajectory can improve the NIB screening yield [
1].
Two PDB-entries were selected for the NIB screening with COX-2. The PDB-entry 3LN1 [
2] (
Figure 1,
Figure 2 and
Figure 5C) is used in the practical example; meanwhile, the PDB-entry 1CX2 [
20] (
Figure 5C) is used as an alternative input for which the applied commands are not shown due to their redundancy. The protein X-ray crystal structure was downloaded directly from the PDB in the terminal (command #5), but it can also be downloaded manually online (e.g.,
https://www.rcsb.org/).
2.4. Protein 3D Structure Editing and Preparation
The extra chains and other non-peptidic residues do not necessarily have to be removed for building NIB models using PANTHER [
3], although their removal can make the process marginally faster. Even though the NIB model generation can be performed without protons added to the protein 3D structure, this can lead to several alternative cavity-based models. This is because certain residues can have alternative protonation states or bond angles for the protons that are responsible for the H-bonding (
Figure 5B) and, thus, depend on the local environment. In fact, one should be mindful on how the added protons affect the charge distribution of the negative image and the eventual docking results. The protons can be added for the target structure and even its cofactors using external software (e.g., REDUCE [
24]), in which case the alternative proton shuffling is omitted during the NIB model generation (
Figure 1). The case-specific protonation of, for example, histidine and aspartic acid residues at the ligand-binding cavity, can be tricky, and, in unclear cases, one should employ protonation prediction algorithms such as PROPKA [
25,
26].
The A chain of the PDB-entry 3LN1 [
2] was selected for the NIB model generation and, for improved computing efficiency, extracted into a separate PDB file (command #6;
Figure 1) where the explicit protons were inserted using the default settings of REDUCE [
24] (command #7;
Figure 1). With COX-2, the outputted bond angles of the protons were visually assessed to be reasonable in BODIL [
5]. Such an evaluation is necessary because, for example, the proton in the hydroxyl group of Ser516 side chain in the COX-2 active site could have an alternative angle that affects the resulting cavity point composition (
Figure 5B).
2.5. Defining the Ligand-Binding Cavity Centroid
The NIB model generation using PANTHER [
3] requires that the ligand-binding cavity location is designated beforehand (
Figure 1A and
Figure 4A). In other words, the user needs to have a concrete idea where the ligand binding should happen to focus on a specific location inside or on the surface of the protein. For this purpose, cavity detection software such as SITEMAP [
27,
28] or POVME 3.0 [
29] can estimate the druggability and dimensions of protein cavities. In any case, the best scenario is to begin the NIB model generation with PANTHER using the centroid coordinates of a bound ligand already included in the PDB-entry. If not applicable, the cavity detection can begin from any arbitrary coordinate point given by the user (-center(s)) or by using any residue atom coordinate present near the cavity center (-basic multipoint). Overall, the centroid selection process is highly similar to choosing the center of radius for any standard docking routine.
The COX-2 inhibitor ligand celecoxib, or CEL, (residue 682 in the A chain;
Figure 1 and
Figure 4D), which is bound at the active site in the PDB-entry 3LN1 [
2], was selected to define the cavity center (A-682) during the NIB model generation with PANTHER [
3] (
Figure 4A,D).
2.6. Generating a Negative Image of the Enzyme’s Ligand-Binding Cavity
The first NIB model is typically generated using the default settings in PANTHER [
3] (
Figure 1), after which it is critically evaluated, and, if needed, the settings are further tweaked. For convenience, especially at the later stages of the NIB model generation, the necessary settings are inserted directly into a PANTHER [
3] input file using a text editor instead of typing and executing them in the terminal, as is done in the example below.
Firstly, the default PANTHER [
3] input file (default.in) is generated (command #8). Secondly, PANTHER [
3] is used to generate a preliminary NIB model (command #9) in the MOL2 format (Model I in
Figure 4B). If the input protein would lack protons (command #
7, not executed), altogether 12 alternative NIB models would be outputted for COX-2. This is because the cavity houses several residues capable of H-bonding, and each proton of the H-bond donor groups, such as the hydroxyl group, is given an alternative position that affects the charge distribution of the model (
Figure 5B).
2.7. Estimating the Negative Image Viability and Tweaking the Settings
There are at least three major concerns regarding the model viability in the NIB screening:
- (1)
The NIB model must be restricted to the area of the cavity that facilitates the ligand binding.
- (2)
The NIB model shape should resemble, however loosely, an envisioned or actual ligand molecule occupying the cavity.
- (3)
The NIB model cavity points must contain crucial charge/electrostatics information required for mimicking the ligand-receptor H-bonding, as the shape similarity alone might not be enough for ensuring rigid docking success (
Figure 5B).
The preliminary model or Model I (command #9) outputted by PANTHER [
3] matches the cavity shape (
Figure 4A versus 4B). It is composed of neutral filler atoms (grey dots in
Figure 4B) and negatively or positively charged cavity points (red or blue dots, respectively, in
Figure 4B) that have the opposite charges in comparison to the protein residues lining the cavity. The partial charges of protein (and possible cofactor) atoms must be pre-defined in a separate file, which contain by default AMBER (Assisted Model Building with Energy Refinement) force field-based charges (a PANTHER library charge.lib). In other words, the charge points face directly those residues capable of either accepting or donating protons in the H-bonds (
Figure 5B). Before performing the similarity comparison with Model I (
Figure 4B), a second model or Model II (
Figure 4E) is generated using a 1.5 Å ligand distance limit (-ldlim) in PANTHER [
3] (command #10). Though the two outputted models are roughly similar, Model II does not expand far away from the space taken by the inhibitor bound at the ligand-binding site in the input PDB-entry (
Figure 4D versus 4E). In comparison, Model I, which was generated without the ligand distance limit but only relies on the 8 Å cavity detection radius, is visibly bulkier than Model II (
Figure 4B versus 4E). This demonstrates that the ligand is loosely bound to the protein, the ligand does not fill the entire space available for binding, or the PANTHER parameterization for the NIB model generation is not optimal.
2.8. Rigid Docking by Aligning Ligands Against the Negative Image
The NIB screening is performed using the similarity comparison algorithm ShaEP [
4] (
Figure 1), which was originally developed for the ligand-based screening. The ligand 3D conformers are geometry optimized or rigidly docked based on the shape/electrostatics against the cavity-based NIB model (
Figure 1). ShaEP [
4] provides a similarity score from 1 to 0 for ranking the compounds from the best to the worst matches against the cavity-based model. Those ligand conformers matching the NIB model best are given the highest score, indicating the highest degree of similarity with the cavity space available for the ligand binding. If the model encompasses the key shape and charge features needed for the ligand binding, in theory, the NIB screening should put forward the best-matching molecules. While the charge can be an important factor for assuring correct H-bonding interactions in the rigid docking (
Figure 4B), the shape is frequently the defining factor in the ligand-receptor complex formation.
The rigid docking is performed using the default settings of ShaEP [
4] (commands #11 and 12). While this basic arrangement works well for COX-2, the yield can be improved with certain targets by either lowering or increasing the charge effect. The NIB screening success for Models I and II (
Figure 4B,E) is estimated using the early enrichment and area under curve (AUC) values (
Table 2 and
Table 3) and the receiver operating characteristics (ROC) curves (
Figure 4C,F) calculated with ROCKER [
6] (commands #13 and 14). The compounds skipped during the ligand preparation or the NIB screening (
Table 1), if any, are not considered in the AUC calculation. The Model II screening produced a higher AUC value than the flexible docking with the DUD set; however, the very early enrichment, or EFd 1%, was not better with either of the models using the OBABEL-generated conformers than what PLANTS produced (
Table 3 versus
Table 4). Notably, the enrichment was higher for both NIB models, surpassing the flexible docking yield, when the rigidly docked single/multiple conformers were generated using, for example, RDKit instead of OBABEL (
Table 3;
Figure 4C,F).
2.9. Feedback Loop—Fine-Tuning the Cavity Detection Settings
Following the rigid docking, one should inspect best-ranked docking poses and fine-tune the NIB models with the benefit of hindsight (
Figure 1). For example, one can extract the 50 best-ranked docking poses (molpicking.bash; commands #15–20) and visualize the predicted binding modes using the preferred 3D viewer (e.g., BODIL) [
5]. With the benchmark test sets, one can apply the trial-and-error approach, where the usefulness of each PANTHER setting generating the model (
Figure 4B,E,H) can be assessed. This sort of training is not possible without verified active and inactive compounds or expert intuition to assess the effect of the applied changes. For COX-2, the DUD/DUD-E test sets [
18,
19] include both active ligands and inactive decoy molecules (
Table 1) for assessing the fitness of the NIB models. The first-tried default settings frequently work well in the NIB screening [
3] (e.g., Model I in
Figure 4B,C); nevertheless, visualization of the models and docking poses and fine-tuning of the cavity detection settings is recommended (-ldlim 1.5 Å was used for Model II in
Figure 4E,F). Better models might be acquired, for example, by varying the centroid position (
Figure 4A), the cavity detection radius (
Figure 4B), and the atomic radii of the protein residues (a PANTHER library rad.lib), the ligand distance limit (
Figure 4E,F) or the filler atom packing method (
Figure 4G).
The third NIB model, or Model III (
Figure 4H), was generated using the otherwise same settings as Model II (
Figure 4E), but the default face-centered cubic (FCC) packing was changed to the less dense body-centered cubic (BCC) method (command #21;
Figure 4G). Model III can also be generated using an input file (final_panther.in in the
Supplementary Material) instead of the elaborate terminal command. Though only the packing method was altered during the cavity-detection phase, the composition and shape of these two negative images differ markedly (
Figure 4E versus 4H). The NIB screening/docking (command #22) and the enrichment analysis (command #23) indicate that Model III does not produce higher EFd 1% than the prior models when the single/multiple ligand 3D conformers were prepared using OBABEL (
Table 2 and
Table 3;
Figure 4B,E). Yet again, the NIB screening results are considerably better for the third model, far surpassing the yield of PLANTS docking, especially, if the conformer generator RDKit was employed (
Figure 4C,F versus 4I;
Table 3 versus
Table 4).
These enrichment metrics (
Table 2 and
Table 3;
Figure 4) indirectly suggest that the rigid docking with the ab initio generated ligand conformers is able to predict the binding poses of the COX-2 active ligands reasonably well. More to the point, the NIB, that ranks the ligands based on the cavity-based similarity, fare better than the ChemPLP scoring in PLANTS (
Table 2 and
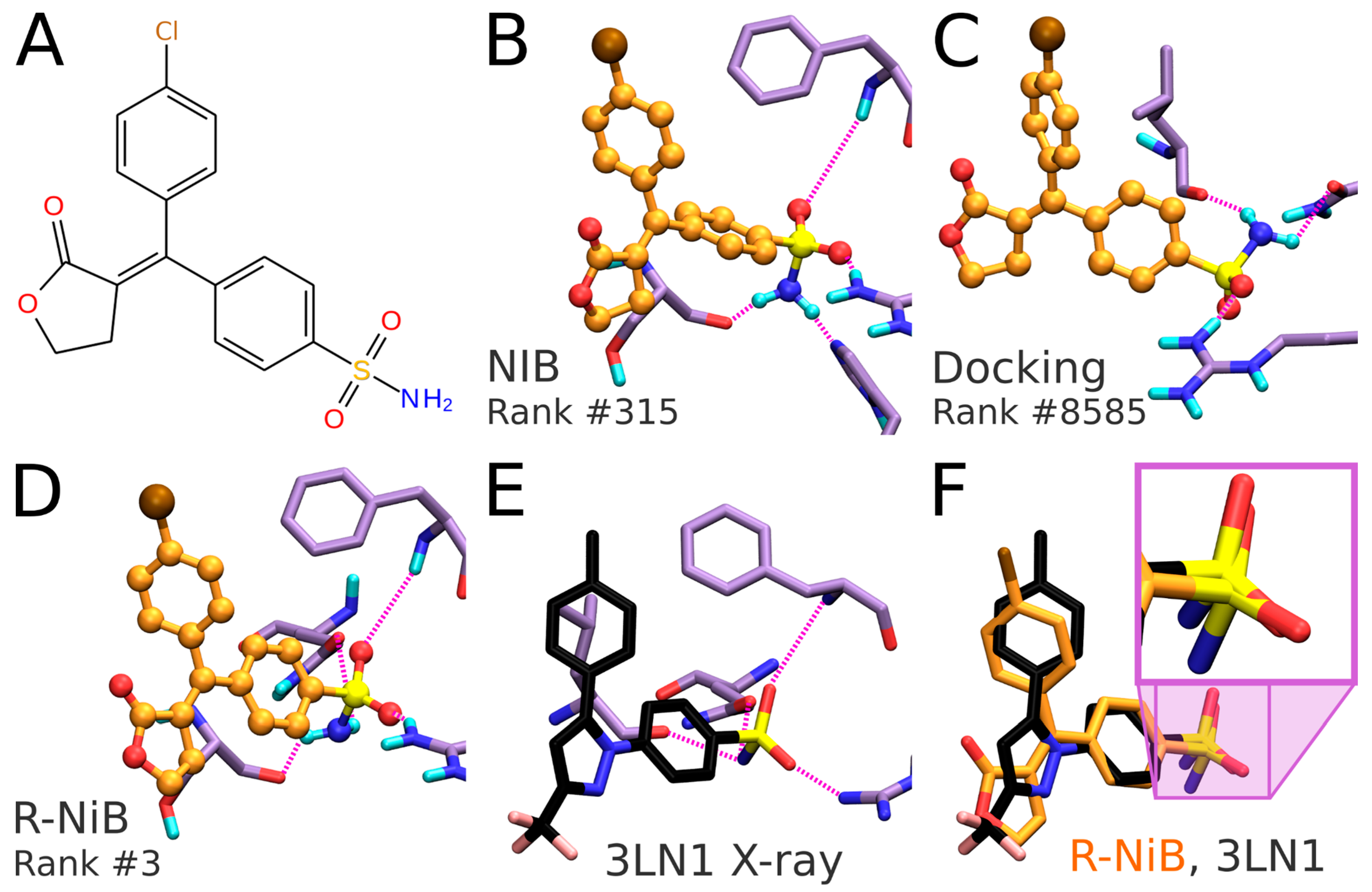
Table 3). As with any docking methodology, it is prudent to compare the predicted poses to experimentally verified binding modes. Here, a close inspection is done for the best-ranked docking pose of an established COX-2 inhibitor included in the DUD set (
Figure 6A,B). The overall alignment of the compound in question is roughly similar between the top-ranked NIB screening and the flexible docking poses (
Figure 6B versus 6C,D), but, more importantly, the NIB proposes a binding pose resembling that of structurally related inhibitor celecoxib (
Figure 6B versus 6E). As a result, the NIB screening provides a lot higher ranking for the inhibitor than the regular flexible docking (NIB rank #315 versus PLANTS rank #8585). The same ranking order, but to a lesser effect, is acquired for the celecoxib (NIB rank #125; PLANTS rank #295).
2.10. Negative Image-Based Rescoring of Flexible Docking Solutions
In addition to the cavity-based rigid docking (
Figure 1 and
Figure 4), the NIB methodology can be used to rescore existing docking solutions for improving the flexible molecular docking performance (
Figure 2) [
16]. The preliminary docking with the target protein can be performed using any flexible docking algorithm such as PLANTS [
17] or AUTODOCK [
30]. Multiple docking poses are outputted and, if need be, converted into the MOL2 format for the rescoring with the R-NiB. Next, the same cavity space used in the flexible docking is again used to generate a NIB model, which encompasses the key shape/charge features needed for the ligand binding, using PANTHER [
3]. Finally, the flexible docking poses are compared against the cavity-based NIB model using ShaEP [
4] without the time-consuming geometry optimization (--noOptimization) that is done during the rigid docking (
Figure 3).
The COX-2 ligands [
18,
19] were docked flexibly using the default settings of PLANTS [
17] after the ligand preparation with LIGPREP in MAESTRO in our previous study [
16]. The used PLANTS input file and the explicit docking poses are included in the
Supplementary Material. Models I–III (and alternative PDB-entry 1CX2-based Models IV–VI) generated for the NIB screening (commands #9, #10, and #21) were directly used with the R-NiB. Altogether, 10 alternative flexible docking poses for each ligand (
Table 1) were rescored using ShaEP [
4] without the geometry optimization (commands #24–26) using the --noopt option. The ShaEP result files containing multiple alternative poses for each rescored compound were trimmed to include only the best-ranked poses for the enrichment calculation (trim_shaep.bash; commands #27–29). The EFd 1%, EFd 5% and even the AUC values (commands #30–32) indicate that the R-NiB improved docking performance for the DUD set (
Table 4). With the more demanding DUD-E set, the first model did not improve the enrichment, but the R-NiB was consistently able to improve the yield with the latter two models (
Table 4).
In theory, the R-NiB or any other rescoring methodology can improve the docking yield (
Table 4), because, while the original docking algorithm samples the “correct” ligand binding poses, its default scoring does not rate these specific poses high enough. Again, this effect is demonstrated by inspecting the predicted binding poses of an established COX-2 inhibitor included in the DUD set (
Figure 6A). The R-NiB selects an alternative docking pose for the inhibitor (
Figure 6D), which remains the best-ranked NIB screening pose (
Figure 6B) regarding the sulfonamide group placement than the one ranked best by PLANTS (
Figure 6C). Notably, the R-NiB ranking of the example compound is substantially higher than what PLANTS or the NIB suggested (R-NiB rank #3 versus NIB/PLANTS rank #315/#8585). By comparing this best-ranked inhibitor pose to structurally similar celecoxib bound at the COX-2 active site (
Figure 6E; PDB: 3LN1 [
2]), it is evident that it is spot on for the sulfonamide (magenta box in
Figure 6F). Again, the R-NiB also provides a far better ranking for the inhibitor celecoxib than the flexible docking or NIB screening (R-NiB rank #42 versus NIB/PLANTS rank #125#295).
Despite this specific example (
Figure 6), the ability of the R-NiB to improve docking performance could also be explained by the low scoring for the inactive decoys that align poorly with or further away from the designated cavity area. In any case, it is always recommended to extract a few of best-ranked docking poses (e.g., ~50–100) to see if the docking or rescoring results make sense at the atomistic level (molpicking.bash; commands #33–35).
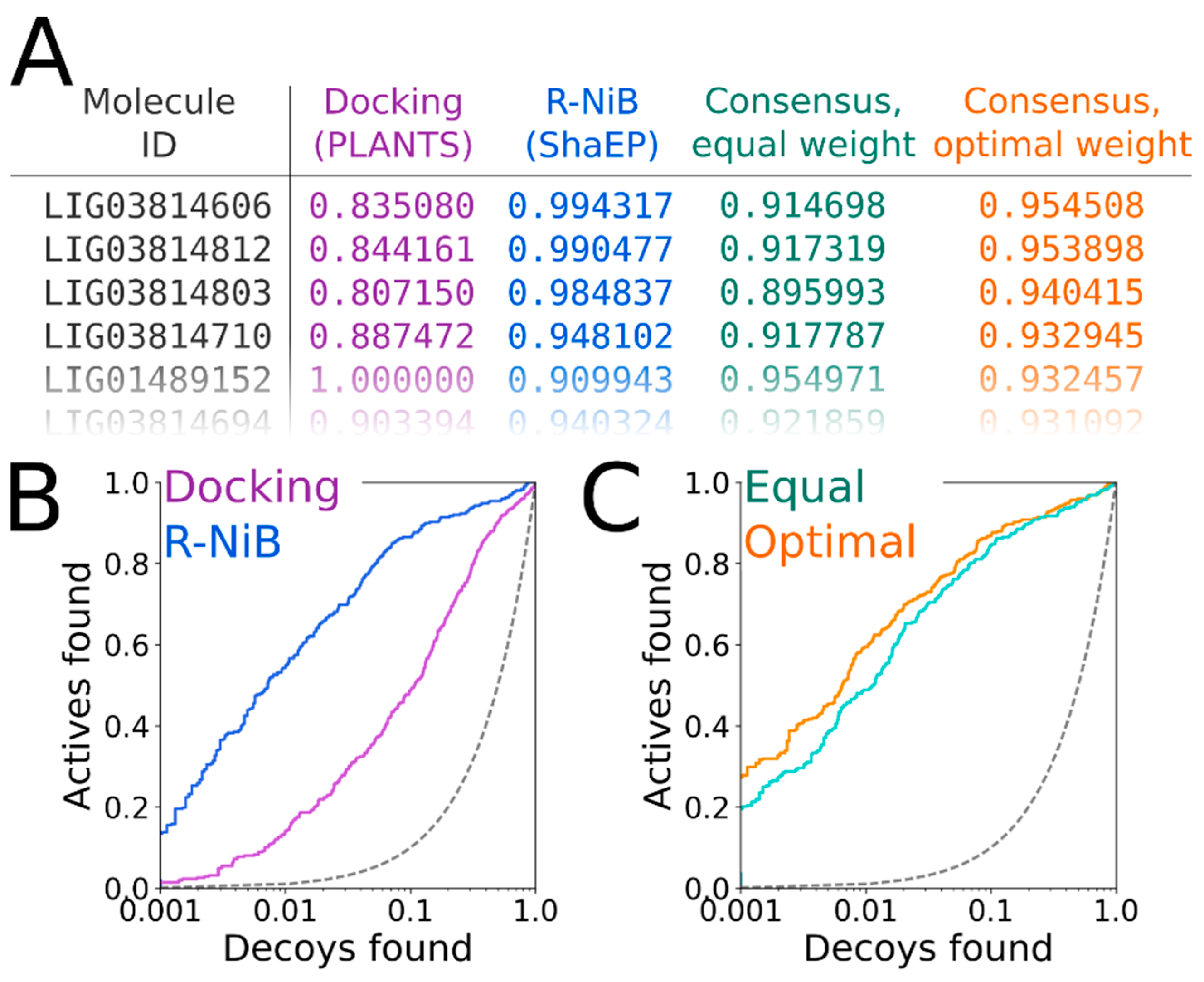
2.11. Consensus Scoring: Balancing the Scoring Functions
The ShaEP scoring can be combined with the original PLANTS scoring in the consensus scoring [
16] (
Figure 7A). The R-NiB scoring relies solely on the shape/electrostatics similarity calculated with ShaEP (weight of 1.00 in
Table 4) and, accordingly, PLANTS is only responsible for sampling the ligand poses. Though the PLANTS docking enrichment is not as high as that of the best NIB or R-NiB results (
Table 3 and
Table 4), the flexible docking poses contain better matching geometries with the target’s cavity than what the ab initio conformer generators produce (
Figure 6B versus 6D). However, the R-NiB performance can be improved in some cases by incorporating the original docking scoring to the ShaEP-based compound ranking. This requires the normalization of the scores outputted by both ShaEP and PLANTS, after which the weight of the two functions is balanced for the optimal effect (
Figure 7A). In general (but not always), applying an equal weight (a weight of 0.50 in
Table 4) between the scoring functions produces better enrichment than what the PLANTS docking or R-NiB produces alone; still, the optimal weight varies for different targets [
16].
The normalization and re-weighting of the PLANTS and ShaEP scoring (
Figure 7A) are performed using a BASH script (consensus.bash; commands #36–38), which is specific for the combined PLANTS and ShaEP usage. The consensus scoring for the DUD docking poses produced better EFd 1% values consistently if the optimal weight was systematically tested, whereas the equal weigh scoring (commands #39–41) improved on the R-NiB yield only with Model I (
Table 4). With the DUD-E set, improvements over the R-NiB were consistent but also moderate with Models I and II (
Table 4), but even a minor up-tick in the early enrichment could have substantial effects for real-life screening experiments. The early enrichment improvements over straight-up R-NiB and PLANTS docking (
Figure 7B) through the consensus scoring are visible in the semi-logarithmic ROC curves of the DUD set with Model II (
Figure 7C) when the optimal weigh was applied (
Table 4).
3. Discussion
The cyclooxygenase 2 (COX-2) benchmarking example shows that the negative image-based (NIB;
Figure 1) [
1] screening or rigid docking is consistently producing higher enrichment than the regular flexible docking (
Table 3 versus
Table 4). The area under curve (AUC) values demonstrate the dominance of NIB over PLANTS [
17] docking with both the DUD [
19] and DUD-E [
18] sets (
Table 3). The NIB has been shown to outperform standard docking regarding the AUC values with a multitude of targets in prior studies [
1,
3,
8]. On a practical level, the early enrichment values (calculated as true positive rates when 1 or 5 % of the decoys have been found) are a better measure of virtual screening success than the AUC values. Hence, it is noteworthy that the highest EFd 1% and EFd 5% values produced by the NIB screening with the COX-2 sets are also higher than those given by the PLANTS docking (
Table 3 versus
Table 4).
The better performance of the cavity-based rigid docking routine over the flexible docking suggests that the shape complementarity is a crucial factor for the COX-2 inhibitor binding, at least with the benchmark test sets. That is not to say that other docking algorithms or PLANTS cannot outperform the NIB methodology in virtual screening experiments on a case-by-case basis [
3]. Both the composition of the test sets and the target’s cavity geometry are bound to lower the effectiveness of the cavity-based rigid docking in comparison to the flexible docking in some cases. Moreover, the COX-2 benchmarking (
Table 2 and
Table 3) indicate that the NIB model composition (
Figure 4B,E,H), the input 3D protein structure conformation (
Figure 4C), and the ligand conformer generation method (
Table 1;
Figure 4C,F,I) profoundly affect the rigid docking success (
Table 3).
The NIB model generation is a straightforward process when the target protein’s ligand-binding site is a well-defined cavity (
Figure 1 and
Figure 4). The charge distribution and dimensions of the model can be improved using different PANTHER [
3] options such as the cavity centroid (center(s), -cent;
Figure 1 and
Figure 4A), cavity detection radius (box radius, -brad;
Figure 1,
Figure 4A and
Figure 5D), and filler atom packing (packing method, -pack;
Figure 4G), as well as via alternative residue protonation (
Figure 4B) or input structure conformation (PDB: 3LN1 versus 1CX2;
Figure 4C;
Table 3). A less direct approach might be needed when dealing with shallow surface pockets, as well as large or even open cavities without clear geometrical limits. To prevent the model protruding too far from the intended cavity area, one can limit the NIB model expansion using an already bound ligand (ligand distance limit, -ldlim;
Figure 4D,E) or other selected residues (basic multipoint, -bmp) inside the cavity or even lining a groove on the protein surface. In addition, the NIB model can be forced to a certain subsection of the cavity by applying the multibox option (-mbox;
Figure 5A), which allows the use of several coordinate points to describe a cavity of irregular or arbitrary shape.
Importantly, the COX-2 example shows that the rigid docking (
Figure 1) is not only dependent on the NIB model shape/electrostatics but that the ligand 3D conformers have a substantial effect (
Table 3). In the case of COX-2, the NIB screening works remarkably well when only a single low-energy 3D conformer is used in the rigid docking for each compound (
Figure 4;
Table 2). In fact, the results suggest that the costly use of multiple conformers (
Figure 3;
Table 3) could improve the NIB screening performance consistently with the DUD set only when the conformers were outputted by the RDKit (
Table 3;
Figure 4). With the more demanding DUD-E set, only the AUC values were improved consistently using the multiple conformers outputted by MARVIN or RDKit in the rigid docking (
Table 3;
Figure 4H,I).
Prior to this study, the ligand 3D conformer generation had been tested thoroughly for the purpose of assessing its effects on the ligand-based screening [
31,
32]; however, the benchmarking (
Table 3) shows that the conformer composition is equally important for the NIB screening (
Figure 1). OBABEL was used in the ligand preparation in the benchmarking example due to its ease of use and open source status (
Table 1). The cavity-based rigid docking, especially using the single low-energy 3D conformers, produced consistently higher AUC values than the PLANTS flexible docking, regardless of the employed ligand preparation method (
Table 3 versus
Table 4). Nevertheless, the NIB screening done using the multiple ligand conformers outputted by OBABEL (
Table 1) produced weaker enrichment than the other software such as RDKit (
Figure 4C,F,I;
Table 3). Though RDKit produced reasonable ligand conformations and excellent NIB screening results with the COX-2 test sets (
Table 3), in practice, its successful usage requires basic knowledge of PYTHON programming and, generally, more effort than the other software (
Table 1).
The effectiveness of the outputted 3D conformers differed between the software in the NIB screening. Their rigid docking performance is undoubtedly case-specific, and one should not draw too far-reaching conclusions based on the COX-2 benchmarking only. For example, the single low-energy conformers worked well with COX-2 (
Figure 4;
Table 2) likely due to the specific composition of the test sets and the flat or unfussy dimensions of the target cavity. The biologically relevant binding poses of the ligands, which are fundamentally sought after in the molecular docking are not necessarily close to the ab initio calculated energetic minima. Thus, the utilization of multiple alternative ligand 3D conformers in the cavity-based rigid docking should also improve the screening yield. The docking results vary significantly depending on the rotatable bond number of the molecules, the target protein’s ligand-binding cavity properties, and, above all, the selected non-default settings. Not all permutations could be tested for the ligand preparation, and it is fully possible that there exist better settings for the tested conformer generators (
Table 1). Nonetheless, the results show that the composition or “quality” of the conformers (
Table 2 and
Table 3) is more important than their sheer quantity (
Table 1) in the NIB screening.
The negative image-based rescoring (R-NiB;
Figure 2) produces better enrichment than the original PLANTS docking (
Table 4;
Figure 7B) [
16] or the standard NIB screening (
Figure 1;
Table 3) with the COX-2 test sets. The improvement is consistent with the DUD set, although the crudest or bulkiest NIB models did not improve the performance with the DUD-E set (Models I and IV in
Table 4 and
Figure 4). Accordingly, the R-NiB produced higher AUC values than the PLANTS docking with both the DUD and DUD-E sets using the alternative models (
Table 4). In addition, the early enrichment improved over the docking with both test sets. The EFd 1% could be improved even further in comparison to the original docking if the scoring from both ShaEP and PLANTS was re-weighted (
Figure 7A) for optimal performance in the consensus scoring (
Table 4;
Figure 7C).
The fact that R-NiB worked better than the NIB screening (
Table 3 versus
Table 4) is not surprising, because, during regular docking, the ligand conformers are sampled and optimized flexibly against the protein cavity. This structure-based sampling intrinsically affects the conformer composition and their placement against the protein’s cavity for the better (
Figure 6). Paradoxically, a clear downside of the R-NiB (
Figure 2) in comparison to the NIB screening (
Figure 1) is the inflated computational cost of the flexible docking sampling prior to the cavity-based rescoring. The rescoring itself is ultrafast (
Figure 3), as no geometry optimization between the template model or the ligand conformers is needed [
16]. The user simply outputs several poses for each docked ligand (e.g.,
n = 10) to have enough explicit solutions to re-rank and improve the docking performance utilizing the cavity’s shape/charge information. The NIB screening (
Figure 1) is faster than the regular docking precisely because the ligand conformers used in the rigid docking have been prepared in advance (
Table 1), and these same ligand sets can be used without bias for all targets [
1,
3,
8]. In contrast, molecular docking, which treats the ligands or even the protein itself flexibly, produces more tailored and target-specific binding modes.
Flexible docking algorithms have been shown to reproduce experimentally-derived ligand binding poses (see, e.g., [
33]), although they might not recognize them in all cases (
Figure 6). Despite the relatively high expense of these computations, one can realistically expect that even the most costly docking simulations and post-processing schemes will become plausible if computing performance continues to improve in the post-silicon era [
34]. Thus, the biggest hurdle of structure-based drug discovery (besides acquiring the relevant protein 3D structures) is not necessarily the ligand pose sampling or the computational efficiency, but the inability of the default docking scoring functions to recognize the high-affinity binding poses and the potent compounds from the vast screening databases [
16,
33]. Consequently, the development of reliable scoring functions and easy-to-use rescoring methodologies such as the R-NiB (
Figure 2) [
16] is needed to supplement the existing docking software.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}