Inhibitory Effects of Quercetin and Its Human and Microbial Metabolites on Xanthine Oxidase Enzyme


 ,
,  , ,
, , 
Abstract
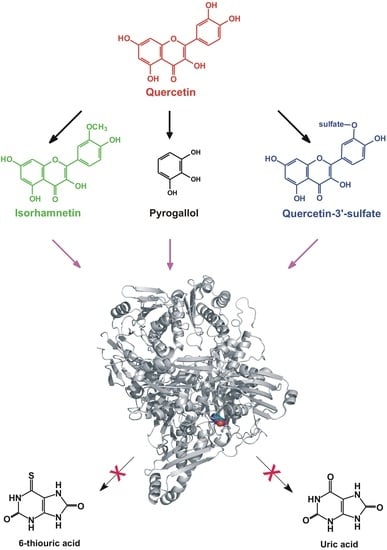
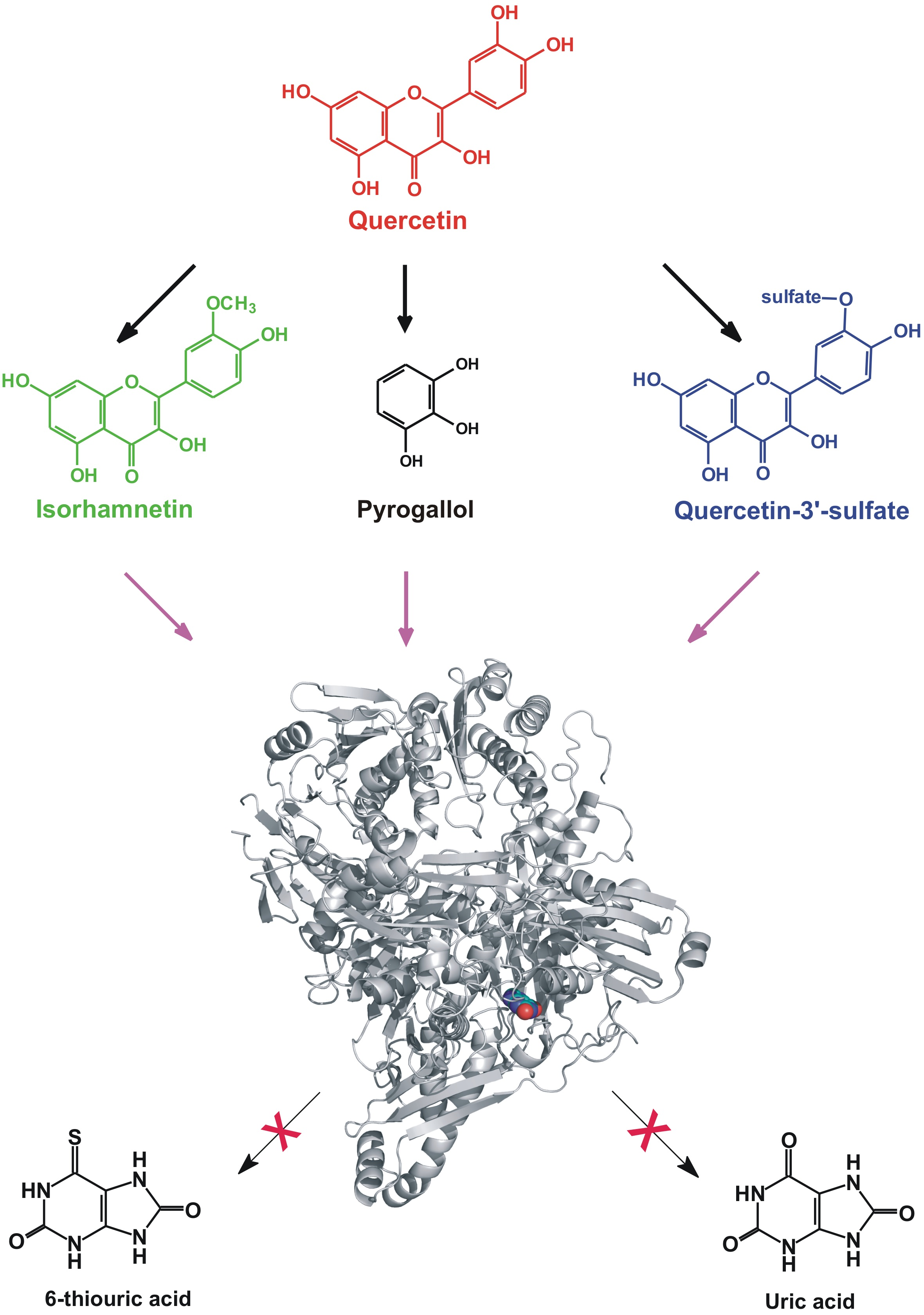
1. Introduction
2. Results
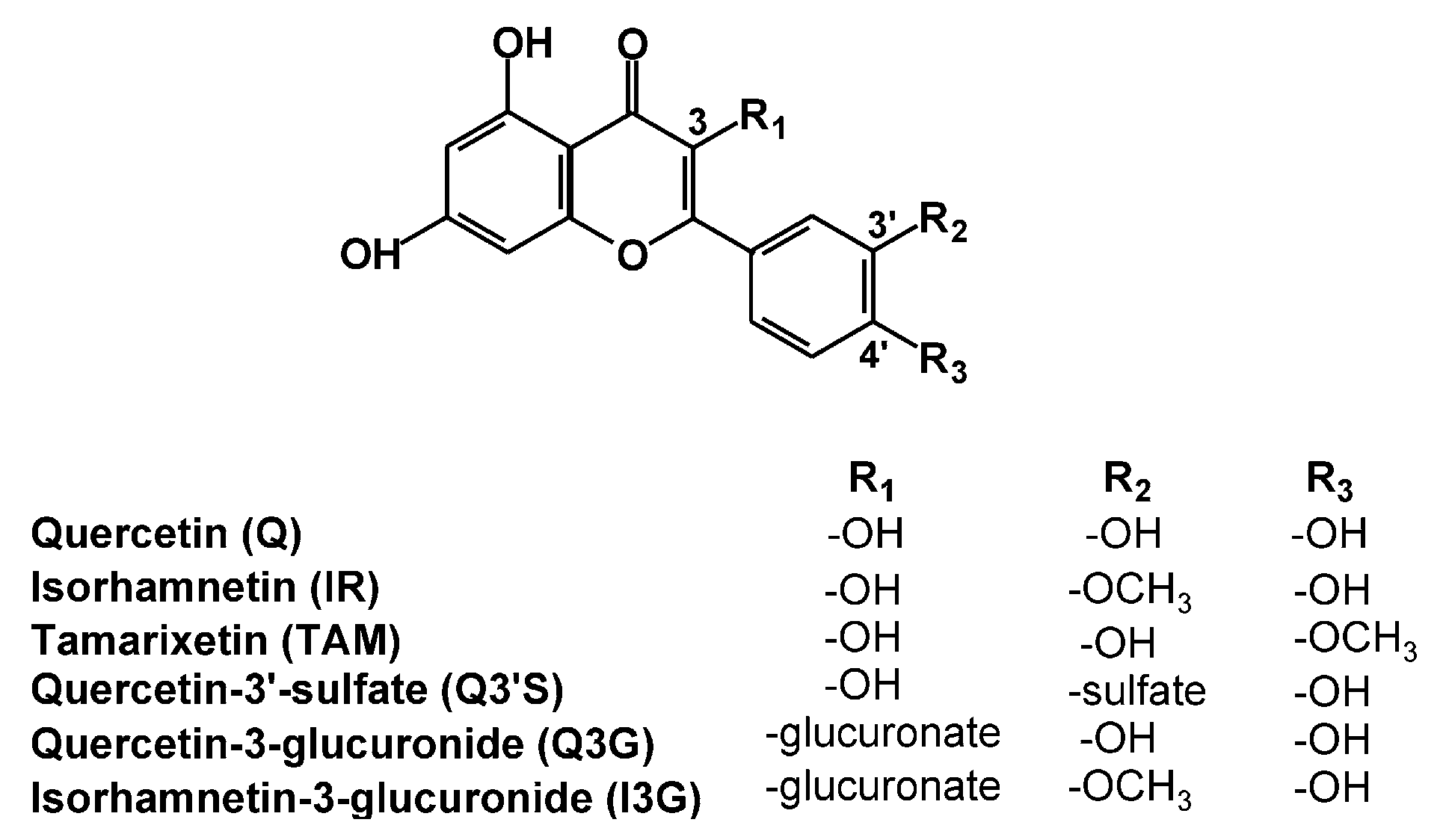
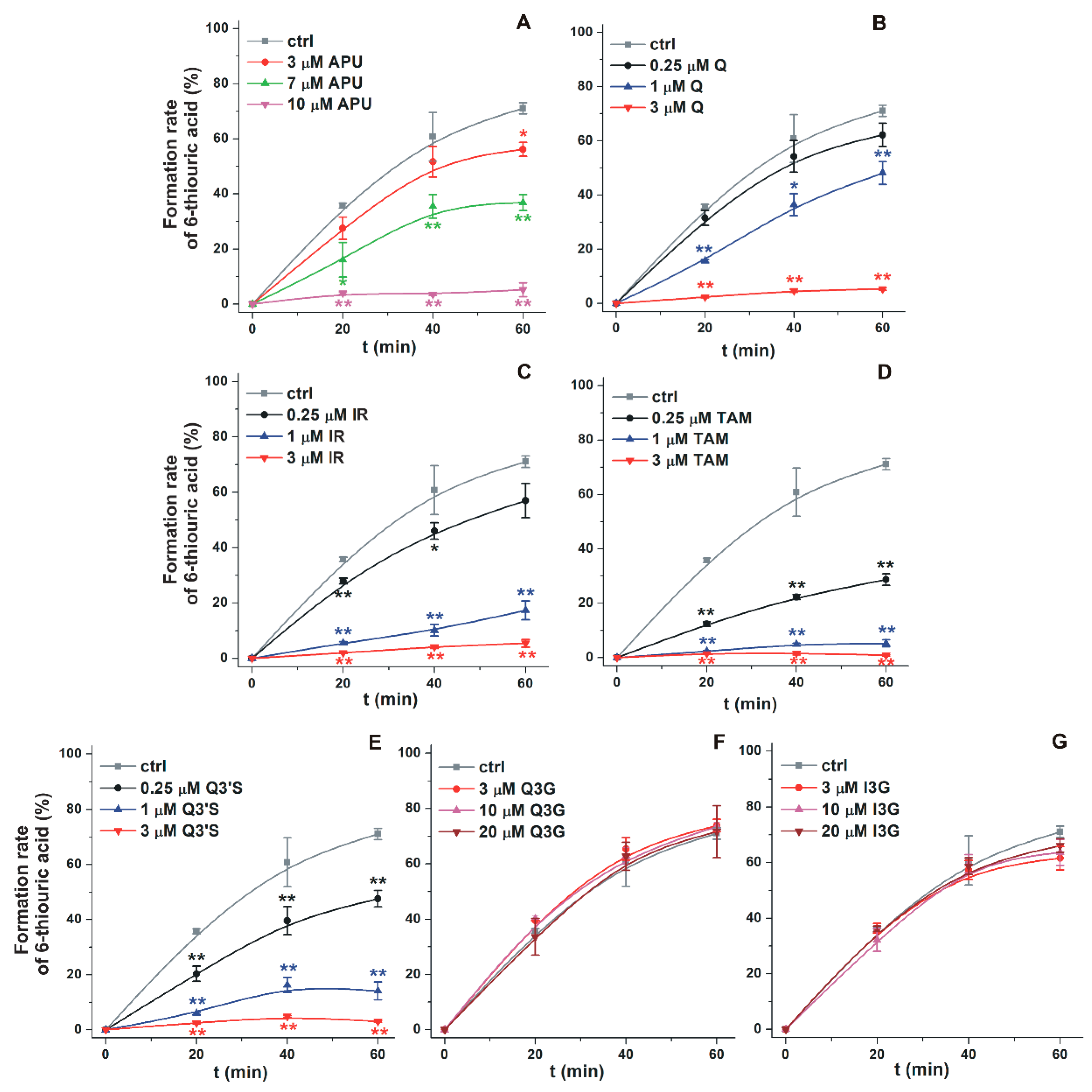
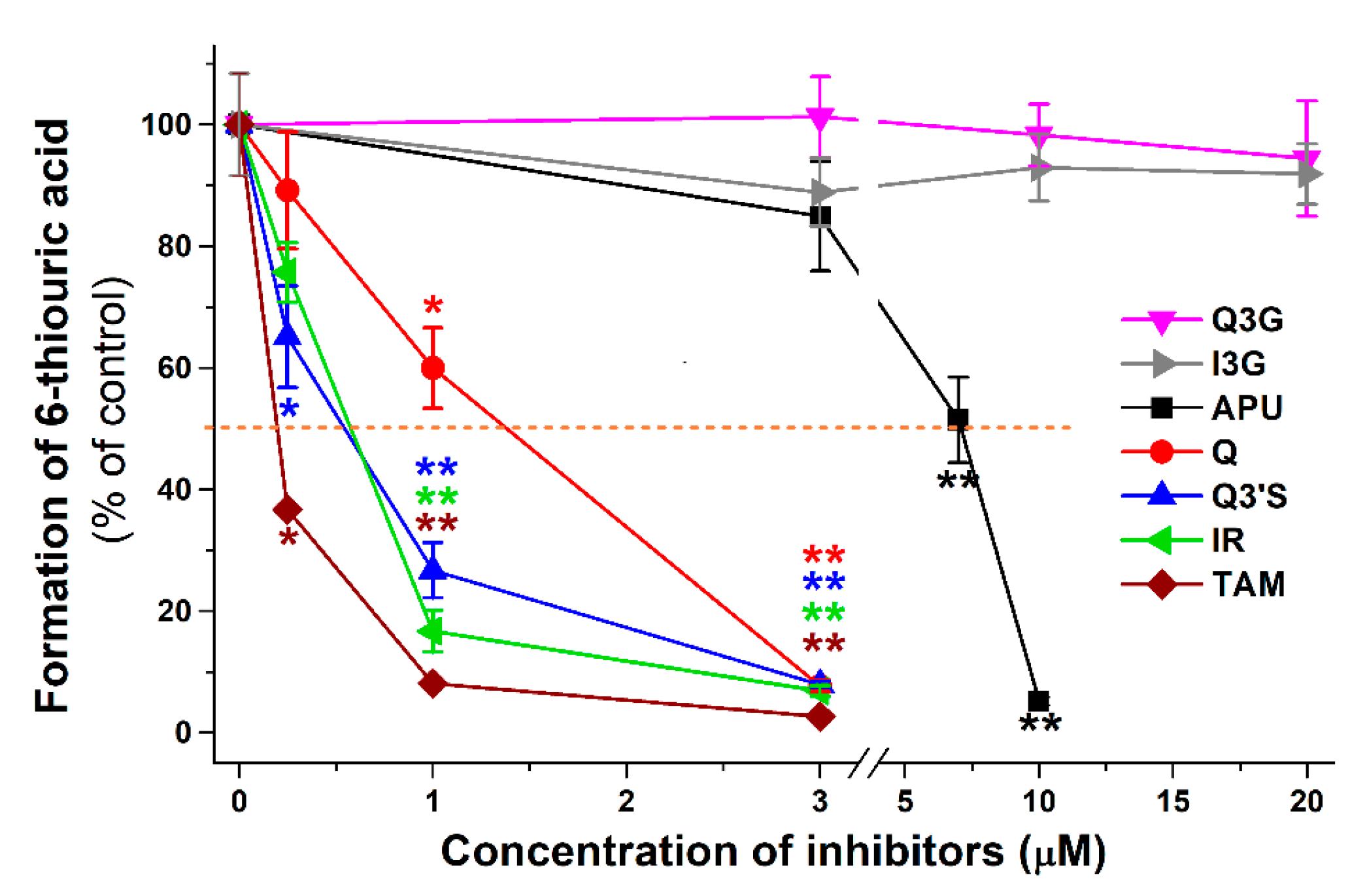
2.1. Inhibitory Effects of Q and Its Human Metabolites on XO-Catalyzed 6-MP Oxidation
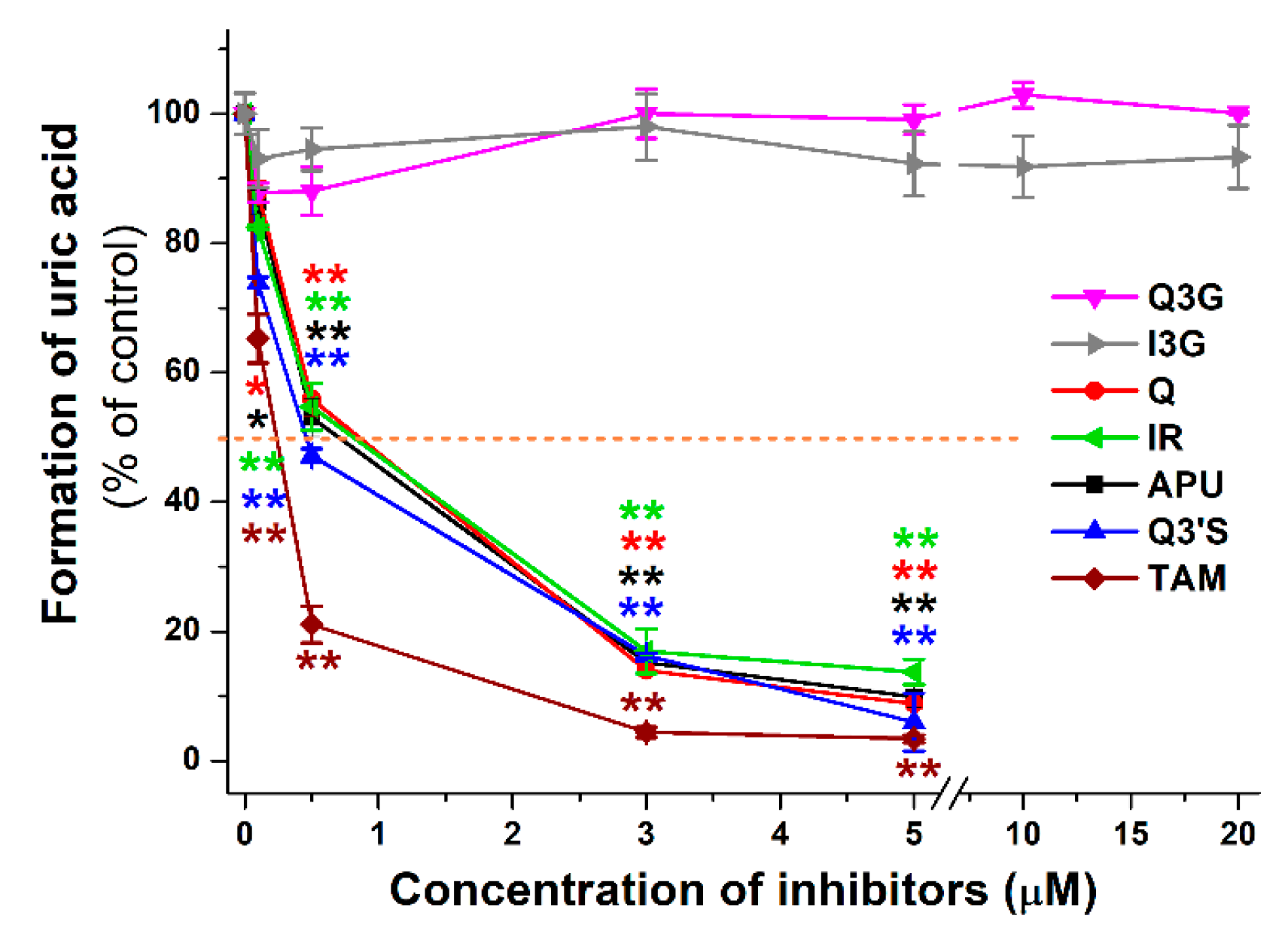
2.2. Inhibitory Effects of Q and Its Human Metabolites on XO-Catalyzed Xanthine Oxidation
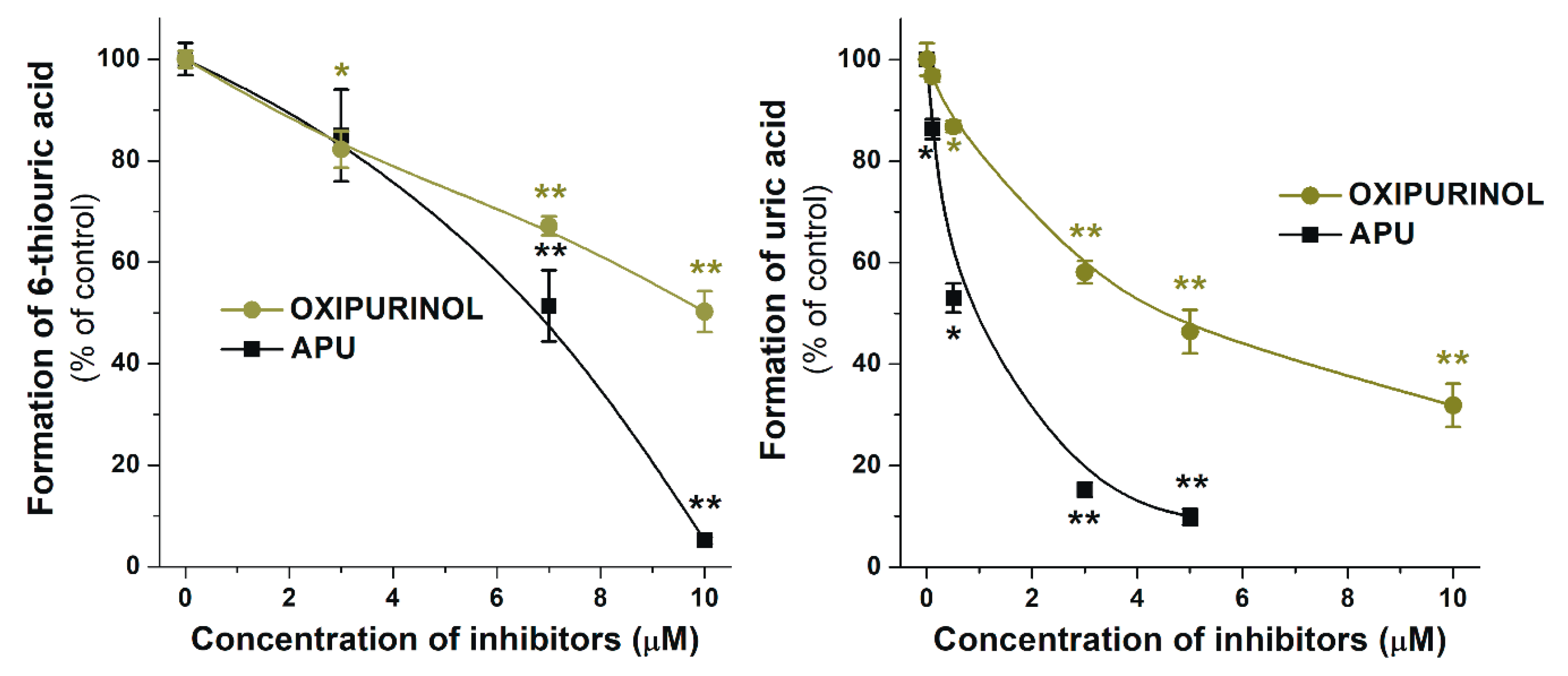
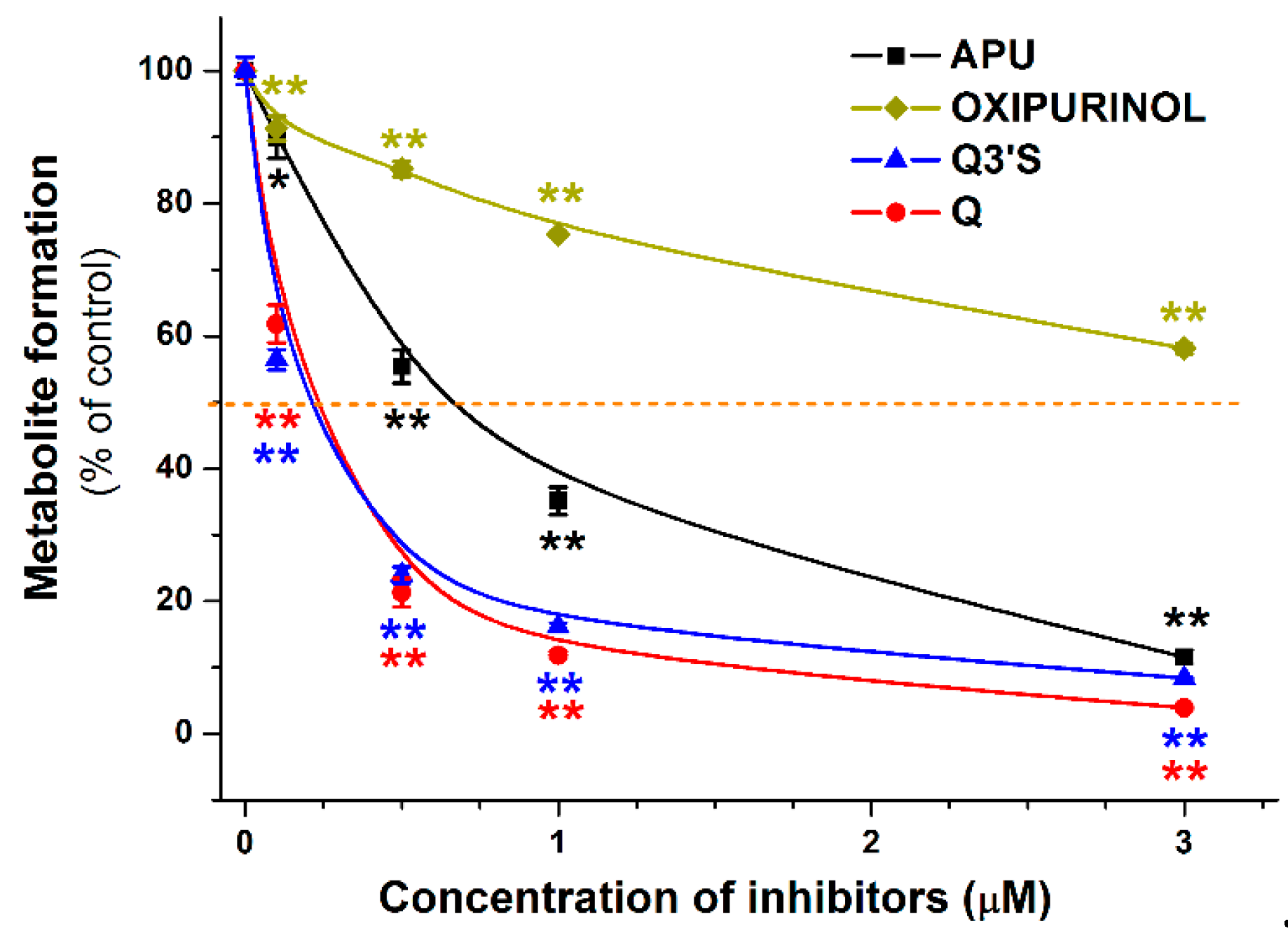
2.3. Inhibitory Effects of Q, Q3′S, APU, and Oxipurinol on XO-Catalyzed Hypoxanthine Oxidation
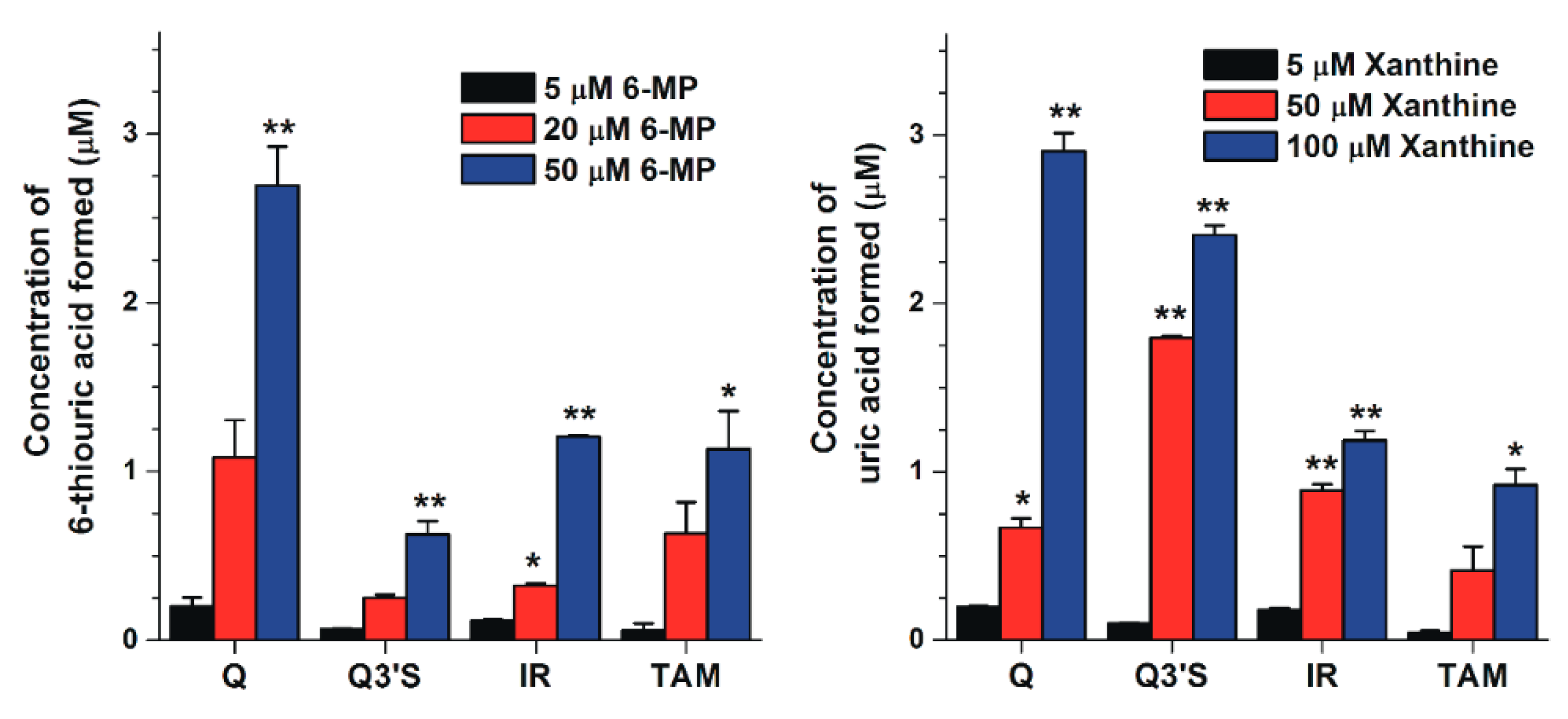
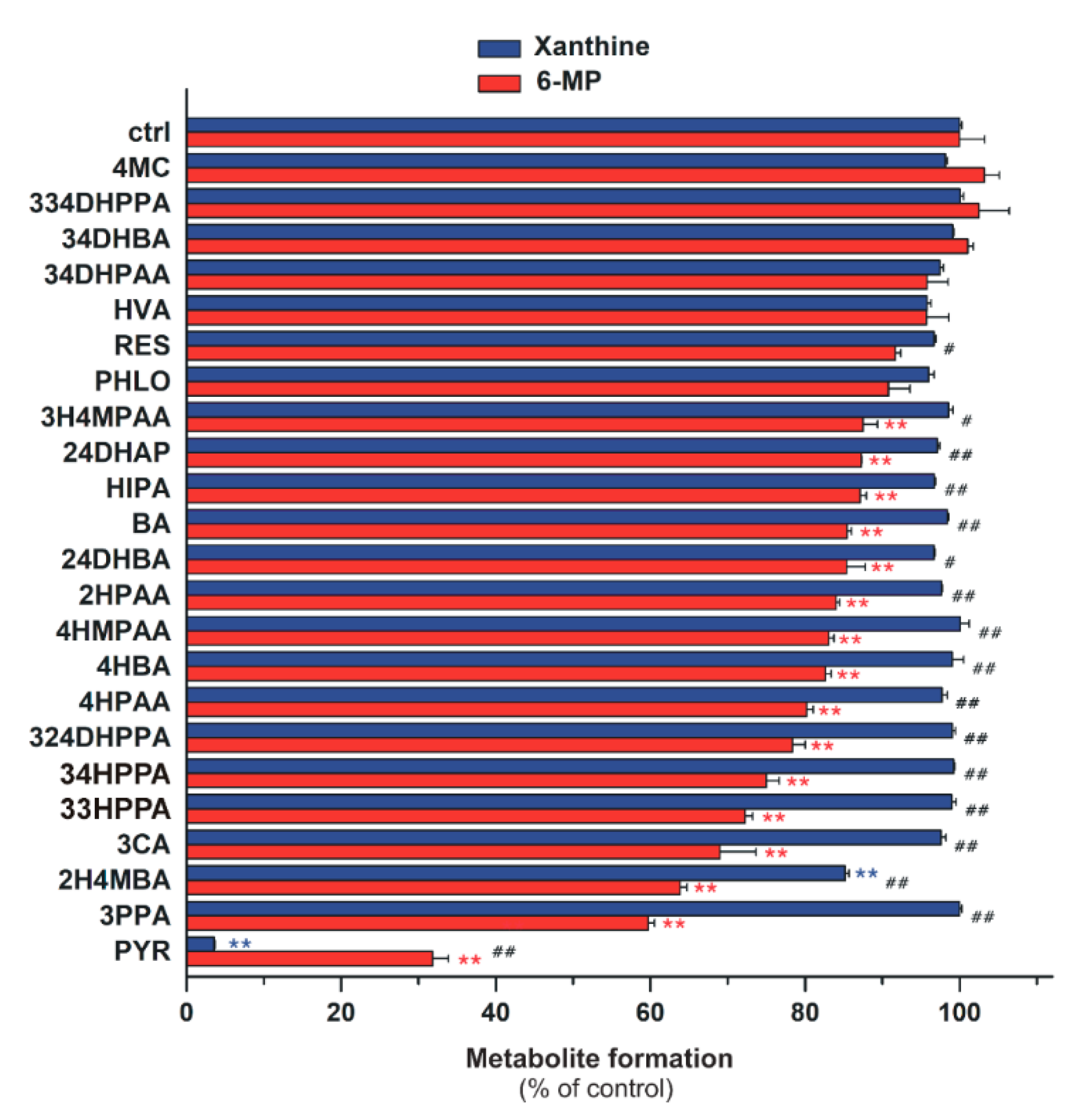
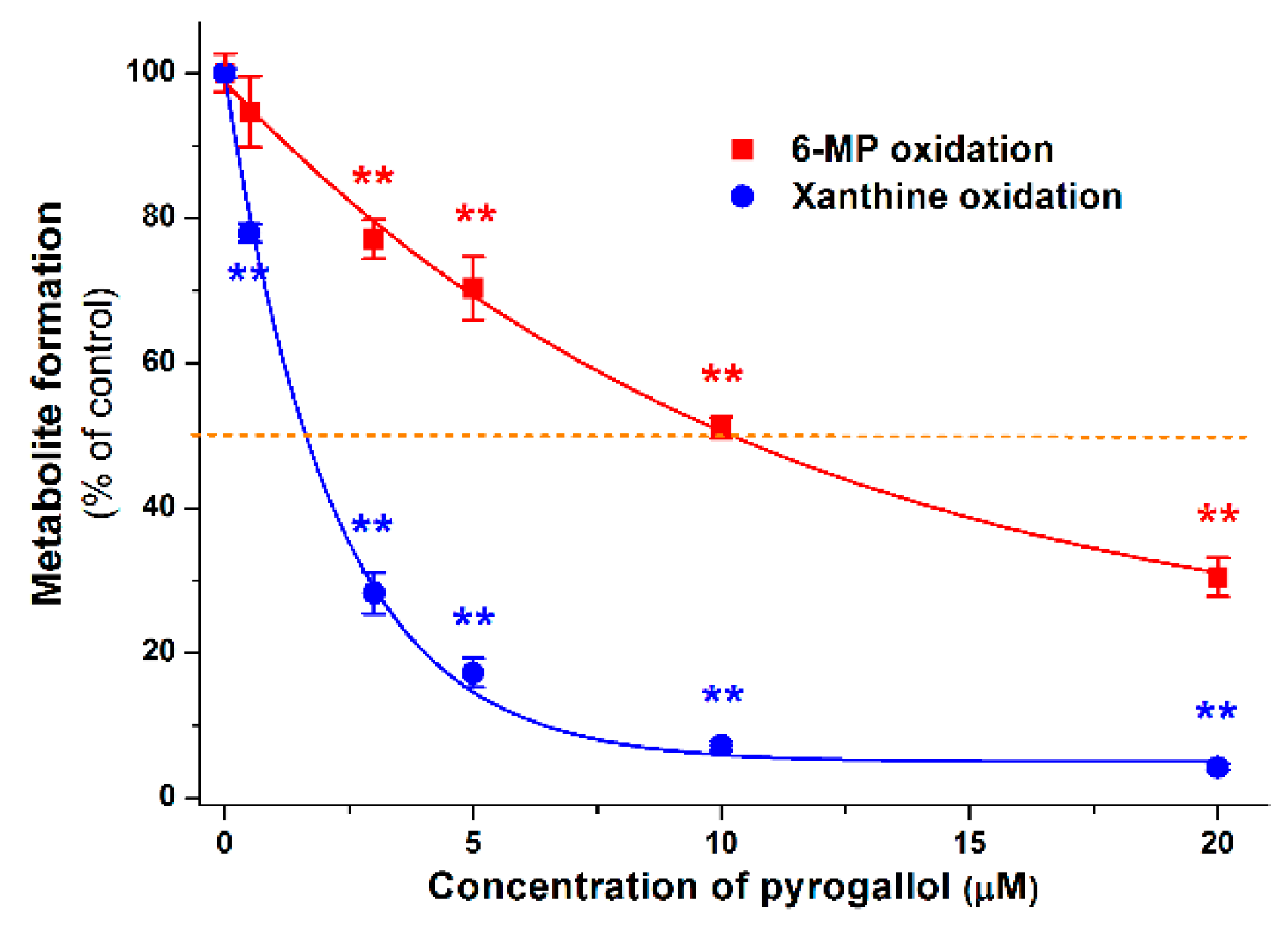
2.4. Inhibitory Effects of the Microbial Metabolites on XO-Catalyzed 6-MP and Xanthine Oxidation
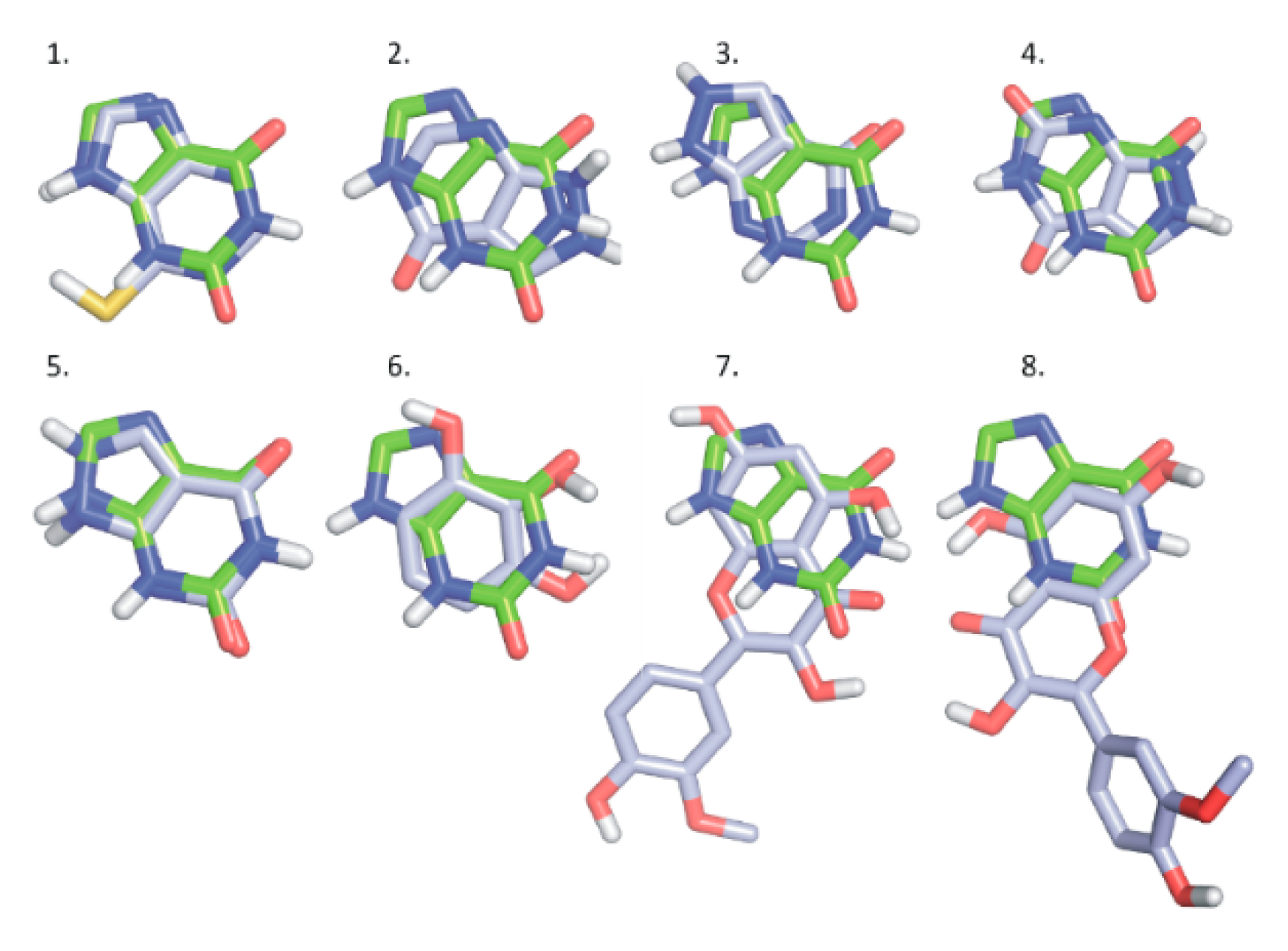
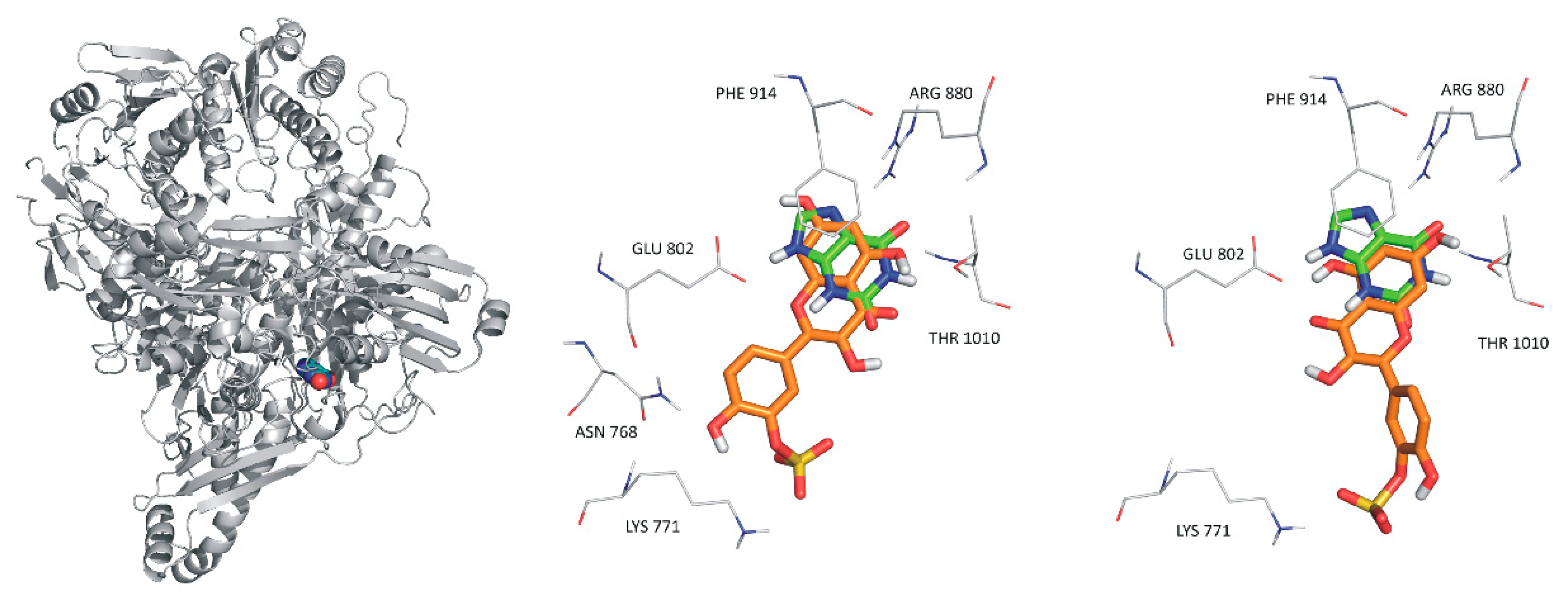
2.5. Modeling Studies
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. XO Assay with 6-MP Substrate
4.3. XO Assay with Xanthine Substrate
4.4. XO Assay with Hypoxanthine Substrate
4.5. HPLC Analyses
4.6. Modeling Studies
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 24DHAP | 2,4-Dihydroxyacetophenon |
| 24DHBA | 2,4-Dihydroxybenzoic acid |
| 2H4MBA | 4-Methoxysalicylic acid |
| 2HPAA | 2-Hydroxyphenylacetic acid |
| 324DHPPA | 3-(2,4-Dihydroxyphenyl)propionic acid |
| 334DHPPA | 3-(3,4-Dihydroxyphenyl)propionic acid |
| 33HPPA | 3-(3-Hydroxyphenyl)propionic acid |
| 34DHBA | 3,4-Dihydroxybenzoic acid |
| 34DHPAA | 3,4-Dihydroxyphenylacetic acid |
| 34HPPA | 3-(4-Hydroxyphenyl)propionic acid |
| 3CA | 3-Coumaric acid |
| 3H4MPAA | 3-Hydroxy-4-methoxyphenylacetic acid |
| 3PPA | 3-Phenylpropionic acid |
| 4HBA | 4-Hydroxybenzoic acid |
| 4HMPAA | 4-(Hydroxymethyl)phenylacetic acid |
| 4MC | 4-Methylcatechol |
| 6-MP | 6-Mercaptopurine |
| 6-TU | 6-Thiouric acid |
| 6-TX | 6-Thioxanthine |
| APU | Allopurinol |
| BA | Benzoic acid |
| HIPA | Hippuric acid |
| HVA | Homovanillic acid |
| I3G | Isorhamnetin-3-glucuronide |
| IR | Isorhamnetin |
| PHLO | Phloroglucinol |
| PYR | Pyrogallol |
| Q | Quercetin |
| Q3G | Quercetin-3-glucuronide |
| Q3′S | Quercetin-3′-sulfate |
| RES | Resorcinol |
| TAM | Tamarixetin |
| XO | Xanthine oxidase |
References
- Formica, J.V.; Regelson, W. Review of the biology of Quercetin and related bioflavonoids. Food Chem. Toxicol. 1995, 12, 1061–1080. [Google Scholar] [CrossRef]
- Hollman, P.C.; De Vries, J.H.; Van Leeuwen, S.D.; Mengellers, M.J.; Katan, M.B. Absorption of dietary quercetin glycosides and quercetin in healthy ileostomy volunteers. Am. J. Clin. Nutr. 1995, 62, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.S. Quercetin. Altern. Med. Rev. 2011, 16, 172–194. [Google Scholar] [PubMed]
- Manach, C.; Texier, O.; Regerat, F.; Agullo, G.; Demigne, C.; Remesy, C. Dietary quercetin is recovered in rat plasma as conjugated derivatives of isorhamnetin and quercetin. J. Nutr. Biochem. 1996, 7, 375–380. [Google Scholar] [CrossRef]
- Terao, J.; Murota, K.; Kawai, Y. Conjugated quercetin glucuronides as bioactive metabolites and precursors of aglycone in vivo. Food Funct. 2011, 2, 11–17. [Google Scholar] [CrossRef]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [PubMed]
- Mullen, W.; Edwards, C.A.; Crozier, A. Absorption, excretion and metabolite profiling of methyl-, glucuronyl-, glucosyl- and sulpho-conjugates of quercetin in human plasma and urine after ingestion of onions. Br. J. Nutr. 2006, 96, 107–116. [Google Scholar] [CrossRef]
- Conquer, J.A.; Maiani, G.; Azzini, E.; Raguzzini, A.; Holub, B.J. Supplementation with quercetin markedly increases plasma quercetin concentration without effect on selected risk factors for heart disease in healthy subjects. J. Nutr. 1998, 128, 593–597. [Google Scholar] [CrossRef]
- Rechner, A.R.; Kuhnle, G.; Bremner, P.; Hubbard, G.P.; Moore, K.P.; Rice-Evans, C.A. The metabolic fate of dietary polyphenols in humans. Free Rad. Biol. Med. 2002, 33, 220–235. [Google Scholar] [CrossRef]
- Aura, A.M. Microbial metabolism of dietary phenolic compounds in the colon. Phytochem. Rev. 2008, 3, 407–429. [Google Scholar] [CrossRef]
- Serra, A.; Macia, A.; Romero, M.P.; Reguant, J.; Ortega, N.; Motilva, M.J. Metabolic pathways of the colonic metabolism of flavonoids (flavonols, flavones and flavanones) and phenolic acids. Food Chem. 2012, 130, 383–393. [Google Scholar] [CrossRef]
- Day, R.O.; Graham, G.G.; Hicks, M.; McLachlan, A.J.; Stocker, S.L.; Williams, K.M. Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin. Pharmacokinet. 2007, 46, 623–644. [Google Scholar] [CrossRef] [PubMed]
- Leong, R.W.; Gearry, R.B.; Sparrow, M.P. Thiopurine hepatotoxicity in inflammatory bowel disease: The role for adding allopurinol. Expert Opin. Drug Saf. 2008, 7, 607–616. [Google Scholar] [CrossRef]
- Galbusera, C.; Orth, P.; Fedida, D.; Spector, T. Superoxide radical production by allopurinol and xanthine oxidase. Biochem. Pharmacol. 2006, 71, 1747–1752. [Google Scholar] [CrossRef]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef]
- McLeod, H.L. Clinically relevant drug-drug interactions in oncology. Br. J. Clin. Pharmacol. 1998, 45, 539–544. [Google Scholar] [CrossRef]
- Lin, C.M.; Chen, C.S.; Chen, C.T.; Liang, Y.C.; Lin, J.K. Molecular modeling of flavonoids that inhibits xanthine oxidase. Biochem. Biophys. Res. Commun. 2002, 294, 162–172. [Google Scholar] [CrossRef]
- Van Hoorn, D.E.C.; Nijveldt, R.J.; Van Leeuwen, P.A.M.; Hofman, Z.; M’Rabet, L.; De Bont, D.B.A.; Van Norren, K. Accurate prediction of xanthine oxidase inhibition based on the structure of flavonoids. Eur. J. Pharmacol. 2002, 451, 111–118. [Google Scholar] [CrossRef]
- Mladenka, P.; Zatloukalová, L.; Filipský, T.; Hrdina, R. Cardiovascular effects of flavonoids are not caused only by direct antioxidant activity. Free Radic. Biol. Med. 2010, 49, 963–975. [Google Scholar] [CrossRef]
- Iio, M.; Moriyama, A.; Matsumoto, Y.; Takaki, N.; Fukumoto, M. Inhibition of Xanthine Oxidase by Flavonoids. Agric. Biol. Chem. 1985, 49, 2173–2176. [Google Scholar] [CrossRef]
- Nagao, A.; Seki, M.; Kobayashi, H. Inhibition of xanthine oxidase by flavonoids. Biosci. Biotechnol. Biochem. 1999, 63, 1787–1790. [Google Scholar] [CrossRef]
- Miron, A.; Aprotosoaie, A.C.; Trifan, A.; Xiao, J. Flavonoids as modulators of metabolic enzymes and drug transporters. Ann. N. Y. Acad. Sci. 2017, 1398, 152–167. [Google Scholar] [CrossRef]
- Poór, M.; Boda, G.; Needs, P.W.; Kroon, P.A.; Lemli, B.; Bencsik, T. Interaction of quercetin and its metabolites with warfarin: Displacement of warfarin from serum albumin and inhibition of CYP2C9 enzyme. Biomed. Pharmacother. 2017, 88, 574–581. [Google Scholar] [CrossRef]
- Cao, H.; Pauff, J.M.; Hille, R. X-ray crystal structure of a xanthine oxidase complex with the flavonoid inhibitor quercetin. J. Nat. Prod. 2014, 77, 1693–1699. [Google Scholar] [CrossRef]
- Cao, H.; Pauff, J.M.; Hille, R. Substrate orientation and catalytic specificity in the action of xanthine oxidase: The sequential hydroxylation of hypoxanthine to uric acid. J. Biol. Chem. 2010, 285, 28044–28053. [Google Scholar] [CrossRef]
- Pauff, J.M.; Cao, H.; Hille, R. Substrate Orientation and Catalysis at the Molybdenum Site in Xanthine Oxidase: CRYSTAL STRUCTURES IN COMPLEX WITH XANTHINE AND LUMAZINE. J. Biol. Chem. 2009, 284, 8760–8770. [Google Scholar] [CrossRef]
- Okamoto, K.; Eger, B.T.; Nishino, T.; Pai, E.F.; Nishino, T. Mechanism of inhibition of xanthine oxidoreductase by allopurinol: Crystal structure of reduced bovine milk xanthine oxidoreductase bound with oxipurinol. Nucleosides Nucleotides Nucleic Acids 2008, 27, 888–893. [Google Scholar] [CrossRef]
- Cos, P.; Ying, L.; Calomne, M.; Hu, J.P.; Cimanga, K.; Van Poel, B.; Pieters, L.; Vlietinck, A.J.; Berghe, D.V. Structure-activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J. Nat. Prod. 1998, 61, 71–76. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, R.; Zhang, G.; Gong, D. Mechanistic insights into the inhibition of quercetin on xanthine oxidase. Int. J. Biol. Macromol. 2018, 112, 405–412. [Google Scholar] [CrossRef]
- Day, A.J.; Bao, Y.; Morgan, M.R.; Williamson, G. Conjugation position of quercetin glucuronides and effect on biological activity. Free Radic. Biol. Med. 2000, 29, 1234–1243. [Google Scholar] [CrossRef]
- Day, A.J.; Mellon, F.; Barron, D.; Sarrazin, G.; Morgan, M.R.; Williamson, G. Human metabolism of dietary flavonoids: Identification of plasma metabolites of quercetin. Free Radic. Res. 2001, 35, 941–952. [Google Scholar] [CrossRef]
- Elion, G.B. Enzymatic and metabolic studies with allopurinol. Ann. Rheum. Dis. 1966, 25, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Spector, T. Inhibition of urate production by allopurinol. Biochem. Pharmacol. 1977, 26, 355–358. [Google Scholar] [CrossRef]
- Zhu, J.X.; Wang, Y.; Kong, L.D.; Yang, C.; Zhang, X. Effects of Biota orientalis extract and its flavonoid constituents, quercetin and rutin on serum uric acid levels in oxonate-induced mice and xanthine dehydrogenase and xanthine oxidase activities in mouse liver. J. Ethnopharmacol. 2004, 93, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, S.; Zhu, M.; Chen, J.; Zhu, X. Effects of Genistein, Apigenin, Quercetin, Rutin and Astilbin on serum uric acid levels and xanthine oxidase activities in normal and hyperuricemic mice. Food Chem. Toxicol. 2011, 49, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Abbey, E.L.; Rankin, J.W. Effect of quercetin supplementation on repeated-sprint performance, xanthine oxidase activity, and inflammation. Int. J. Sport Nutr. Exerc. Metab. 2011, 21, 91–96. [Google Scholar] [CrossRef]
- Boots, A.W.; Drent, M.; De Boer, V.C.; Bast, A.; Haenen, G.R. Quercetin reduces markers of oxidative stress and inflammation in sarcoidosis. Clin. Nutr. 2011, 30, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Williamson, G. Quercetin lowers plasma uric acid in pre-hyperuricaemic males: A randomised, double-blinded, placebo-controlled, cross-over trial. Br. J. Nutr. 2016, 115, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.A.; Henson, D.A.; Nieman, D.C.; Austin, M.D. A 12-week supplementation with quercetin does not affect natural killer cell activity, granulocyte oxidative burst activity or granulocyte phagocytosis in female human subjects. Br. J. Nutr. 2010, 104, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Turnheim, K.; Krivanek, P.; Oberbauer, R. Pharmacokinetics and pharmacodynamics of allopurinol in elderly and young subjects. Br. J. Clin. Pharmacol. 1999, 48, 501–509. [Google Scholar] [CrossRef]
- Cialdella-Kam, L.; Nieman, D.C.; Sha, W.; Meaney, M.P.; Knab, A.M.; Shanely, R.A. Dose-response to 3 months of quercetin-containing supplements on metabolite and quercetin conjugate profile in adults. Br. J. Nutr. 2013, 109, 1923–1933. [Google Scholar] [CrossRef] [PubMed]
- De Santi, C.; Pietrabissa, A.; Mosca, F.; Pacifici, G.M. Methylation of quercetin and fisetin, flavonoids widely distributed in edible vegetables, fruits and wine, by human liver. Int. J. Clin. Pharmacol. Ther. 2002, 40, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Vida, R.G.; Fittler, A.; Somogyi-Végh, A.; Poór, M. Dietary quercetin supplements: Assessment of online product informations and quantitation of quercetin in the products by high performance liquid chromatography. Phytother. Res. 2019. Accepted manuscript. [Google Scholar] [CrossRef]
- Pimpão, R.C.; Ventura, M.R.; Ferreira, R.B.; Williamson, G.; Santos, C.N. Phenolic sulfates as new and highly abundant metabolites in human plasma after ingestion of a mixed berry fruit purée. Br. J. Nutr. 2015, 113, 454–463. [Google Scholar] [CrossRef]
- Needs, P.W.; Kroon, P.A. Convenient synthesis of metabolically important glucuronides and sulfates. Tetrahedron 2006, 62, 6862–6868. [Google Scholar] [CrossRef]
- Mei, D.A.; Gross, G.J.; Nithipatikom, K. Simultaneous determination of adenosine, inosine, hypoxanthine, xanthine, and uric acid in microdialysis samples using microbore column high-performance liquid chromatography with a diode array detector. Anal. Biochem. 1996, 238, 34–39. [Google Scholar] [CrossRef]
- Hawwa, A.F.; Millership, J.S.; Collier, P.S.; McElnay, J.C. Development and validation of an HPLC method for the rapid and simultaneous determination of 6-mercaptopurine and four of its metabolites in plasma and red blood cells. J. Pharm. Biomed. Anal. 2009, 49, 401–409. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; Wang, J.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Stewart, J.J.P. MOPAC: A semiempirical molecular orbital program. Computer-Aided Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Com. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Compound | 6-MP Oxidation | Xanthine Oxidation | IC50 (6-MP)/IC50 (Xanthine) | ||||
|---|---|---|---|---|---|---|---|
| IC50 (μM) | IC50(rel) | α | IC50 (μM) | IC50(rel) | α | ||
| APU (positive ctrl) | 7.00 | 1.40 | 1.00 | 0.60 | 0.12 | 1.00 | 11.67 |
| Q | 1.40 | 0.28 | 0.20 | 0.80 | 0.16 | 1.33 | 1.75 |
| Q3′S | 0.50 | 0.10 | 0.07 | 0.40 | 0.08 | 0.67 | 1.25 |
| IR | 0.60 | 0.12 | 0.09 | 0.70 | 0.14 | 1.17 | 0.86 |
| TAM | 0.20 | 0.04 | 0.03 | 0.20 | 0.04 | 0.33 | 1.00 |
| Q3G | >20.0 | >4.0 | - | >20.0 | >4.0 | - | - |
| I3G | >20.0 | >4.0 | - | >20.0 | >4.0 | - | - |
| PYR | 10.10 | 2.02 | 1.44 | 1.80 | 0.36 | 3.00 | 5.61 |
| Test Compound | IC50 (μM) | IC50(rel) | α |
|---|---|---|---|
| APU (positive ctrl) | 0.66 | 0.13 | 1.00 |
| Q | 0.24 | 0.05 | 0.36 |
| Q3′S | 0.21 | 0.04 | 0.32 |
| Oxipurinol | >3.00 | >0.60 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohos, V.; Pánovics, A.; Fliszár-Nyúl, E.; Schilli, G.; Hetényi, C.; Mladěnka, P.; Needs, P.W.; Kroon, P.A.; Pethő, G.; Poór, M. Inhibitory Effects of Quercetin and Its Human and Microbial Metabolites on Xanthine Oxidase Enzyme. Int. J. Mol. Sci. 2019, 20, 2681. https://doi.org/10.3390/ijms20112681
Mohos V, Pánovics A, Fliszár-Nyúl E, Schilli G, Hetényi C, Mladěnka P, Needs PW, Kroon PA, Pethő G, Poór M. Inhibitory Effects of Quercetin and Its Human and Microbial Metabolites on Xanthine Oxidase Enzyme. International Journal of Molecular Sciences. 2019; 20(11):2681. https://doi.org/10.3390/ijms20112681
Chicago/Turabian StyleMohos, Violetta, Attila Pánovics, Eszter Fliszár-Nyúl, Gabriella Schilli, Csaba Hetényi, Přemysl Mladěnka, Paul W. Needs, Paul A. Kroon, Gábor Pethő, and Miklós Poór. 2019. "Inhibitory Effects of Quercetin and Its Human and Microbial Metabolites on Xanthine Oxidase Enzyme" International Journal of Molecular Sciences 20, no. 11: 2681. https://doi.org/10.3390/ijms20112681
APA StyleMohos, V., Pánovics, A., Fliszár-Nyúl, E., Schilli, G., Hetényi, C., Mladěnka, P., Needs, P. W., Kroon, P. A., Pethő, G., & Poór, M. (2019). Inhibitory Effects of Quercetin and Its Human and Microbial Metabolites on Xanthine Oxidase Enzyme. International Journal of Molecular Sciences, 20(11), 2681. https://doi.org/10.3390/ijms20112681