Leptin-induced Trafficking of KATP Channels: A Mechanism to Regulate Pancreatic β-cell Excitability and Insulin Secretion


{kind=link}
Abstract
1. Introduction
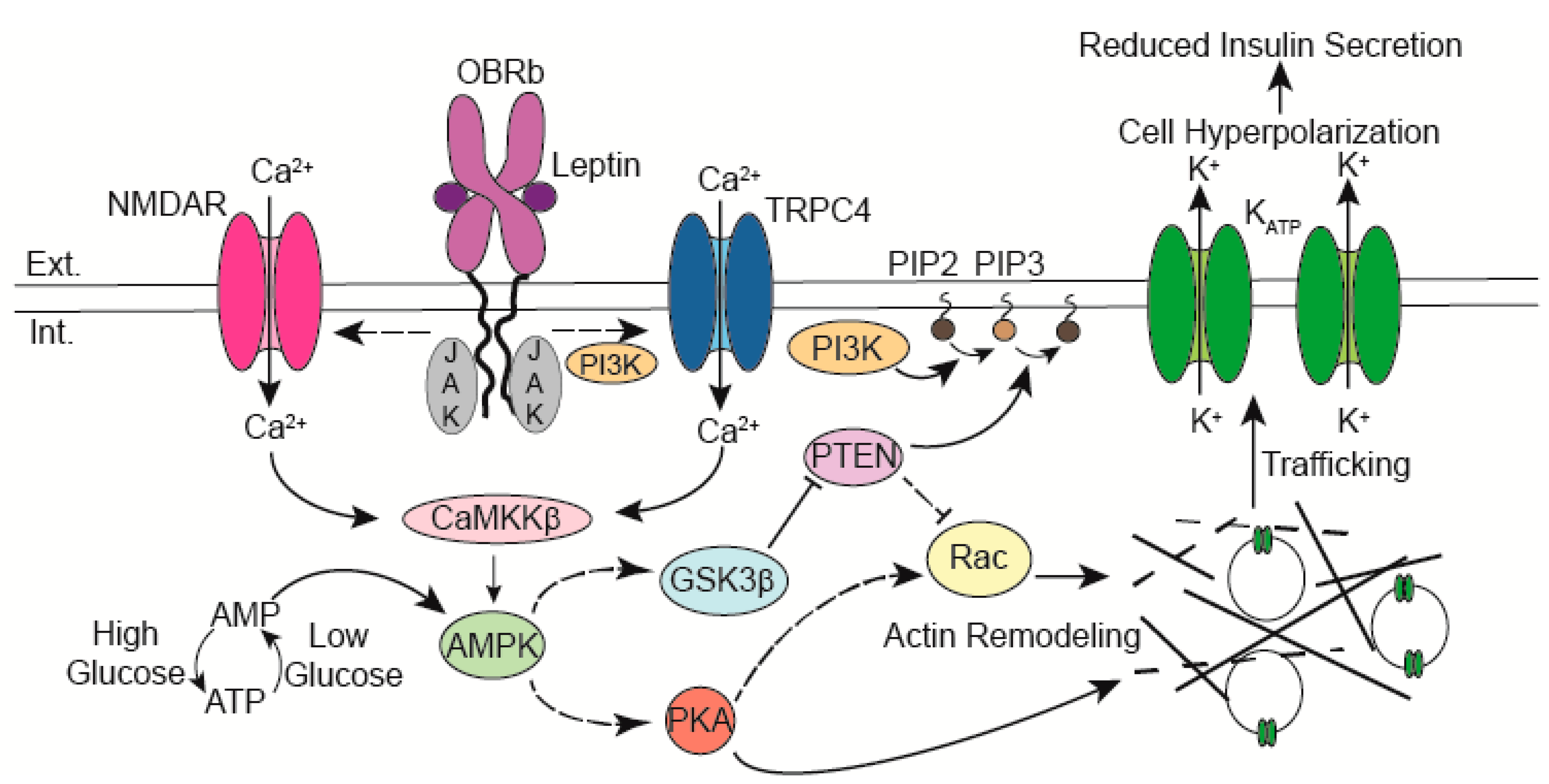
2. Leptin Increases KATP Channel Conductance in β-Cells
3. Leptin Increases KATP Channel Surface Density
4. KATP Channel Trafficking Signaling Molecules
4.1. AMPK
4.2. CaMKKβ
4.3. PI3 Kinase and TRPC4 Channels
4.4. NMDA Receptors
4.5. Actin Remodeling Molecules
4.5.1. PKA
4.5.2. PTEN
5. The Role of Glucose and its Interplay with Leptin in KATP Channel Trafficking Regulation
6. Future Questions
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dubuc, P.U. The development of obesity, hyperinsulinemia, and hyperglycemia in ob/ob mice. Metabolism 1976, 25, 1567–1574. [Google Scholar] [CrossRef]
- Dubuc, P.U.; Willis, P.L. Postweaning development of diabetes in ob/ob mice. Metabolism 1979, 28, 633–640. [Google Scholar] [CrossRef]
- Pelleymounter, M.A.; Cullen, M.J.; Baker, M.B.; Hecht, R.; Winters, D.; Boone, T.; Collins, F. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 1995, 269, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Kieffer, T.J.; Heller, R.S.; Habener, J.F. Leptin Receptors Expressed on Pancreatic β-Cells. Biochem. Biophys. Res. Commun. 1996, 224, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, T.J.; Heller, R.S.; Leech, C.A.; Holz, G.G.; Habener, J.F. Leptin Suppression of Insulin Secretion by the Activation of ATP-Sensitive K+ Channels in Pancreatic β-Cells. Diabetes 1997, 46, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Liu, Y.L.; Cawthorne, M.A.; Morton, N.M.; Davenport, M. Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes 1997, 46, 313–316. [Google Scholar] [CrossRef]
- Fehmann, H.C.; Peiser, C.; Bode, H.P.; Stamm, M.; Staats, P.; Hedetoft, C.; Lang, R.E.; Göke, B. Leptin: A potent inhibitor of insulin secretion. Peptides 1997, 18, 1267–1273. [Google Scholar] [CrossRef]
- Ookuma, K.; Ookuma, M.; York, D.A. Effects of Leptin on Insulin Secretion from Isolated Rat Pancreatic Islets. Diabetes 1998, 47, 219–223. [Google Scholar] [CrossRef]
- Kulkarni, R.N.; Wang, Z.L.; Wang, R.M.; Hurley, J.D.; Smith, D.M.; Ghatei, M.A.; Withers, D.J.; Gardiner, J.V.; Bailey, C.J.; Bloom, S.R. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J. Clin. Investig. 1997, 100, 2729–2736. [Google Scholar] [CrossRef]
- Seufert, J.; Kieffer, T.J.; Leech, C.A.; Holz, G.G.; Moritz, W.; Ricordi, C.; Habener, J.F. Leptin suppression of insulin secretion and gene expression in human pancreatic islets: Implications for the development of adipogenic diabetes mellitus. J. Clin. Endocrinol. Metab. 1999, 84, 670–676. [Google Scholar] [CrossRef]
- Kieffer, T.J.; Habener, J.F. The adipoinsular axis: Effects of leptin on pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E1–E14. [Google Scholar] [CrossRef]
- Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Rorsman, P. KATP channels and islet hormone secretion: New insights and controversies. Nat. Rev. Endocrinol. 2013, 9, 660–669. [Google Scholar] [CrossRef]
- Martin, G.M.; Chen, P.C.; Devaraneni, P.; Shyng, S.L. Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Front. Physiol. 2013, 4, 1–16. [Google Scholar] [CrossRef]
- Zhao, A.Z.; Bornfeldt, K.E.; Beavo, J.A. Leptin inhibits insulin secretion by activation of phosphodiesterase 3B. J. Clin. Investig. 1997, 102, 869–873. [Google Scholar] [CrossRef]
- Morioka, T.; Dishinger, J.F.; Reid, K.R.; Liew, C.W.; Zhang, T.; Inaba, M.; Kennedy, R.T.; Kulkarni, R.N. Enhanced GLP-1- and sulfonylurea-induced insulin secretion in islets lacking leptin signaling. Mol. Endocrinol. 2012, 26, 967–976. [Google Scholar] [CrossRef][Green Version]
- Harvey, J.; McKenna, F.; Herson, P.S.; Spanswick, D.; Ashford, M.L.J. Leptin activates ATP-sensitive potassium channels in the rat insulin-secreting cell line, CRI-G1. J. Physiol. 1997, 504, 527–535. [Google Scholar] [CrossRef]
- Hiraoka, J.; Hosoda, K.; Ogawa, Y.; Ikeda, K.; Nara, Y.; Masuzaki, H.; Takaya, K.; Nakagawa, K.; Mashimo, T.; Sawamura, M.; et al. Augmentation ofobese(ob) Gene Expression and Leptin Secretion in Obese Spontaneously Hypertensive Rats (Obese SHR or Koletsky Rats). Biochem. Biophys. Res. Commun. 1997, 231, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, TL.; et al. Serum Immunoreactive-Leptin Concentrations in Normal-Weight and Obese Humans. Endocrinologist 1996, 6, 349. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.; McKay, N.G.; Walker, K.S.; van der Kaay, J.; Downes, C.P.; Ashford, M.L. Essential Role of Phosphoinositide 3-Kinase in Leptin-induced K ATP Channel Activation in the Rat CRI-G1 Insulinoma Cell Line. J. Biol. Chem. 2000, 275, 4660–4669. [Google Scholar] [CrossRef]
- Ning, K.; Miller, L.C.; Laidlaw, H.A.; Burgess, L.A.; Perera, N.M.; Downes, C.P.; Leslie, N.R.; Ashford, M.L. A novel leptin signalling pathway via PTEN inhibition in hypothalamic cell lines and pancreatic beta-cells. EMBO J. 2006, 25, 2377–2387. [Google Scholar] [CrossRef]
- Ning, K.; Miller, L.C.; Laidlaw, H.A.; Watterson, K.R.; Gallagher, J.; Sutherland, C.; Ashford, M.L.J. Leptin-dependent phosphorylation of PTEN mediates actin restructuring and activation of ATP-sensitive K+ channels. J. Biol. Chem. 2009, 284, 9331–9340. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Terzic, A.; Kurachi, Y. Actin microfilament disrupters enhance K(ATP) channel opening in patches from guinea-pig cardiomyocytes. J. Physiol. 1996, 492, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.; Hardy, S.C.; Irving, A.J.; Ashford, M.L. Leptin activation of ATP-sensitive K+ (KATP) channels in rat CRI-G1 insulinoma cells involves disruption of the actin cytoskeleton. J. Physiol. 2000, 527, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Ryu, S.-Y.; Yu, W.-J.; Han, Y.E.; Ji, Y.-S.; Oh, K.; Sohn, J.-W.; Lim, A.; Jeon, J.-P.; Lee, H. Leptin promotes KATP channel trafficking by AMPK signaling in pancreatic -cells. Proc. Natl. Acad. Sci. USA 2013, 110, 12673–12678. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-C.; Kryukova, Y.N.; Shyng, S.-L. Leptin Regulates K ATP Channel Trafficking in Pancreatic β-Cells by a Signaling Mechanism Involving AMP-activated Protein Kinase (AMPK) and cAMP-dependent Protein Kinase (PKA). J. Biol. Chem. 2013, 288, 34098–34109. [Google Scholar] [CrossRef] [PubMed]
- Hohmeier, H.E.; Mulder, H.; Chen, G.; Henkel-Rieger, R.; Prentki, M.; Newgard, C.B. Isolation of INS-1-derived cell lines with robust ATP- sensitive K+ channel-dependent and -independent glucose- stimulated insulin secretion. Diabetes 2000, 49, 424–430. [Google Scholar] [CrossRef]
- Wu, Y.; Fortin, D.A.; Cochrane, V.A.; Chen, P.-C.; Shyng, S.-L. NMDA receptors mediate leptin signaling and regulate potassium channel trafficking in pancreatic β-cells. J. Biol. Chem. 2017, 292, 15512–15524. [Google Scholar] [CrossRef]
- Wu, Y.; Shyng, S.L.; Chen, P.C. Concerted trafficking regulation of Kv2.1 and KATP channels by leptin in pancreatic β-cells. J. Biol. Chem. 2015, 290, 29676–29690. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Lim, A.; Park, S.-H.; Sohn, J.-W.; Jeon, J.-H.; Park, J.-H.; Song, D.-K.; Lee, S.-H.; Ho, W.-K. Glucose Deprivation Regulates KATP Channel Trafficking via AMP-Activated Protein Kinase in Pancreatic β-Cells. Diabetes 2009, 58, 2813–2819. [Google Scholar] [CrossRef]
- Beall, C.; Piipari, K.; Al-Qassab, H.; Smith, M.A.; Parker, N.; Carling, D.; Viollet, B.; Withers, D.J.; Ashford, M.L.J. Loss of AMP-activated protein kinase α2 subunit in mouse β-cells impairs glucose-stimulated insulin secretion and inhibits their sensitivity to hypoglycaemia. Biochem. J. 2010, 429, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.; Peroni, O.; Fryer, L.; Müller, C.; Carling, D.; Kahn, B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Alquier, T.; Furukawa, H.; Kim, Y.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferré, P.; Birnbaum, M.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574. [Google Scholar] [CrossRef]
- Marcelo, K.L.; Means, A.R.; York, B. The Ca2+ /Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718. [Google Scholar] [CrossRef]
- Marcelo, K.L.; Ribar, T.; Means, C.; Tsimelzon, A.; Stevens, R.; Ilkayeva, O.; Bain, J.; Hilsenbeck, S.; Newgard, C.; Means, A.; et al. Research Resource: Roles for Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CaMKK2) in Systems Metabolism. Mol. Endocrinol. 2016, 30, 557–572. [Google Scholar] [CrossRef]
- Hong, D.; Choi, I.; Son, Y.; Kim, D.; Na, S.; Jung, W.; Yoon, Y.; Park, W. The effect of PI3 kinase inhibitor LY294002 on voltage-dependent K+ channels in rabbit coronary arterial smooth muscle cells. Life Sci. 2013, 92, 916–922. [Google Scholar] [CrossRef]
- Iacobucci, G.J.; Popescu, G.K. NMDA receptors: linking physiological output to biophysical operation. Nat. Rev. Neurosci. 2017, 18, 236–249. [Google Scholar] [CrossRef]
- Shanley, L.J.; Irving, A.J.; Harvey, J. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 2001, 21, RC186. [Google Scholar] [CrossRef] [PubMed]
- Otter, S.; Lammert, E. Exciting Times for Pancreatic Islets: Glutamate Signaling in Endocrine Cells. Trends Endocrinol. Metab. 2016, 27, 177–188. [Google Scholar] [CrossRef]
- Marquard, J.; Otter, S.; Welters, A.; Stirban, A.; Fischer, A.; Eglinger, J.; Herebian, D.; Kletke, O.; Klemen, M.; Stožer, A.; et al. Characterization of pancreatic NMDA receptors as possible drug targets for diabetes treatment. Nat. Med. 2015, 21, 363–372. [Google Scholar] [CrossRef]
- Mairet-Coello, G.; Courchet, J.; Pieraut, S.; Courchet, V.; Maximov, A.; Polleux, F. The CAMKK2-AMPK Kinase Pathway Mediates the Synaptotoxic Effects of Aβ Oligomers through Tau Phosphorylation. Neuron 2013, 78, 94–108. [Google Scholar] [CrossRef]
- Cabrera, O.; Jacques-Silva, M.; Speier, S.; Yang, S.; Köhler, M.; Fachado, A.; Vieira, E.; Zierath, J.; Kibbey, R.; Berman, D.; et al. Glutamate is a positive autocrine signal for glucagon release. Cell Metab. 2008, 7, 545–554. [Google Scholar] [CrossRef]
- Meunier, F.A.; Gutiérrez, L.M. Captivating New Roles of F-Actin Cortex in Exocytosis and Bulk Endocytosis in Neurosecretory Cells. Trends Neurosci. 2016, 39, 605–613. [Google Scholar] [CrossRef]
- Klussmann, E.; Maric, K.; Wiesner, B.; Beyermann, M.; Rosenthal, W. Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 1999, 274, 4934–4938. [Google Scholar] [CrossRef] [PubMed]
- Klussman, E.; Nedvetsky, P.; Tamma, G.; Beulshausen, S.; Valenti, G.; Rosenthal, W. Regulation of aquaporin-2 trafficking. Handb. Exp. Pharm. 2009, 133–157. [Google Scholar]
- Butterworth, M.B.; Frizzell, R.A.; Johnson, J.P.; Peters, K.W.; Edinger, R.S. PKA-dependent ENaC trafficking requires the SNARE-binding protein complexin. Am. J. Physiol. Ren. Physiol. 2005, 289, F969–F977. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Narine, A.; Tulis, D.A. Inhibition of vascular smooth muscle growth via signaling crosstalk between AMP-activated protein kinase and cAMP-dependent protein kinase. Front. Physiol. 2012, 3, 409. [Google Scholar] [CrossRef] [PubMed]
- Kobashigawa, L.C.; Xu, Y.C.; Padbury, J.F.; Tseng, Y.-T.; Yano, N. Metformin Protects Cardiomyocyte from Doxorubicin Induced Cytotoxicity through an AMP-Activated Protein Kinase Dependent Signaling Pathway: An In Vitro Study. PLoS ONE 2014, 9, e104888. [Google Scholar] [CrossRef]
- Johanns, M.; Lai, Y.; Hsu, M.; Jacobs, R.; Vertommen, D.; Van Sande, J.; Dumont, J.; Woods, A.; Carling, D.; Hue, L.; et al. AMPK antagonizes hepatic glucagon-stimulated cyclic AMP signalling via phosphorylation-induced activation of cyclic nucleotide phosphodiesterase 4B. Nat. Commun. 2016, 7, 10856. [Google Scholar] [CrossRef]
- Nadella, K.; Saji, M.; Jacob, N.; Pavel, E.; Ringel, M.; Kirscner, L. Regulation of actin function by protein kinase A-mediated phosphorylation of Limk1. EMBO Rep. 2009, 10, 599–605. [Google Scholar] [CrossRef]
- Bachmann, V.; Riml, A.; Huber, R.; Baillie, G.; Liedl, K.; Valovka, T.; Stefan, E. Reciprocal regulation of PKA and Rac signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 8531–8536. [Google Scholar] [CrossRef]
- Park, S.H.; Ho, W.K.; Jeon, J.H. AMPK regulates KATP channel trafficking via PTEN inhibition in leptin-treated pancreatic β-cells. Biochem. Biophys. Res. Commun. 2013, 440, 539–544. [Google Scholar] [CrossRef]
- Han, Y.; Lim, A.; Park, S.; Chang, S.; Lee, S.; Ho, W. Rac-mediated actin remodeling and myosin II are involved in KATP channel trafficking in pancreatic β-cells. Exp. Mol. Med. 2015, 47, e190. [Google Scholar] [CrossRef][Green Version]
- Chi, X.; Wang, S.; Huang, Y.; Stamnes, M.; Chen, J.-L. Roles of rho GTPases in intracellular transport and cellular transformation. Int. J. Mol. Sci. 2013, 14, 7089–7108. [Google Scholar] [CrossRef]
- Han, J.; Luby-Phelps, K.; Das, B.; Shu, X.; Xia, Y.; Mosteller, R.; Krishna, U.; Falck, J.; White, M.; Broek, D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science 1998, 279, 558–560. [Google Scholar] [CrossRef]
- Fleming, I.N.; Gray, A.; Downes, C.P. Regulation of the Rac1-specific exchange factor Tiam1 involves both phosphoinositide 3-kinase-dependent and -independent components. Biochem. J. 2000, 351, 173–182. [Google Scholar] [CrossRef]
- Shinohara, M.; Terada, Y.; Iwamatsu, A.; Shinohara, A.; Mochizuki, N.; Higuchi, M.; Gotoh, Y.; Ihara, S.; Nagata, S.; Itoh, H.; et al. SWAP-70 is a guanine-nucleotide-exchange factor that mediates signalling of membrane ruffling. Nature 2002, 416, 759–763. [Google Scholar] [CrossRef]
- Papadopulos, A.; Tomatis, V.M.; Kasula, R.; Meunier, F.A. The Cortical Acto-Myosin Network: From Diffusion Barrier to Functional Gateway in the Transport of Neurosecretory Vesicles to the Plasma Membrane. Front. Endocrinol. 2013, 4, 153. [Google Scholar] [CrossRef]
- Yang, S.; Wenna, N.; Yu, J.; Yang, G.; Qiu, H.; Yu, L.; Juntti-Berggren, L.; Köhler, M.; Berggren, P. Glucose Recruits KATP Channels via Non-Insulin-Containing Dense-Core Granules. Cell Metab. 2007, 6, 217–228. [Google Scholar] [CrossRef]
- Han, Y.; Chun, J.; Kwon, M.; Ji, Y.; Jeong, M.; Kim, H.; Park, S.; Rah, J.; Kang, J.; Lee, S.; et al. Endocytosis of K ATP Channels Drives Glucose-Stimulated Excitation of Pancreatic β Cells. Cell Rep. 2018, 22, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Lin, Y.; Yan, F.; Pratt, E.; Shyng, S. Kir6.2 Mutations Associated with Neonatal Diabetes Reduce Expression of ATP-Sensitive K+ channels: Implications in Disease Mechanism and Sulfonylurea Therapy. Diabetes 2006, 55, 1738–1746. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ashcroft, F.M.; Puljung, M.C.; Vedovato, N. Neonatal Diabetes and the KATP Channel: From Mutation to Therapy. Trends Endocrinol Metab. 2017, 28, 377–387. [Google Scholar] [CrossRef]
- Balamurugan, K.; Kavitha, B.; Yang, Z.; Mohan, V.; Radha, V.; Shyng, S. Functional characterization of activating mutations in the sulfonylurea receptor 1 (ABCC8) causing neonatal diabetes mellitus in Asian Indian children. Pediatr. Diabetes 2019, 20, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Pinney, S.; MacMullen, C.; Becker, S.; Lin, Y.; Hanna, C.; Thornton, P.; Ganguly, A.; Shyng, S.; Stanley, C. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J. Clin. Investig. 2008, 118, 2877–2886. [Google Scholar] [CrossRef]
- Macmullen, C.; Zhou, Q.; Snider, K.; Tewson, P.; Becker, S.; Aziz, A.; Ganguly, A.; Shyng, S.; Stanley, C. Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the β-cell sulfonylurea receptor SUR1. Diabetes 2011, 60, 1797–1804. [Google Scholar] [CrossRef]
- Rorsman, P.; Braun, M. Regulation of Insulin Secretion in Human Pancreatic Islets. Annu. Rev. Physiol. 2003, 75, 155–179. [Google Scholar] [CrossRef]
- Geng, X.; Li, L.; Watkins, S.; Robbins, P.D.; Drain, P. The insulin secretory granule is the major site of K(ATP) channels of the endocrine pancreas. Diabetes 2003, 52, 767–776. [Google Scholar] [CrossRef][Green Version]
- Varadi, A.; Grant, A.; McCormack, M.; Nicolson, T.; Magistri, M.; Mitchel, K.; Halestrap, A.; Yuan, H.; Schwappach, B.; Rutter, G. Intracellular ATP-sensitive K+ channels in mouse pancreatic beta cells: against a role in organelle cation homeostasis. Diabetologia 2006, 49, 1567–1577. [Google Scholar] [CrossRef]
- Covey, S.; Wideman, R.; McDonald, C.; Unniappan, S.; Huynh, F.; Asadi, A.; Speck, M.; Webber, T.; Chua, S.; Kieffer, T. The pancreatic beta cell is a key site for mediating the effects of leptin on glucose homeostasis. Cell Metab. 2006, 4, 291–302. [Google Scholar] [CrossRef]
- Gray, S.L.; Donald, C.; Jetha, A.; Covey, S.D.; Kieffer, T.J. Hyperinsulinemia precedes insulin resistance in mice lacking pancreatic beta-cell leptin signaling. Endocrinology 2010, 151, 4178–4186. [Google Scholar] [CrossRef]
- Morioka, T.; Asilmaz, E.; Hu, J.; Dishinger, J.; Kurpad, A.; Elias, C.; Li, H.; Elmquist, J.; Kennedy, R.; Kulkarni, R. Disruption of leptin receptor expression in the pancreas directly affects β cell growth and function in mice. J. Clin. Investig. 2007, 117, 2860–2868. [Google Scholar] [CrossRef]
- Soedling, H.; Hodson, D.; Adrianssens, A.; Gribble, F.; Reimann, F.; Trapp, S.; Rutter, G. Limited impact on glucose homeostasis of leptin receptor deletion from insulin- or proglucagon- expressing cells. Mol. Metab. 2015, 4, 619–630. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cochrane, V.; Shyng, S.-L. Leptin-induced Trafficking of KATP Channels: A Mechanism to Regulate Pancreatic β-cell Excitability and Insulin Secretion. Int. J. Mol. Sci. 2019, 20, 2660. https://doi.org/10.3390/ijms20112660
Cochrane V, Shyng S-L. Leptin-induced Trafficking of KATP Channels: A Mechanism to Regulate Pancreatic β-cell Excitability and Insulin Secretion. International Journal of Molecular Sciences. 2019; 20(11):2660. https://doi.org/10.3390/ijms20112660
Chicago/Turabian StyleCochrane, Veronica, and Show-Ling Shyng. 2019. "Leptin-induced Trafficking of KATP Channels: A Mechanism to Regulate Pancreatic β-cell Excitability and Insulin Secretion" International Journal of Molecular Sciences 20, no. 11: 2660. https://doi.org/10.3390/ijms20112660
APA StyleCochrane, V., & Shyng, S.-L. (2019). Leptin-induced Trafficking of KATP Channels: A Mechanism to Regulate Pancreatic β-cell Excitability and Insulin Secretion. International Journal of Molecular Sciences, 20(11), 2660. https://doi.org/10.3390/ijms20112660