Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy


Abstract
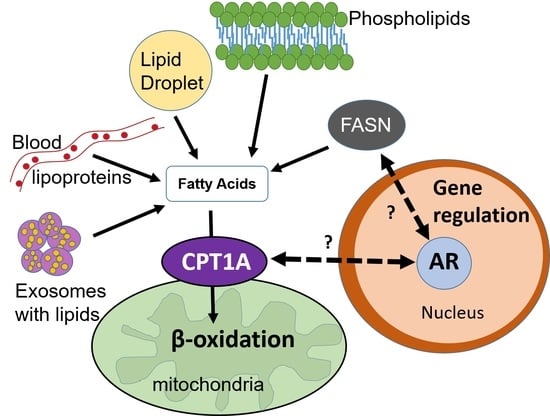
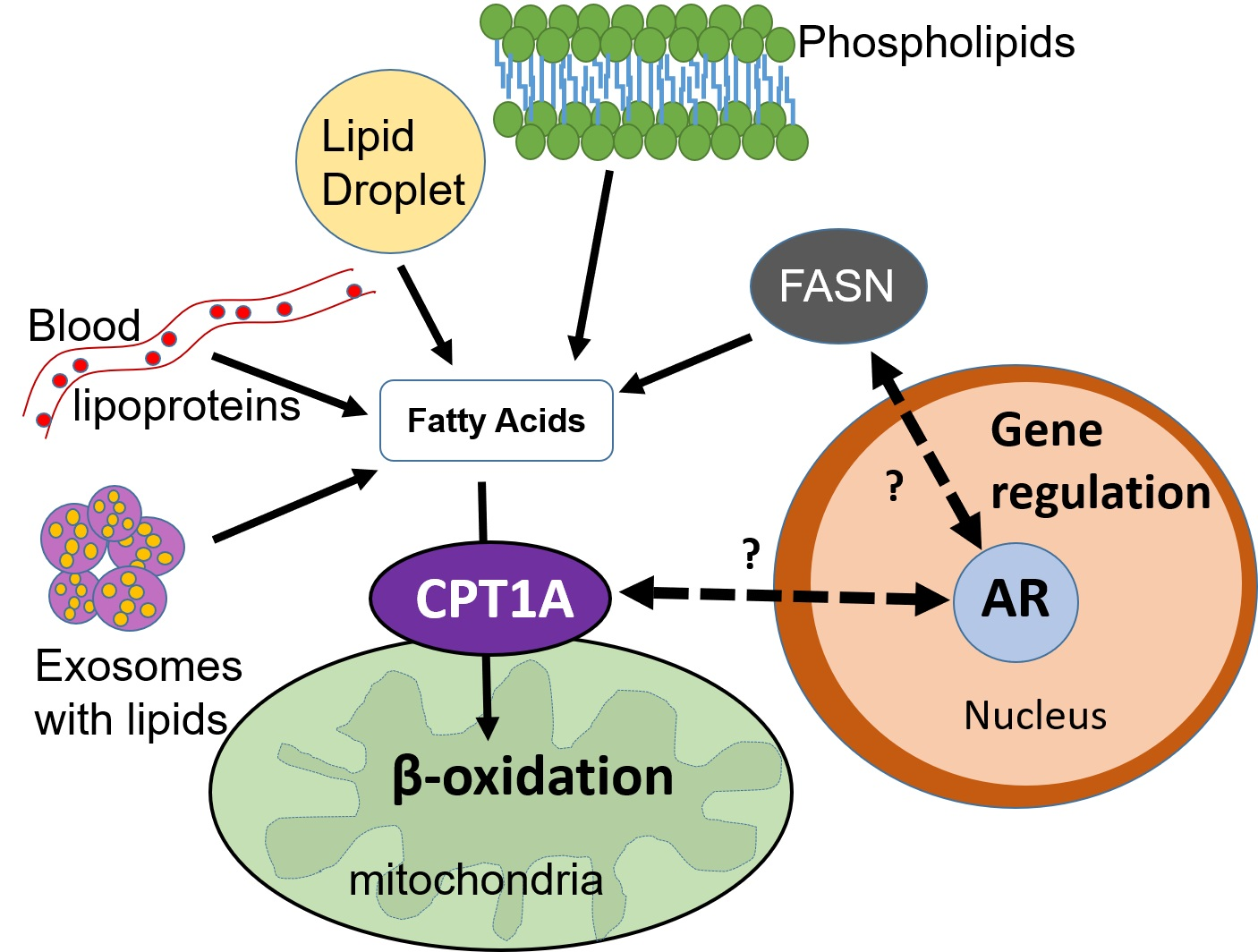
1. Introduction
2. Prostate Cancer and Lipid Metabolism
2.1. Lipid Synthesis and Uptake
2.2. Lipid Breakdown and Oxidation
3. Endocrine Therapy Challenges in Prostate Cancer
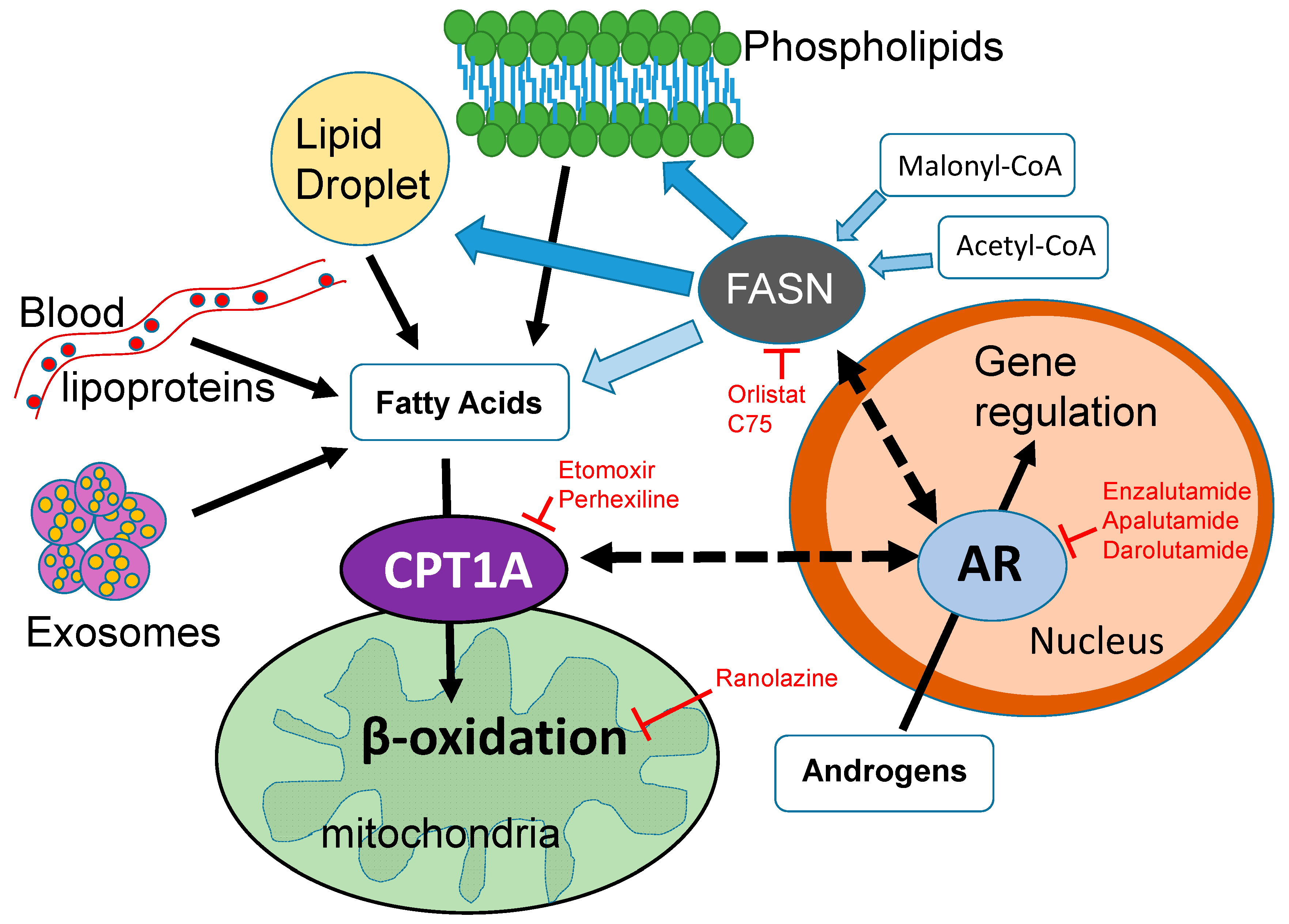
4. Therapeutic Opportunities Targeting Lipid Metabolism and Androgen Signaling
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Attard, G.; Parker, C.; Eeles, R.A.; Schroder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- HUGGINS, C. Prostatic cancer treated by orchiectomy; the five year results. J. Am. Med. Assoc. 1946, 131, 576–581. [Google Scholar] [CrossRef]
- Chan, S.C.; Dehm, S.M. Constitutive activity of the androgen receptor. Adv. Pharmacol. 2014, 70, 327–366. [Google Scholar] [PubMed]
- Karantanos, T.; Evans, C.P.; Tombal, B.; Thompson, T.C.; Montironi, R.; Isaacs, W.B. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur. Urol. 2015, 67, 470–479. [Google Scholar] [CrossRef]
- Braga-Basaria, M.; Dobs, A.S.; Muller, D.C.; Carducci, M.A.; John, M.; Egan, J.; Basaria, S. Metabolic syndrome in men with prostate cancer undergoing long-term androgen-deprivation therapy. J. Clin. Oncol. 2006, 24, 3979–3983. [Google Scholar] [CrossRef]
- Dockery, F.; Bulpitt, C.J.; Agarwal, S.; Donaldson, M.; Rajkumar, C. Testosterone suppression in men with prostate cancer leads to an increase in arterial stiffness and hyperinsulinaemia. Clin. Sci. 2003, 104, 195–201. [Google Scholar] [CrossRef]
- Saylor, P.J.; Karoly, E.D.; Smith, M.R. Prospective study of changes in the metabolomic profiles of men during their first three months of androgen deprivation therapy for prostate cancer. Clin. Cancer Res. 2012, 18, 3677–3685. [Google Scholar] [CrossRef]
- Smith, M.R.; Finkelstein, J.S.; McGovern, F.J.; Zietman, A.L.; Fallon, M.A.; Schoenfeld, D.A.; Kantoff, P.W. Changes in body composition during androgen deprivation therapy for prostate cancer. J. Clin. Endocrinol. Metab. 2002, 87, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, J.V.; Brusselmans, K.; Verhoeven, G. Increased lipogenesis in cancer cells: New players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Putluri, V.; Arnold, J.M.; Tsouko, E.; Maity, S.; Roberts, J.M.; Coarfa, C.; Frigo, D.E.; Putluri, N.; Sreekumar, A.; et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget 2015, 6, 31997–32012. [Google Scholar] [CrossRef]
- Sonn, G.A.; Aronson, W.; Litwin, M.S. Impact of diet on prostate cancer: A review. Prostate Cancer Prostatic Dis. 2005, 8, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Gronberg, H. Prostate cancer epidemiology. Lancet 2003, 361, 859–864. [Google Scholar] [CrossRef]
- Ngo, T.H.; Barnard, R.J.; Cohen, P.; Freedland, S.; Tran, C.; deGregorio, F.; Elshimali, Y.I.; Heber, D.; Aronson, W.J. Effect of isocaloric low-fat diet on human LAPC-4 prostate cancer xenografts in severe combined immunodeficient mice and the insulin-like growth factor axis. Clin. Cancer Res. 2003, 9, 2734–2743. [Google Scholar] [PubMed]
- Pour, P.M.; Groot, K.; Kazakoff, K.; Anderson, K.; Schally, A.V. Effects of high-fat diet on the patterns of prostatic cancer induced in rats by N-nitrosobis(2-oxopropyl)amine and testosterone. Cancer Res. 1991, 51, 4757–4761. [Google Scholar] [PubMed]
- Kuhajda, F.P. Fatty acid synthase and cancer: New application of an old pathway. Cancer Res. 2006, 66, 5977–5980. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Van Veldhoven, P.P.; Esquenet, M.; Heyns, W.; Verhoeven, G. Androgens markedly stimulate the accumulation of neutral lipids in the human prostatic adenocarcinoma cell line LNCaP. Endocrinology 1996, 137, 4468–4474. [Google Scholar] [CrossRef]
- Suburu, J.; Chen, Y.Q. Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012, 98, 1–10. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid Catabolism via CPT1 as a Therapeutic Target for Prostate Cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef]
- Zadra, G.; Priolo, C.; Patnaik, A.; Loda, M. New strategies in prostate cancer: Targeting lipogenic pathways and the energy sensor AMPK. Clin. Cancer Res. 2010, 16, 3322–3328. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef]
- Abate-Shen, C. Prostate Cancer Metastasis—Fueled by Fat? N. Engl. J. Med. 2018, 378, 1643–1645. [Google Scholar] [CrossRef] [PubMed]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Hitz, C.A.; Gijon, M.A.; Bergman, B.C.; Eckel, R.H.; Jacobsen, B.M. Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel. Mol. Cell Endocrinol. 2012, 363, 111–121. [Google Scholar] [CrossRef]
- Potcoava, M.C.; Futia, G.L.; Aughenbaugh, J.; Schlaepfer, I.R.; Gibson, E.A. Raman and coherent anti-Stokes Raman scattering microscopy studies of changes in lipid content and composition in hormone-treated breast and prostate cancer cells. J. Biomed. Opt. 2014, 19, 111605. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulos, A.A.; Kalyvianaki, K.; Castanas, E.; Kampa, M. Eicosanoids in prostate cancer. Cancer Metastasis Rev. 2018, 37, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wilson, K.M.; Stampfer, M.J.; Willett, W.C.; Giovannucci, E.L. A 24-year prospective study of dietary alpha-linolenic acid and lethal prostate cancer. Int. J. Cancer 2018, 142, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Allott, E.H.; Arab, L.; Su, L.J.; Farnan, L.; Fontham, E.T.; Mohler, J.L.; Bensen, J.T.; Steck, S.E. Saturated fat intake and prostate cancer aggressiveness: Results from the population-based North Carolina-Louisiana Prostate Cancer Project. Prostate Cancer Prostatic Dis. 2017, 20, 48–54. [Google Scholar] [CrossRef]
- Downer, M.K.; Batista, J.L.; Mucci, L.A.; Stampfer, M.J.; Epstein, M.M.; Hakansson, N.; Wolk, A.; Johansson, J.E.; Andren, O.; Fall, K.; et al. Dairy intake in relation to prostate cancer survival. Int. J. Cancer 2017, 140, 2060–2069. [Google Scholar] [CrossRef]
- Aucoin, M.; Cooley, K.; Knee, C.; Fritz, H.; Balneaves, L.G.; Breau, R.; Fergusson, D.; Skidmore, B.; Wong, R.; Seely, D. Fish-Derived Omega-3 Fatty Acids and Prostate Cancer: A Systematic Review. Integr. Cancer Ther. 2017, 16, 32–62. [Google Scholar] [CrossRef]
- Saxena, R.; Yang, C.; Rao, M.; Turaga, R.C.; Garlapati, C.; Gundala, S.R.; Myers, K.; Ghareeb, A.; Bhattarai, S.; Kamalinia, G.; et al. Preclinical Development of a Nontoxic Oral Formulation of Monoethanolamine, a Lipid Precursor, for Prostate Cancer Treatment. Clin. Cancer Res. 2017, 23, 3781–3793. [Google Scholar] [CrossRef]
- Heir, T.; Falk, R.S.; Robsahm, T.E.; Sandvik, L.; Erikssen, J.; Tretli, S. Cholesterol and prostate cancer risk: A long-term prospective cohort study. BMC Cancer 2016, 16, 643. [Google Scholar] [CrossRef] [PubMed]
- Khankari, N.K.; Murff, H.J.; Zeng, C.; Wen, W.; Eeles, R.A.; Easton, D.F.; Kote-Jarai, Z.; Al Olama, A.A.; Benlloch, S.; Muir, K.; et al. Polyunsaturated fatty acids and prostate cancer risk: A Mendelian randomisation analysis from the PRACTICAL consortium. Br. J. Cancer 2016, 115, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Van Blarigan, E.L.; Kenfield, S.A.; Yang, M.; Sesso, H.D.; Ma, J.; Stampfer, M.J.; Chan, J.M.; Chavarro, J.E. Fat intake after prostate cancer diagnosis and mortality in the Physicians’ Health Study. Cancer Causes Control 2015, 26, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Lovegrove, C.; Ahmed, K.; Challacombe, B.; Khan, M.S.; Popert, R.; Dasgupta, P. Systematic review of prostate cancer risk and association with consumption of fish and fish-oils: Analysis of 495,321 participants. Int. J. Clin. Pract. 2015, 69, 87–105. [Google Scholar] [CrossRef]
- Lippi, G.; Mattiuzzi, C. Fried food and prostate cancer risk: Systematic review and meta-analysis. Int. J. Food Sci. Nutr. 2015, 66, 587–589. [Google Scholar] [CrossRef]
- Richman, E.L.; Kenfield, S.A.; Chavarro, J.E.; Stampfer, M.J.; Giovannucci, E.L.; Willett, W.C.; Chan, J.M. Fat intake after diagnosis and risk of lethal prostate cancer and all-cause mortality. JAMA Intern. Med. 2013, 173, 1318–1326. [Google Scholar] [CrossRef]
- Epstein, M.M.; Kasperzyk, J.L.; Mucci, L.A.; Giovannucci, E.; Price, A.; Wolk, A.; Hakansson, N.; Fall, K.; Andersson, S.O.; Andren, O. Dietary fatty acid intake and prostate cancer survival in Orebro County, Sweden. Am. J. Epidemiol. 2012, 176, 240–252. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Howell, A.; Pestell, R.G.; Tanowitz, H.B.; Sotgia, F.; Lisanti, M.P. Stromal-epithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int. J. Biochem. Cell Biol. 2011, 43, 1045–1051. [Google Scholar] [CrossRef]
- Singh, M.; Jha, R.; Melamed, J.; Shapiro, E.; Hayward, S.W.; Lee, P. Stromal androgen receptor in prostate development and cancer. Am. J. Pathol. 2014, 184, 2598–2607. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Tousignant, K.D.; Rockstroh, A.; Taherian Fard, A.; Lehman, M.L.; Wang, C.; McPherson, S.J.; Philp, L.K.; Bartonicek, N.; Dinger, M.E.; Nelson, C.C.; et al. Lipid Uptake Is an Androgen-Enhanced Lipid Supply Pathway Associated with Prostate Cancer Disease Progression and Bone Metastasis. Mol. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Samovski, D.; Sun, J.; Pietka, T.; Gross, R.W.; Eckel, R.H.; Su, X.; Stahl, P.D.; Abumrad, N.A. Regulation of AMPK activation by CD36 links fatty acid uptake to beta-oxidation. Diabetes 2015, 64, 353–359. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Dheeraj, A.; Agarwal, C.; Schlaepfer, I.R.; Raben, D.; Singh, R.; Agarwal, R.; Deep, G. A novel approach to target hypoxic cancer cells via combining beta-oxidation inhibitor etomoxir with radiation. Hypoxia 2018, 6, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J.T.; Melbye, M. Cancer patterns in Inuit populations. Lancet Oncol. 2008, 9, 892–900. [Google Scholar] [CrossRef]
- Frazier-Wood, A.C.; Aslibekyan, S.; Absher, D.M.; Hopkins, P.N.; Sha, J.; Tsai, M.Y.; Tiwari, H.K.; Waite, L.L.; Zhi, D.; Arnett, D.K. Methylation at CPT1A locus is associated with lipoprotein subfraction profiles. J. Lipid Res. 2014, 55, 1324–1330. [Google Scholar] [CrossRef]
- Alfaqih, M.A.; Nelson, E.R.; Liu, W.; Safi, R.; Jasper, J.S.; Macias, E.; Geradts, J.; Thompson, J.W.; Dubois, L.G.; Freeman, M.R.; et al. CYP27A1 Loss Dysregulates Cholesterol Homeostasis in Prostate Cancer. Cancer Res. 2017, 77, 1662–1673. [Google Scholar] [CrossRef]
- Wettstein, M.S.; Saba, K.; Umbehr, M.H.; Murtola, T.J.; Fankhauser, C.D.; Adank, J.P.; Hofmann, M.; Sulser, T.; Hermanns, T.; Moch, H.; et al. Prognostic Role of Preoperative Serum Lipid Levels in Patients Undergoing Radical Prostatectomy for Clinically Localized Prostate Cancer. Prostate 2017, 77, 549–556. [Google Scholar] [CrossRef]
- Wolny-Rokicka, E.I.; Tukiendorf, A.; Wydmanski, J.; Zembron-Lacny, A. The Comparison and Estimation of the Prognostic Value of Lipid Profiles in Patients with Prostate Cancer Depends on Cancer Stage Advancement. Am. J. Mens. Health 2017, 11, 1745–1751. [Google Scholar] [CrossRef]
- Valentino, A.; Calarco, A.; Di Salle, A.; Finicelli, M.; Crispi, S.; Calogero, R.A.; Riccardo, F.; Sciarra, A.; Gentilucci, A.; Galderisi, U.; et al. Deregulation of MicroRNAs mediated control of carnitine cycle in prostate cancer: Molecular basis and pathophysiological consequences. Oncogene 2017, 36, 6030–6040. [Google Scholar] [CrossRef]
- Bull, C.J.; Bonilla, C.; Holly, J.M.; Perks, C.M.; Davies, N.; Haycock, P.; Yu, O.H.; Richards, J.B.; Eeles, R.; Easton, D.; et al. Blood lipids and prostate cancer: A Mendelian randomization analysis. Cancer Med. 2016, 5, 1125–1136. [Google Scholar] [CrossRef]
- Ma, H.Q.; Cui, L.H.; Li, C.C.; Yu, Z.; Piao, J.M. Effects of Serum Triglycerides on Prostate Cancer and Breast Cancer Risk: A Meta-Analysis of Prospective Studies. Nutr. Cancer 2016, 68, 1073–1082. [Google Scholar] [CrossRef]
- Zapata, D.; Howard, L.E.; Allott, E.H.; Hamilton, R.J.; Goldberg, K.; Freedland, S.J. Is PSA related to serum cholesterol and does the relationship differ between black and white men? Prostate 2015, 75, 1877–1885. [Google Scholar] [CrossRef]
- Li, Y.; Liang, C.; Huang, J. Serum lipid profiles and aggressive prostate cancer. Asian J. Androl. 2015, 17, 336. [Google Scholar] [CrossRef]
- Allott, E.H.; Howard, L.E.; Cooperberg, M.R.; Kane, C.J.; Aronson, W.J.; Terris, M.K.; Amling, C.L.; Freedland, S.J. Serum lipid profile and risk of prostate cancer recurrence: Results from the SEARCH database. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2349–2356. [Google Scholar] [CrossRef]
- Llorente, A.; Skotland, T.; Sylvanne, T.; Kauhanen, D.; Rog, T.; Orlowski, A.; Vattulainen, I.; Ekroos, K.; Sandvig, K. Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim. Biophys. Acta 2013, 1831, 1302–1309. [Google Scholar] [CrossRef]
- Skotland, T.; Ekroos, K.; Kauhanen, D.; Simolin, H.; Seierstad, T.; Berge, V.; Sandvig, K.; Llorente, A. Molecular lipid species in urinary exosomes as potential prostate cancer biomarkers. Eur. J. Cancer 2017, 70, 122–132. [Google Scholar] [CrossRef]
- Record, M.; Poirot, M.; Silvente-Poirot, S. Emerging concepts on the role of exosomes in lipid metabolic diseases. Biochimie 2014, 96, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Nambiar, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P.; et al. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015, 6, 22836–22856. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Glode, L.M.; Hitz, C.A.; Pac, C.T.; Boyle, K.E.; Maroni, P.; Deep, G.; Agarwal, R.; Lucia, S.M.; Cramer, S.D.; et al. Inhibition of Lipid Oxidation Increases Glucose Metabolism and Enhances 2-Deoxy-2-[F]Fluoro-D-Glucose Uptake in Prostate Cancer Mouse Xenografts. Mol. Imaging Biol. 2015, 17, 529–538. [Google Scholar] [CrossRef]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Deep, G.; Schlaepfer, I.R. Aberrant Lipid Metabolism Promotes Prostate Cancer: Role in Cell Survival under Hypoxia and Extracellular Vesicles Biogenesis. Int. J. Mol. Sci. 2016, 17, 1061. [Google Scholar] [CrossRef]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Harper, M.E.; Antoniou, A.; Villalobos-Menuey, E.; Russo, A.; Trauger, R.; Vendemelio, M.; George, A.; Bartholomew, R.; Carlo, D.; Shaikh, A.; et al. Characterization of a novel metabolic strategy used by drug-resistant tumor cells. FASEB J. 2002, 16, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.S.; Smift, A.L.; Croteau, N.J.; Ferrick, D.A.; Wu, M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim. Biophys. Acta 2011, 1807, 726–734. [Google Scholar] [CrossRef]
- Wang, Y.N.; Zeng, Z.L.; Lu, J.; Wang, Y.; Liu, Z.X.; He, M.M.; Zhao, Q.; Wang, Z.X.; Li, T.; Lu, Y.X.; et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene 2018, 37, 6025–6040. [Google Scholar] [CrossRef]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Flaig, T.W.; Salzmann-Sullivan, M.; Su, L.J.; Zhang, Z.; Joshi, M.; Gijon, M.A.; Kim, J.; Arcaroli, J.J.; Van Bokhoven, A.; Lucia, M.S.; et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 2017, 8, 56051. [Google Scholar] [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Camarda, R.; Williams, J.; Goga, A. In vivo Reprogramming of Cancer Metabolism by MYC. Front. Cell Dev. Biol. 2017, 5, 35. [Google Scholar] [CrossRef]
- Hubbard, G.K.; Mutton, L.N.; Khalili, M.; McMullin, R.P.; Hicks, J.L.; Bianchi-Frias, D.; Horn, L.A.; Kulac, I.; Moubarek, M.S.; Nelson, P.S.; et al. Combined MYC Activation and Pten Loss Are Sufficient to Create Genomic Instability and Lethal Metastatic Prostate Cancer. Cancer Res. 2016, 76, 283–292. [Google Scholar] [CrossRef]
- Priolo, C.; Loda, M. Untargeted metabolomics for profiling oncogene-specific metabolic signatures of prostate cancer. Mol. Cell Oncol. 2015, 2, e1001197. [Google Scholar] [CrossRef]
- Giunchi, F.; Fiorentino, M.; Loda, M. The Metabolic Landscape of Prostate Cancer. Eur. Urol. Oncol. 2019, 2, 28–36. [Google Scholar] [CrossRef]
- Davies, A.; Conteduca, V.; Zoubeidi, A.; Beltran, H. Biological Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus 2019, 5, 147–154. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Dehm, S.M.; Tindall, D.J. Alternatively spliced androgen receptor variants. Endocr. Relat. Cancer 2011, 18, R183–R196. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de, W.R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Rapozzi, V.; Ragno, D.; Guerrini, A.; Ferroni, C.; della Pietra, E.; Cesselli, D.; Castoria, G.; Di Donato, M.; Saracino, E.; Benfenati, V.; et al. Androgen Receptor Targeted Conjugate for Bimodal Photodynamic Therapy of Prostate Cancer in Vitro. Bioconjug. Chem. 2015, 26, 1662–1671. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Vidal, A.C.; Freedland, S.J. Obesity and Prostate Cancer: A Focused Update on Active Surveillance, Race, and Molecular Subtyping. Eur. Urol. 2017, 72, 78–83. [Google Scholar] [CrossRef]
- Vidal, A.C.; Howard, L.E.; de Hoedt, A.; Kane, C.J.; Terris, M.K.; Aronson, W.J.; Cooperberg, M.R.; Amling, C.L.; Freedland, S.J. Obese patients with castration-resistant prostate cancer may be at a lower risk of all-cause mortality: Results from the Shared Equal Access Regional Cancer Hospital (SEARCH) database. BJU Int. 2018, 122, 76–82. [Google Scholar] [CrossRef]
- Heemers, H.V.; Verhoeven, G.; Swinnen, J.V. Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol. Endocrinol. 2006, 20, 2265–2277. [Google Scholar] [CrossRef]
- Migita, T.; Ruiz, S.; Fornari, A.; Fiorentino, M.; Priolo, C.; Zadra, G.; Inazuka, F.; Grisanzio, C.; Palescandolo, E.; Shin, E.; et al. Fatty acid synthase: A metabolic enzyme and candidate oncogene in prostate cancer. J. Natl. Cancer Inst. 2009, 101, 519–532. [Google Scholar] [CrossRef]
- Butler, L.M.; Centenera, M.M.; Swinnen, J.V. Androgen control of lipid metabolism in prostate cancer: Novel insights and future applications. Endocr. Relat. Cancer 2016, 23, R219–R227. [Google Scholar] [CrossRef]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene 2014, 33, 5251. [Google Scholar] [CrossRef]
- Little, J.L.; Wheeler, F.B.; Fels, D.R.; Koumenis, C.; Kridel, S.J. Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Res. 2007, 67, 1262–1269. [Google Scholar] [CrossRef]
- Kong, Y.; Cheng, L.; Mao, F.; Zhang, Z.; Zhang, Y.; Farah, E.; Bosler, J.; Bai, Y.; Ahmad, N.; Kuang, S.; et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 2018, 293, 14328–14341. [Google Scholar] [CrossRef]
- Han, W.; Gao, S.; Barrett, D.; Ahmed, M.; Han, D.; Macoska, J.A.; He, H.H.; Cai, C. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 2018, 37, 710–721. [Google Scholar] [CrossRef]
- Iglesias-Gato, D.; Wikstrom, P.; Tyanova, S.; Lavallee, C.; Thysell, E.; Carlsson, J.; Hagglof, C.; Cox, J.; Andren, O.; Stattin, P.; et al. The Proteome of Primary Prostate Cancer. Eur. Urol. 2016, 69, 942–952. [Google Scholar] [CrossRef]
- Iglesias-Gato, D.; Thysell, E.; Tyanova, S.; Crnalic, S.; Santos, A.; Lima, T.S.; Geiger, T.; Cox, J.; Widmark, A.; Bergh, A.; et al. The Proteome of Prostate Cancer Bone Metastasis Reveals Heterogeneity with Prognostic Implications. Clin. Cancer Res. 2018, 24, 5433–5444. [Google Scholar] [CrossRef]
- Itkonen, H.M.; Brown, M.; Urbanucci, A.; Tredwell, G.; Ho Lau, C.; Barfeld, S.; Hart, C.; Guldvik, I.J.; Takhar, M.; Heemers, H.V.; et al. Lipid degradation promotes prostate cancer cell survival. Oncotarget 2017, 8, 38264–38275. [Google Scholar] [CrossRef]
- Kim, S.H.; Singh, S.V. D,L-Sulforaphane causes transcriptional repression of androgen receptor in human prostate cancer cells. Mol. Cancer Ther. 2009, 8, 1946–1954. [Google Scholar] [CrossRef]
- Singh, K.B.; Kim, S.H.; Hahm, E.R.; Pore, S.K.; Jacobs, B.L.; Singh, S.V. Prostate Cancer Chemoprevention by Sulforaphane in a Preclinical Mouse Model is Associated with Inhibition of Fatty Acid Metabolism. Carcinogenesis 2018, 39, 826–837. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Lipids | Association Found | Reference |
|---|---|---|
| Dietary linoleic and alpha-linoleic acids | Metabolism at cellular level produces eicosanoids, some of which possibly function as tumor suppressors in PCa | Eicosanoids in prostate cancer. Cancer Metastasis Rev. 2018. Review. [27]. |
| Dietary alpha-linoleic acid (ALA) | Intake of ALA (mainly through mayonnaise consumption) associated with increased risk of lethal PCa prior to February 1994, when PSA testing began. | A 24-year prospective study of dietary α-linolenic acid and lethal prostate cancer. Int. J. Cancer 2018. [28] |
| Saturated fats | High saturated fat intake associated with increased PCa aggressiveness | Saturated fat intake and prostate cancer aggressiveness: results from the population-based North Carolina-Louisiana Prostate Cancer Project. Prostate Cancer Prostatic Dis. 2017. [29] |
| High-fat milk | High-fat milk intake associated with PCa progression (in localized PCa patients) | Dairy intake in relation to prostate cancer survival. Int J Cancer. 2017. [30] |
| Omega 3 FA (fish-derived) | Higher Omega 3 intake may be associated with decreased PCa mortality | Fish-Derived Omega-3 Fatty Acids and Prostate Cancer: A Systematic Review. Integr Cancer Ther. 2017. [31] |
| Monoethanolamine/lipid precursor | Anti-cancer activity of monoethanolamine evident and has clinic-use potential | Preclinical Development of a Nontoxic Oral Formulation of Monoethanolamine, a Lipid Precursor, for Prostate Cancer Treatment. Clin Cancer Res. 2017. [32] |
| Cholesterol | Low cholesterol, low BMI, high physical activity associated with higher PCa risk **conflict with current PCa recommendations | Cholesterol and prostate cancer risk: a long-term prospective cohort study. BMC Cancer. 2016. [33] |
| PUFA | Risk reductions observed for long-chain PUFA, short-chain PUFA, linoleic acid, and ALA in men under 62; increased risk for LA in men over 62. | Polyunsaturated fatty acids and prostate cancer risk: a Mendelian randomisation analysis from the PRACTICAL consortium. Br J Cancer. 2016. [34]. |
| Saturated fat, vegetable fat | For non-metastatic PCa, saturated fat intake may increase risk of death, while vegetable fat intake may lower it | Fat intake after prostate cancer diagnosis and mortality in the Physicians’ Health Study. Cancer Causes Control. 2015. [35]. |
| Fish and fish oils | Fish and fish oil consumption not consistently associated with reduction in PCa incidence, aggressiveness, and mortality | Systematic review of prostate cancer risk and association with consumption of fish and fish-oils: analysis of 495,321 participants. Int J Clin Pract. 2015. [36]. |
| Fried food | Larger intake of fried food associated with 35% increased risk of PCa | Fried food and prostate cancer risk: systematic review and meta-analysis. Int J Food Sci Nutr. 2015. Review. [37]. |
| Animal vs. vegetable fat | Potential benefit of vegetable fat for PCa-specific outcomes | Fat intake after diagnosis and risk of lethal prostate cancer and all-cause mortality. JAMA Intern Med. 2013. [38]. |
| SAFA, MUFA, PUFA | Balanced fat consumption diet may reduce the risk of PCa and prevent progression | Lipids and prostate cancer. Prostaglandins Other Lipid Mediat. 2012. Review. [19] |
| Short chain fatty acids | High intake of total fat and certain saturated fatty acids may worsen PCa survival. | Dietary fatty acid intake and prostate cancer survival in Örebro County, Sweden. Am J Epidemiol. 2012. [39]. |
| Lipid(s) Investigated | Association Found | Reference |
|---|---|---|
| Cholesterol | CYP27A1 (PCa cellular cholesterol sensor) significantly contributes to PCa pathogenesis | CYP27A1 Loss Dysregulates Cholesterol Homeostasis in Prostate Cancer. Cancer Res. 2017. [50] |
| Total cholesterol, LDL, HDL, and triglycerides | High LDL associated with longer recurrence-free survival | Prognostic Role of Preoperative Serum Lipid Levels in Patients Undergoing Radical Prostatectomy for Clinically Localized Prostate Cancer. Prostate. 2017. [51]. |
| Total cholesterol, LDL, HDL, and triglycerides | Upon irradiation with external beam therapy, LDL/HDL ratio in palliative subjects shows significant difference when compared to locoregional subjects | The Comparison and Estimation of the Prognostic Value of Lipid Profiles in Patients With Prostate Cancer Depends on Cancer Stage Advancement. Am J Mens Health. 2017. [52]. |
| Carnitine cycle (long-chain FA) | Carnitine cycle is indicated as a primary regulator of adaptive metabolic reprogramming in PCa cells | Deregulation of MicroRNAs mediated control of carnitine cycle in prostate cancer: molecular basis and pathophysiological consequences. Oncogene. 2017. [53]. |
| LDL and triglycerides | Weak evidence of higher LDL and triglycerides increasing PCa risk | Blood lipids and prostate cancer: a Mendelian randomization analysis. Cancer Med. 2016. [54]. |
| Serum triglyceride | Serum triglyceride levels not associated with PCa risk | Effects of Serum Triglycerides on Prostate Cancer and Breast Cancer Risk: A Meta-Analysis of Prospective Studies. Nutr Cancer. 2016. [55]. |
| Serum cholesterol, LDL | Total cholesterol and LDL correlated with PSA levels in cancer-free white males (prior to statin treatment) | Is PSA related to serum cholesterol and does the relationship differ between black and white men? Prostate. 2015. [56]. |
| Total cholesterol, LDL, and triglycerides | High cholesterol associated with increased risk lymph node metastasis; high LDL levels predict high Gleason scores | Serum lipid profiles and aggressive prostate cancer. Asian J Androl. 2015. [57]. |
| Total cholesterol, LDL, HDL, triglycerides | In dyslipidemia patients, elevated cholesterol associated with increased PCa recurrence, while elevated HDL associated with decreased PCa recurrence. Elevated triglycerides associated with increased PCa recurrence in general; elevated LDL not found to be associated with PCa recurrence | Serum lipid profile and risk of prostate cancer recurrence: Results from the SEARCH database. Cancer Epidemiol Biomarkers Prev. 2014. [58]. |
| Sphingolipids,, cholesterol, and phosphatidylserine | These lipids are enriched in the PC-3 exosomes and could be used as PCa biomarkers | Molecular lipidomics of exosomes released by PC-3 prostate cancer cells. Biochim Biophys Acta. 2013. [59]. |
| Phosphatidylserine, sphingolipids | The highest significance was shown for phosphatidylserine and lactosylceramide, which showed the highest patient-to-control ratio | Molecular lipid species in urinary exosomes as potential prostate cancer biomarkers. Eur J Cancer 2017. [60]. |
| Eicosanoids, fatty acids, and cholesterol | Exosomes are enriched in cholesterol and sphingomyelin and their accumulation in cells might modulate recipient cell homeostasis. | Exosomes as new vesicular lipid transporters involved in cell-cell communication and various pathophysiologies. Biochim Biophys Acta 2014. [61] |
| Triglycerides | Exosomes from PCa cells enriched in triglycerides | Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015. [62] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoykova, G.E.; Schlaepfer, I.R. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. Int. J. Mol. Sci. 2019, 20, 2626. https://doi.org/10.3390/ijms20112626
Stoykova GE, Schlaepfer IR. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. International Journal of Molecular Sciences. 2019; 20(11):2626. https://doi.org/10.3390/ijms20112626
Chicago/Turabian StyleStoykova, Gergana E., and Isabel R. Schlaepfer. 2019. "Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy" International Journal of Molecular Sciences 20, no. 11: 2626. https://doi.org/10.3390/ijms20112626
APA StyleStoykova, G. E., & Schlaepfer, I. R. (2019). Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. International Journal of Molecular Sciences, 20(11), 2626. https://doi.org/10.3390/ijms20112626