Hepatic Osteodystrophy—Molecular Mechanisms Proposed to Favor Its Development

 ,
, 


Abstract
1. Hepatic Osteodystrophy—Definition and Prevalence
2. Limitations of Current Diagnostic Tools for HOD
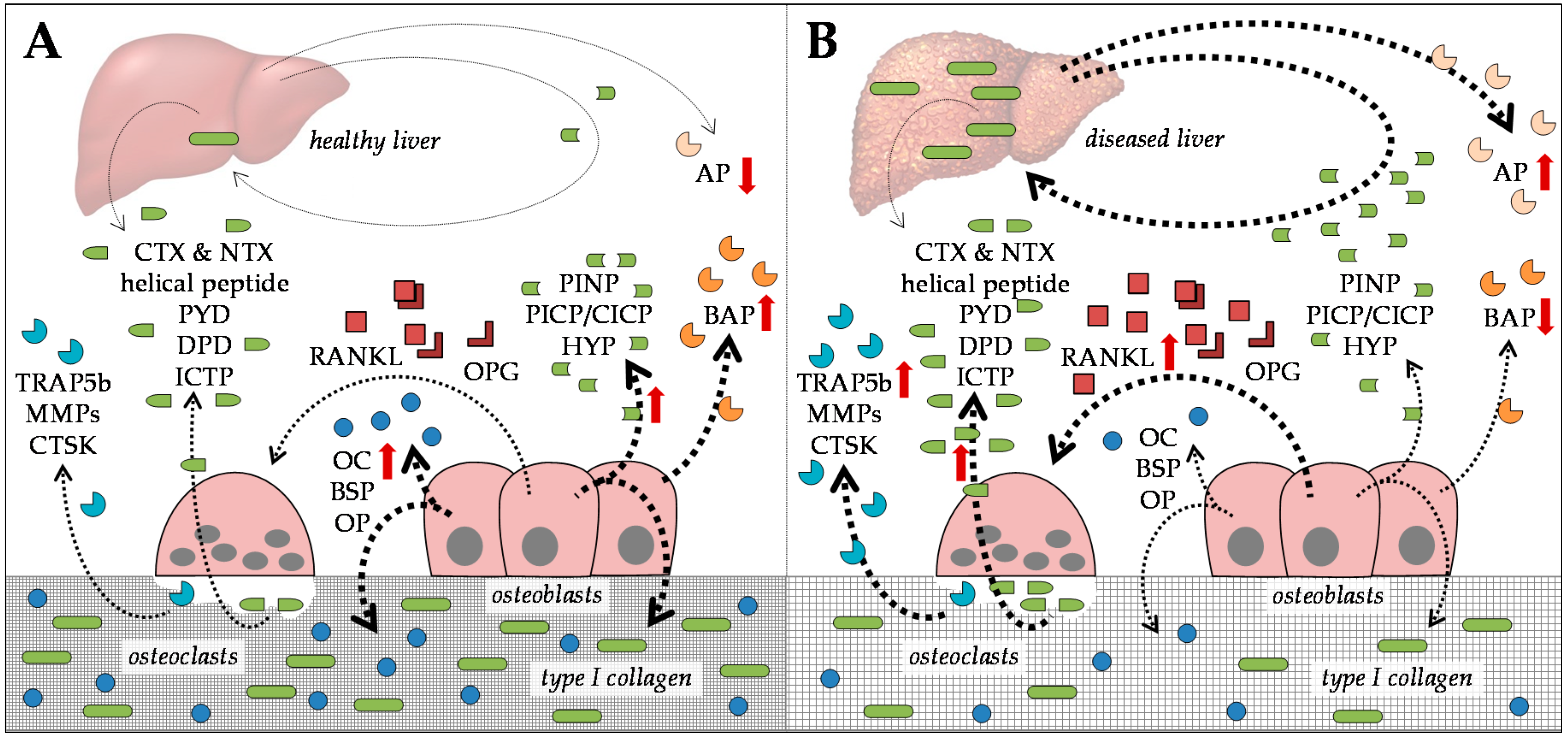
2.1. Serum Markers for Bone Formation
2.2. Serum Markers for Bone Degradation
3. Common Risk Factors Favoring the Development of HOD
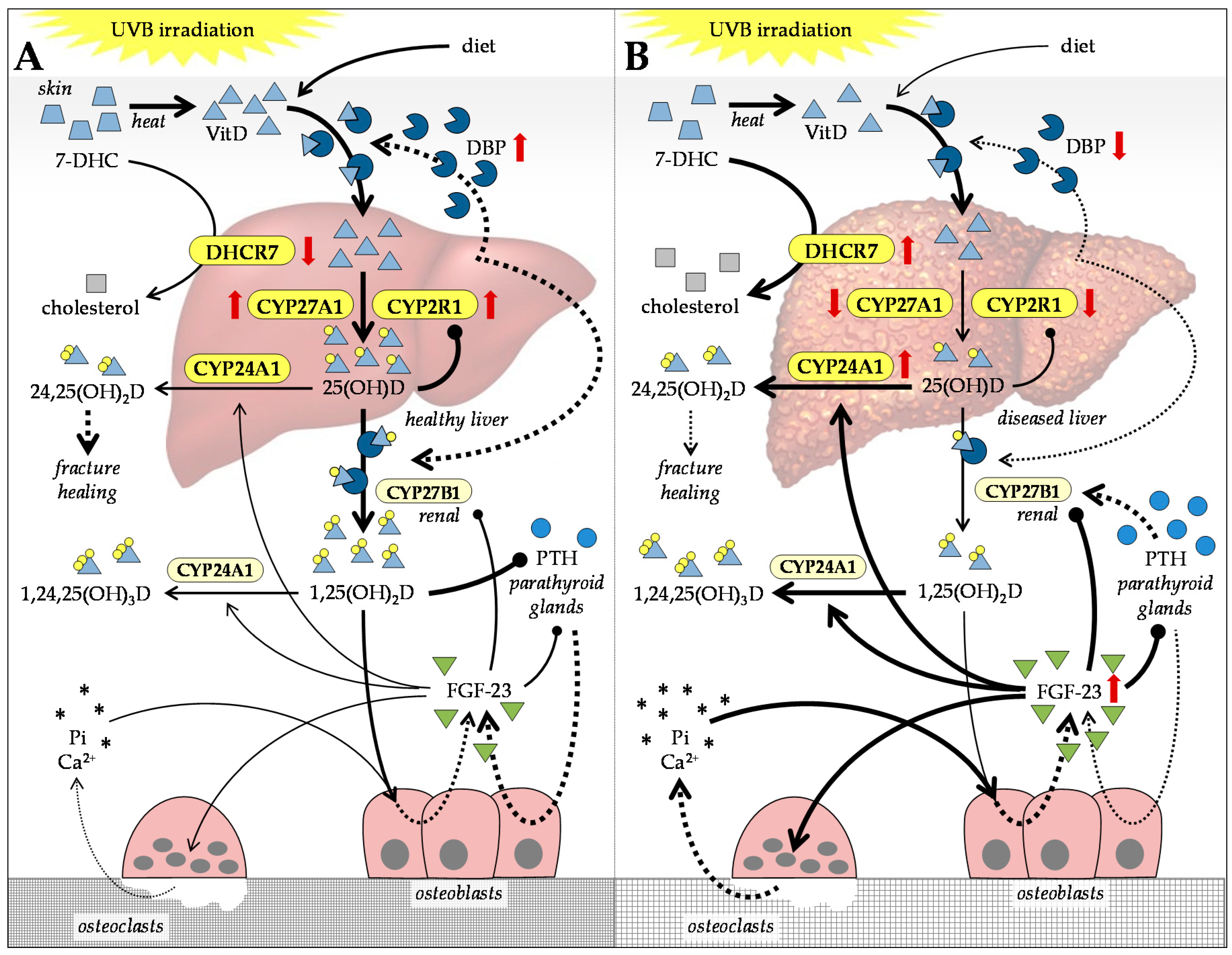
4. Alterations in Vitamin D and Calcium in Patients with CLD
4.1. Vitamin D Metabolism in Patients with CLD
4.2. VitD-Dependent Cellular Effects Affected in Patients with CLD
4.3. Balancing VitD Levels in Patients with CLD
4.4. Feedback Mechanisms Regulating VitD Levels in Patients with CLD
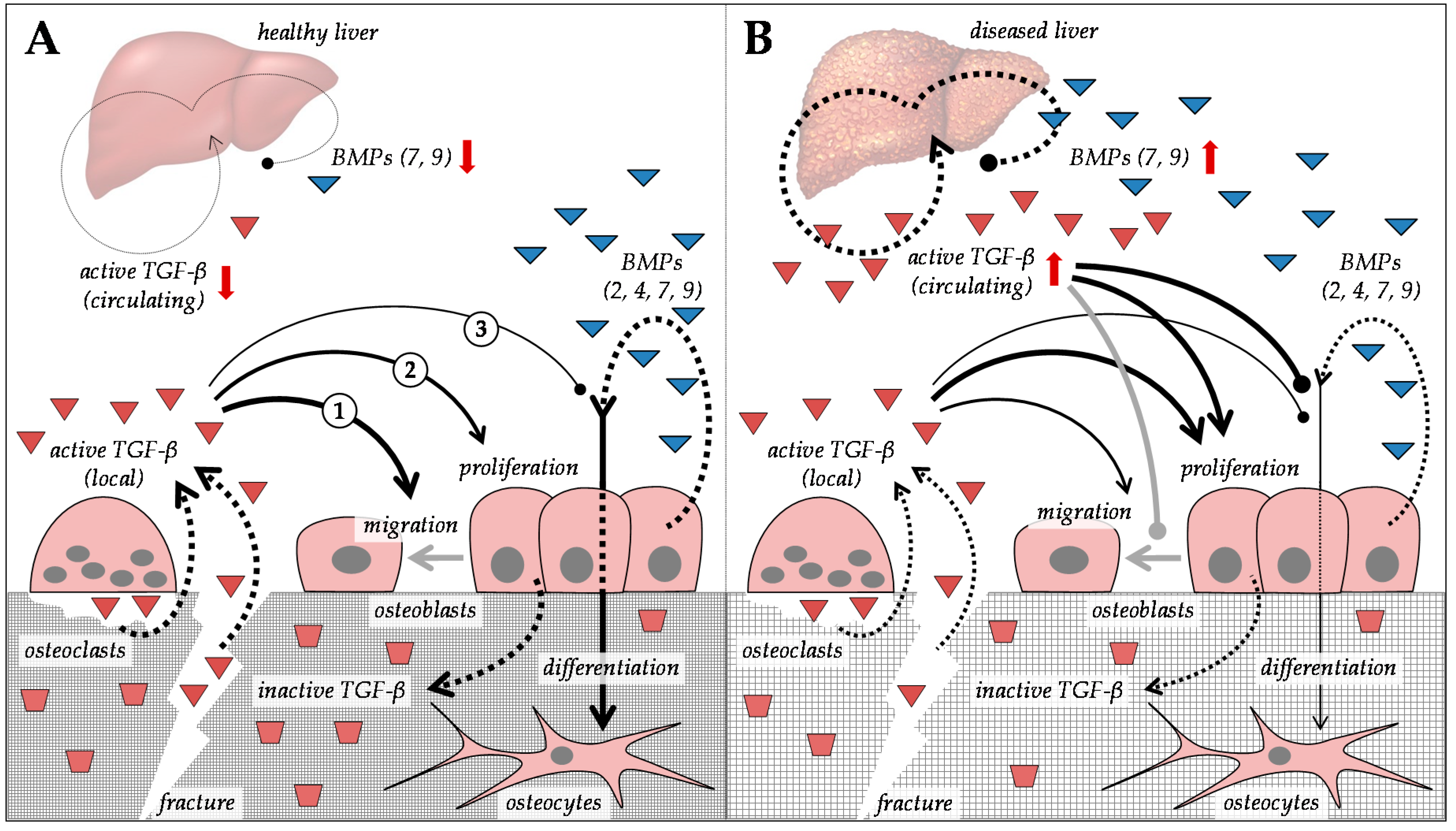
5. Alterations in Transforming Growth Factor-β Superfamily in Patients with CLD
5.1. Regulation of Extracellular Matrix Proteins by Members of the Transforming Growth Factor-β Superfamily
5.2. Regulation of TGF-β and BMP Signaling
5.3. HDACs as Possible Secondary Regulators for HOD
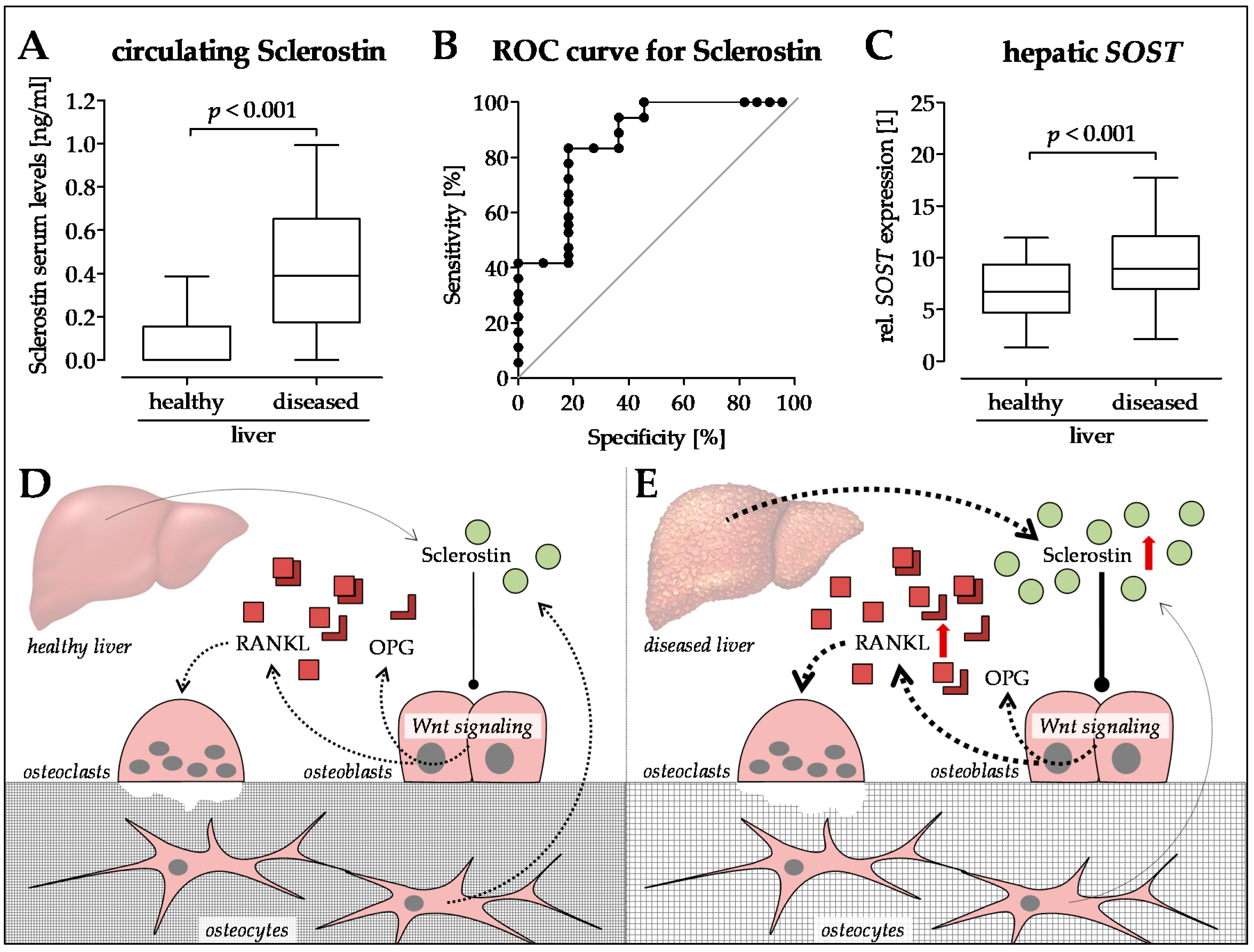
6. Sclerostin—A New Player in the Game?
7. Summary and Outlook
Author Contributions
Conflicts of Interest
Abbreviations
| 1,24,25(OH)D | 1,24,25-trihydroxyvitamin D |
| 1,25(OH)D | 1,25-dihydroxyvitamin D, also called calcitriol |
| 24,25(OH)D | 24,25-dihydroxyvitamin D |
| 25(OH)D | 25-hydroxyvitamin D, also called calcidiol |
| 7-DHC | 7-dehydrocholesterol |
| BAMBI | BMP and activin receptor membrane bound inhibitor |
| BAP | bone-specific alkaline phosphatase |
| BMD | bone mineral density |
| BMI | body mass index |
| BMP | bone morphogenetic protein |
| BSP | bone sialoprotein |
| CLD | chronic liver disease |
| CTSK | cathepsin K |
| CTX | C-telopeptide crosslinks of type I collagen |
| CYP24A1 | 25-hydroxyvitamin D 24-hydroxylase |
| CYP27A1 | sterol 27-hydroxylase |
| CYP27B1 | 25-hydroxyvitamin D-1α-hydroxylase |
| CYP2R1 | vitamin D-25-hydroxylase |
| CYP7A1 | cholesterol 7α-hydroxylase |
| DBP | vitamin-D-binding protein GC |
| DHCR7 | 7-dehydrocholesterol reductase |
| DKK1 | dickkopf 1 |
| DKK2 | dickkopf 2 |
| DPD | desoxypyridinolin |
| ECM | extracellular matrix |
| ELISA | enzyme-linked immunosorbent assay |
| EIA | (solid-phase) enzyme immunoassay |
| FGF | fibroblast growth factors |
| GH | growth hormone |
| HBV | hepatitis B virus |
| HCV | hepatitis C virus |
| HDAC | histone deacetylase |
| HOD | hepatic osteodystrophy |
| HYP | hydroxyprolin |
| ICTP | type I collagen cross-linked C-telopeptide |
| IGF-1 | insulin-like growth factor 1 |
| M-CSF | macrophage colony-stimulating factor |
| MMP | matrix metalloproteinase |
| MSC | mesenchymal stem/stromal cell |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NTX | N-telopeptide crosslinks of type I collagen |
| OC | osteocalcin |
| OLT | orthotopic liver transplantation |
| OP | osteopontin |
| OPG | osteoprotegerin |
| PBC | primary biliary cirrhosis |
| PDGF | platelet-derived growth factor |
| Pi | inorganic phosphate |
| PICP | type I collagen C-terminal propeptide |
| PINP | type I collagen N-terminal propeptide |
| PPARγ | peroxisome proliferator-activated receptor γ |
| PSC | primary sclerosing cholangitis |
| PTH | parathyroid hormone |
| PYD | pyridinolin |
| RANKL | receptor activator of nuclear factor kappa B ligand |
| RIA | radioimmunoassay |
| SARA | Smad anchor for receptor activation |
| Ski | v-ski sarcoma viral oncogene homolog |
| SnoN | Ski-like oncogene |
| TGF-β | transforming growth forming factor-β |
| TRAP5b | tartrate-resistant acid phosphatase isoform 5b |
| UVB | ultraviolet B |
| VDR | vitamin D receptor |
| VRDE | vitamin D response element |
| VitD | vitamin D |
| VitD2 | vitamin D2, also called ergocalciferol |
| VitD3 | vitamin D3, also called cholecalciferol |
| VitK | vitamin K |
Appendix A
| “hepatic osteodystrophy” | 145 hits |
| “liver disease” and “bone metabolism” | 80 hits |
| “liver disease” and “osteopenia” | 137 hits |
| “liver disease” and “osteoporosis” | 447 hits |
| “liver disease” and “fractures” | 225 hits |
| “hepatitis” and “bone metabolism” | 39 hits |
| “hepatitis” and “osteopenia” | 354 hits |
| “hepatitis” and “osteoporosis” | 340 hits |
| “hepatitis” and “fractures” | 193 hits |
| “liver fibrosis” or “liver cirrhosis” and “bone metabolism” | 48 hits |
| “liver fibrosis” or “liver cirrhosis” and “osteopenia” | 460 hits |
| “liver fibrosis” or “liver cirrhosis” and “osteoporosis” | 375 hits |
| “liver fibrosis” or “liver cirrhosis” and “fractures” | 162 hits |
References
- Angulo, P.; Grandison, G.A.; Fong, D.G.; Keach, J.C.; Lindor, K.D.; Bjornsson, E.; Koch, A. Bone disease in patients with primary sclerosing cholangitis. Gastroenterology 2011, 140, 180–188. [Google Scholar] [CrossRef]
- Guanabens, N.; Cerda, D.; Monegal, A.; Pons, F.; Caballeria, L.; Peris, P.; Pares, A. Low bone mass and severity of cholestasis affect fracture risk in patients with primary biliary cirrhosis. Gastroenterology 2010, 138, 2348–2356. [Google Scholar] [CrossRef] [PubMed]
- Nakchbandi, I.A.; van der Merwe, S.W. Current understanding of osteoporosis associated with liver disease. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 660–670. [Google Scholar] [CrossRef]
- Diamond, T.; Stiel, D.; Lunzer, M.; Wilkinson, M.; Roche, J.; Posen, S. Osteoporosis and skeletal fractures in chronic liver disease. Gut 1990, 31, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Wibaux, C.; Legroux-Gerot, I.; Dharancy, S.; Boleslawski, E.; Declerck, N.; Canva, V.; Mathurin, P.; Pruvot, F.R.; Cortet, B. Assessing bone status in patients awaiting liver transplantation. Jt. Bone Spine 2010, 78, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Liu, C.J.; Chen, T.J.; Chu, C.J.; Lin, H.C.; Lee, F.Y.; Su, T.P.; Lu, C.L. Increased incidence of orthopedic fractures in cirrhotic patients: A nationwide population-based study. J. Hepatol. 2013, 58, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Larramona, G.; Lucendo, A.J.; Gonzalez-Castillo, S.; Tenias, J.M. Hepatic osteodystrophy: An important matter for consideration in chronic liver disease. World J. Hepatol. 2011, 3, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M.; Loperto, I.; Camera, S.; Cossiga, V.; Di Somma, C.; Colao, A.; Caporaso, N.; Morisco, F. Osteoporosis across chronic liver disease. Osteoporos. Int. 2016, 27, 1967–1977. [Google Scholar] [CrossRef]
- Goel, V.; Kar, P. Hepatic osteodystrophy. Trop. Gastroenterol. 2010, 31, 82–86. [Google Scholar]
- Orsini, L.G.; Pinheiro, M.M.; Castro, C.H.; Silva, A.E.; Szejnfeld, V.L. Bone mineral density measurements, bone markers and serum vitamin d concentrations in men with chronic non-cirrhotic untreated hepatitis c. PLoS ONE 2013, 8, e81652. [Google Scholar] [CrossRef]
- El-Husseini, A.; Sabry, A.; Hassan, R.; Sobh, M. Effect of chronic hepatitis c virus infection on bone mineral density in pediatric renal transplant recipients. Saudi J. Kidney Dis. Transpl. 2013, 24, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, N.S.; Tomar, M.; Chawla, Y.K.; Bhadada, S.K.; Khandelwal, N.; Dhiman, R.K.; Duseja, A.; Bhansali, A. Hepatic osteodystrophy is common in patients with noncholestatic liver disease. Dig. Dis. Sci. 2011, 56, 3323–3327. [Google Scholar] [CrossRef]
- Keller, S.; Ittrich, H.; Schramm, C.; Lohse, A.W.; Amling, M.; Adam, G.; Yamamura, J. Diffusion-weighted mri for detection of hepatic osteodystrophy in primary sclerosing cholangitis: A comparison study with dual-energy X-ray absorptiometry. Jpn. J. Radiol. 2016, 34, 677–683. [Google Scholar] [CrossRef]
- Menon, K.V.; Angulo, P.; Weston, S.; Dickson, E.R.; Lindor, K.D. Bone disease in primary biliary cirrhosis: Independent indicators and rate of progression. J. Hepatol. 2001, 35, 316–323. [Google Scholar] [CrossRef]
- Mounach, A.; Ouzzif, Z.; Wariaghli, G.; Achemlal, L.; Benbaghdadi, I.; Aouragh, A.; Bezza, A.; El Maghraoui, A. Primary biliary cirrhosis and osteoporosis: A case-control study. J. Bone Min. Metab. 2008, 26, 379–384. [Google Scholar] [CrossRef]
- Lindor, K.D.; Janes, C.H.; Crippin, J.S.; Jorgensen, R.A.; Dickson, E.R. Bone disease in primary biliary cirrhosis: Does ursodeoxycholic acid make a difference? Hepatology 1995, 21, 389–392. [Google Scholar] [PubMed]
- Guanabens, N.; Pares, A.; Marinoso, L.; Brancos, M.A.; Piera, C.; Serrano, S.; Rivera, F.; Rodes, J. Factors influencing the development of metabolic bone disease in primary biliary cirrhosis. Am. J. Gastroenterol. 1990, 85, 1356–1362. [Google Scholar] [PubMed]
- Danford, C.J.; Trivedi, H.D.; Papamichael, K.; Tapper, E.B.; Bonder, A. Osteoporosis in primary biliary cholangitis. World J. Gastroenterol. 2018, 24, 3513–3520. [Google Scholar] [CrossRef] [PubMed]
- Savic, Z.; Damjanov, D.; Curic, N.; Kovacev-Zavisic, B.; Hadnadjev, L.; Novakovic-Paro, J.; Nikolic, S. Vitamin d status, bone metabolism and bone mass in patients with alcoholic liver cirrhosis. Bratisl. Lek. Listy 2014, 115, 573–578. [Google Scholar] [CrossRef]
- Malik, P.; Gasser, R.W.; Kemmler, G.; Moncayo, R.; Finkenstedt, G.; Kurz, M.; Fleischhacker, W.W. Low bone mineral density and impaired bone metabolism in young alcoholic patients without liver cirrhosis: A cross-sectional study. Alcohol. Clin. Exp. Res. 2009, 33, 375–381. [Google Scholar] [CrossRef]
- Diamond, T.; Stiel, D.; Lunzer, M.; Wilkinson, M.; Posen, S. Ethanol reduces bone formation and may cause osteoporosis. Am. J. Med. 1989, 86, 282–288. [Google Scholar] [CrossRef]
- Pardee, P.E.; Dunn, W.; Schwimmer, J.B. Non-alcoholic fatty liver disease is associated with low bone mineral density in obese children. Aliment. Pharm. Ther. 2012, 35, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kim, K.J.; Rhee, Y.; Lim, S.K. Significant liver fibrosis assessed using liver transient elastography is independently associated with low bone mineral density in patients with non-alcoholic fatty liver disease. PLoS ONE 2017, 12, e0182202. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Z.; Xu, Q.M.; Wu, X.X.; Cai, C.; Zhang, L.J.; Shi, K.Q.; Shi, H.Y.; Li, L.J. The combined effect of nonalcoholic fatty liver disease and metabolic syndrome on osteoporosis in postmenopausal females in eastern china. Int. J. Endocrinol. 2018, 2018, 2314769. [Google Scholar] [CrossRef] [PubMed]
- Diamond, T.; Stiel, D.; Posen, S. Osteoporosis in hemochromatosis: Iron excess, gonadal deficiency, or other factors? Ann. Intern. Med. 1989, 110, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Sinigaglia, L.; Fargion, S.; Fracanzani, A.L.; Binelli, L.; Battafarano, N.; Varenna, M.; Piperno, A.; Fiorelli, G. Bone and joint involvement in genetic hemochromatosis: Role of cirrhosis and iron overload. J. Rheumatol. 1997, 24, 1809–1813. [Google Scholar]
- Guggenbuhl, P.; Deugnier, Y.; Boisdet, J.F.; Rolland, Y.; Perdriger, A.; Pawlotsky, Y.; Chales, G. Bone mineral density in men with genetic hemochromatosis and hfe gene mutation. Osteoporos. Int. 2005, 16, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Van de Moortele, M.; Gotthardt, D.N.; Pfeiffenberger, J.; Seessle, J.; Ullrich, E.; Gielen, E.; Borghs, H.; Adriaens, E.; Stremmel, W.; et al. Bone demineralisation in a large cohort of wilson disease patients. J. Inherit. Metab. Dis. 2015, 38, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Quemeneur, A.S.; Trocello, J.M.; Ea, H.K.; Ostertag, A.; Leyendecker, A.; Duclos-Vallee, J.C.; de Vernejoul, M.C.; Woimant, F.; Liote, F. Bone status and fractures in 85 adults with wilson’s disease. Osteoporos. Int. 2014, 25, 2573–2580. [Google Scholar] [CrossRef] [PubMed]
- Selimoglu, M.A.; Ertekin, V.; Doneray, H.; Yildirim, M. Bone mineral density of children with wilson disease: Efficacy of penicillamine and zinc therapy. J. Clin. Gastroenterol. 2008, 42, 194–198. [Google Scholar] [CrossRef]
- Hegedus, D.; Ferencz, V.; Lakatos, P.L.; Meszaros, S.; Lakatos, P.; Horvath, C.; Szalay, F. Decreased bone density, elevated serum osteoprotegerin, and beta-cross-laps in wilson disease. J. Bone Min. Res. 2002, 17, 1961–1967. [Google Scholar] [CrossRef]
- Angulo, P.; Therneau, T.M.; Jorgensen, A.; DeSotel, C.K.; Egan, K.S.; Dickson, E.R.; Hay, J.E.; Lindor, K.D. Bone disease in patients with primary sclerosing cholangitis: Prevalence, severity and prediction of progression. J. Hepatol. 1998, 29, 729–735. [Google Scholar] [CrossRef]
- Duarte, M.P.; Farias, M.L.; Coelho, H.S.; Mendonca, L.M.; Stabnov, L.M.; do Carmo d Oliveira, M.; Lamy, R.A.; Oliveira, D.S. Calcium-parathyroid hormone-vitamin d axis and metabolic bone disease in chronic viral liver disease. J. Gastroenterol. Hepatol. 2001, 16, 1022–1027. [Google Scholar] [CrossRef]
- Auletta, M.; Nuzzo, V.; Esposito, A.; Antonello, S.; Lupoli, G.; Federico, F.; De Puente, A. Osteoporosis in men: A study in patients affected by chronic non-advanced liver disease. Clin. Cases Min. Bone Metab. 2005, 2, 25–28. [Google Scholar]
- Hofmann, W.P.; Kronenberger, B.; Bojunga, J.; Stamm, B.; Herrmann, E.; Bucker, A.; Mihm, U.; von Wagner, M.; Zeuzem, S.; Sarrazin, C. Prospective study of bone mineral density and metabolism in patients with chronic hepatitis c during pegylated interferon alpha and ribavirin therapy. J. Viral. Hepat. 2008, 15, 790–796. [Google Scholar]
- Lin, J.C.; Hsieh, T.Y.; Wu, C.C.; Chen, P.J.; Chueh, T.H.; Chang, W.K.; Chu, H.C. Association between chronic hepatitis c virus infection and bone mineral density. Calcif. Tissue Int. 2012, 91, 423–429. [Google Scholar] [CrossRef]
- Lai, J.C.; Shoback, D.M.; Zipperstein, J.; Lizaola, B.; Tseng, S.; Terrault, N.A. Bone mineral density, bone turnover, and systemic inflammation in non-cirrhotics with chronic hepatitis c. Dig. Dis. Sci. 2015, 60, 1813–1819. [Google Scholar] [CrossRef]
- Huang, Z.; Wei, H.; Cheng, C.; Yang, S.; Wang, J.; Liu, X. Low bone mineral density in chronic hepatitis b virus infection: A case-control study. Pak. J. Med. Sci. 2017, 33, 457–461. [Google Scholar] [CrossRef]
- Bering, T.; Diniz, K.G.D.; Coelho, M.P.P.; Vieira, D.A.; Soares, M.M.S.; Kakehasi, A.M.; Correia, M.; Teixeira, R.; Queiroz, D.M.M.; Rocha, G.A.; et al. Association between pre-sarcopenia, sarcopenia, and bone mineral density in patients with chronic hepatitis c. J. Cachexia Sarcopenia Muscle 2018, 9, 255–268. [Google Scholar] [CrossRef]
- Gallego-Rojo, F.J.; Gonzalez-Calvin, J.L.; Munoz-Torres, M.; Mundi, J.L.; Fernandez-Perez, R.; Rodrigo-Moreno, D. Bone mineral density, serum insulin-like growth factor i, and bone turnover markers in viral cirrhosis. Hepatology 1998, 28, 695–699. [Google Scholar] [CrossRef]
- George, J.; Ganesh, H.K.; Acharya, S.; Bandgar, T.R.; Shivane, V.; Karvat, A.; Bhatia, S.J.; Shah, S.; Menon, P.S.; Shah, N. Bone mineral density and disorders of mineral metabolism in chronic liver disease. World J. Gastroenterol. 2009, 15, 3516–3522. [Google Scholar] [CrossRef]
- Goubraim, R.; Kabbaj, N.; Salihoun, M.; Chaoui, Z.; Nya, M.; Amrani, N. Metabolic bone disease in viral cirrhosis: A prospective study. ISRN Hepatol. 2013, 2013, 276563. [Google Scholar] [CrossRef] [PubMed]
- Karoli, Y.; Karoli, R.; Fatima, J.; Manhar, M. Study of hepatic osteodystrophy in patients with chronic liver disease. J. Clin. Diagn. Res. 2016, 10, OC31–OC34. [Google Scholar] [CrossRef]
- Spencer, H.; Rubio, N.; Rubio, E.; Indreika, M.; Seitam, A. Chronic alcoholism. Frequently overlooked cause of osteoporosis in men. Am. J. Med. 1986, 80, 393–397. [Google Scholar] [CrossRef]
- Gonzalez-Calvin, J.L.; Garcia-Sanchez, A.; Bellot, V.; Munoz-Torres, M.; Raya-Alvarez, E.; Salvatierra-Rios, D. Mineral metabolism, osteoblastic function and bone mass in chronic alcoholism. Alcohol Alcohol. 1993, 28, 571–579. [Google Scholar]
- Kim, S.; Koga, T.; Isobe, M.; Kern, B.E.; Yokochi, T.; Chin, Y.E.; Karsenty, G.; Taniguchi, T.; Takayanagi, H. Stat1 functions as a cytoplasmic attenuator of runx2 in the transcriptional program of osteoblast differentiation. Genes Dev. 2003, 17, 1979–1991. [Google Scholar] [CrossRef] [PubMed]
- Neyeloff, J.L.; Fuchs, S.C.; Moreira, L.B. Meta-analyses and forest plots using a microsoft excel spreadsheet: Step-by-step guide focusing on descriptive data analysis. BMC Res. Notes 2012, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J. Posttransplantation bone disease. Transplantation 2005, 79, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Hamburg, S.M.; Piers, D.A.; van den Berg, A.P.; Slooff, M.J.; Haagsma, E.B. Bone mineral density in the long term after liver transplantation. Osteoporos. Int. 2000, 11, 600–606. [Google Scholar] [CrossRef]
- Loria, I.; Albanese, C.; Giusto, M.; Galtieri, P.A.; Giannelli, V.; Lucidi, C.; Di Menna, S.; Pirazzi, C.; Corradini, S.G.; Mennini, G.; et al. Bone disorders in patients with chronic liver disease awaiting liver transplantation. Transpl. Proc. 2010, 42, 1191–1193. [Google Scholar] [CrossRef]
- Guichelaar, M.M.; Kendall, R.; Malinchoc, M.; Hay, J.E. Bone mineral density before and after olt: Long-term follow-up and predictive factors. Liver Transpl. 2006, 12, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.E. Osteoporosis in liver diseases and after liver transplantation. J. Hepatol. 2003, 38, 856–865. [Google Scholar] [CrossRef]
- Ulivieri, F.M.; Silva, B.C.; Sardanelli, F.; Hans, D.; Bilezikian, J.P.; Caudarella, R. Utility of the trabecular bone score (tbs) in secondary osteoporosis. Endocrine 2014, 47, 435–448. [Google Scholar] [CrossRef]
- Sheu, A.; Diamond, T. Bone mineral density: Testing for osteoporosis. Aust. Prescr. 2016, 39, 35–39. [Google Scholar] [CrossRef]
- Coates, P. Bone turnover markers. Aust. Fam. Physician 2013, 42, 285–287. [Google Scholar]
- Nishizawa, Y.; Ohta, H.; Miura, M.; Inaba, M.; Ichimura, S.; Shiraki, M.; Takada, J.; Chaki, O.; Hagino, H.; Fujiwara, S.; et al. Guidelines for the use of bone metabolic markers in the diagnosis and treatment of osteoporosis (2012 edition). J. Bone Min. Metab. 2013, 31, 1–15. [Google Scholar] [CrossRef]
- Szulc, P. The role of bone turnover markers in monitoring treatment in postmenopausal osteoporosis. Clin. Biochem. 2012, 45, 907–919. [Google Scholar] [CrossRef]
- Szulc, P.; Delmas, P.D. Biochemical markers of bone turnover: Potential use in the investigation and management of postmenopausal osteoporosis. Osteoporos. Int. 2008, 19, 1683–1704. [Google Scholar] [CrossRef]
- Liu, T.; Wang, X.; Karsdal, M.A.; Leeming, D.J.; Genovese, F. Molecular serum markers of liver fibrosis. Biomark. Insights 2012, 7, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef]
- Naim, A.; Pan, Q.; Baig, M.S. Matrix metalloproteinases (mmps) in liver diseases. J. Clin. Exp. Hepatol. 2017, 7, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Hajiabbasi, A.; Shafaghi, A.; Fayazi, H.S.; Shenavar Masooleh, I.; Hedayati Emami, M.H.; Ghavidel Parsa, P.; Amir Maafi, A. The factors affecting bone density in cirrhosis. Hepat. Mon. 2015, 15, e26871. [Google Scholar] [CrossRef]
- Asomaning, K.; Bertone-Johnson, E.R.; Nasca, P.C.; Hooven, F.; Pekow, P.S. The association between body mass index and osteoporosis in patients referred for a bone mineral density examination. J. Womens Health 2006, 15, 1028–1034. [Google Scholar] [CrossRef]
- Collier, J. Bone disorders in chronic liver disease. Hepatology 2007, 46, 1271–1278. [Google Scholar] [CrossRef]
- Hay, J.E.; Guichelaar, M.M. Evaluation and management of osteoporosis in liver disease. Clin. Liver Dis. 2005, 9, 747–766. [Google Scholar] [CrossRef]
- Guanabens, N.; Pares, A.; Ros, I.; Caballeria, L.; Pons, F.; Vidal, S.; Monegal, A.; Peris, P.; Rodes, J. Severity of cholestasis and advanced histological stage but not menopausal status are the major risk factors for osteoporosis in primary biliary cirrhosis. J. Hepatol. 2005, 42, 573–577. [Google Scholar] [CrossRef]
- Gonzalez-Reimers, E.; Alvisa-Negrin, J.; Santolaria-Fernandez, F.; Candelaria Martin-Gonzalez, M.; Hernandez-Betancor, I.; Fernandez-Rodriguez, C.M.; Vina-Rodriguez, J.; Gonzalez-Diaz, A. Vitamin d and nutritional status are related to bone fractures in alcoholics. Alcohol Alcohol. 2011, 46, 148–155. [Google Scholar] [CrossRef]
- Santori, C.; Ceccanti, M.; Diacinti, D.; Attilia, M.L.; Toppo, L.; D’Erasmo, E.; Romagnoli, E.; Mascia, M.L.; Cipriani, C.; Prastaro, A.; et al. Skeletal turnover, bone mineral density, and fractures in male chronic abusers of alcohol. J. Endocrinol. Investig. 2008, 31, 321–326. [Google Scholar] [CrossRef]
- Sampson, H.W. Alcohol, osteoporosis, and bone regulating hormones. Alcohol. Clin. Exp. Res. 1997, 21, 400–403. [Google Scholar] [CrossRef]
- Laitinen, K.; Valimaki, M. Alcohol and bone. Calcif. Tissue Int. 1991, 49 (Suppl. 1), S70–S73. [Google Scholar] [CrossRef]
- Maran, A.; Zhang, M.; Spelsberg, T.C.; Turner, R.T. The dose-response effects of ethanol on the human fetal osteoblastic cell line. J. Bone Min. Res. 2001, 16, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, K.M.; Karlsson, C.; Ahlborg, H.G.; Valdimarsson, O.; Ljunghall, S.; Obrant, K.J. Bone turnover responses to changed physical activity. Calcif. Tissue Int. 2003, 72, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.A.; McCulloch, R.G. Bone tissue and physical activity. Can. J. Sport Sci. 1990, 15, 229–239. [Google Scholar] [PubMed]
- Scharf, J.G.; Schmitz, F.; Frystyk, J.; Skjaerbaek, C.; Moesus, H.; Blum, W.F.; Ramadori, G.; Hartmann, H. Insulin-like growth factor-i serum concentrations and patterns of insulin-like growth factor binding proteins in patients with chronic liver disease. J. Hepatol. 1996, 25, 689–699. [Google Scholar] [CrossRef]
- Foresta, C.; Schipilliti, M.; Ciarleglio, F.A.; Lenzi, A.; D’Amico, D. Male hypogonadism in cirrhosis and after liver transplantation. J. Endocrinol. Investig. 2008, 31, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Veldurthy, V.; Wei, R.; Oz, L.; Dhawan, P.; Jeon, Y.H.; Christakos, S. Vitamin d, calcium homeostasis and aging. Bone Res. 2016, 4, 16041. [Google Scholar] [CrossRef]
- Christakos, S.; Dhawan, P.; Porta, A.; Mady, L.J.; Seth, T. Vitamin d and intestinal calcium absorption. Mol. Cell. Endocrinol. 2011, 347, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Jamil, Z.; Arif, S.; Khan, A.; Durrani, A.A.; Yaqoob, N. Vitamin d deficiency and its relationship with child-pugh class in patients with chronic liver disease. J. Clin. Transl. Hepatol. 2018, 6, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Li, J.; Wang, J.H.; Habib, S.; Wei, W.; Sun, S.J.; Strobel, H.W.; Jia, J.D. Vitamin d serum level is associated with child-pugh score and metabolic enzyme imbalances, but not viral load in chronic hepatitis b patients. Medicine 2016, 95, e3926. [Google Scholar] [CrossRef]
- Corey, R.L.; Whitaker, M.D.; Crowell, M.D.; Keddis, M.T.; Aqel, B.; Balan, V.; Byrne, T.; Carey, E.; Douglas, D.D.; Harrison, M.E.; et al. Vitamin d deficiency, parathyroid hormone (pth) levels, and bone disease among patients with end stage liver disease (esld) and normal serum creatinine awaiting liver transplantation (lt). Clin. Transpl. 2014, 28, 579–584. [Google Scholar] [CrossRef]
- Garingarao, C.J.; Paz-Pacheco, E.; Jimeno, C.A. Primary hyperparathyroidism from a probable ectopic parathyroid adenoma with severe skeletal disease and vitamin d deficiency. BMJ Case Rep. 2014, 2014, bcr2014203716. [Google Scholar] [CrossRef]
- Nishiguchi, S.; Shimoi, S.; Kurooka, H.; Tamori, A.; Habu, D.; Takeda, T.; Kubo, S. Randomized pilot trial of vitamin k2 for bone loss in patients with primary biliary cirrhosis. J. Hepatol. 2001, 35, 543–545. [Google Scholar] [CrossRef]
- Levy, C.; Lindor, K.D. Management of osteoporosis, fat-soluble vitamin deficiencies, and hyperlipidemia in primary biliary cirrhosis. Clin. Liver Dis. 2003, 7, 901–910. [Google Scholar] [CrossRef]
- Plaza, S.M.; Lamson, D.W. Vitamin k2 in bone metabolism and osteoporosis. Altern. Med. Rev. 2005, 10, 24–35. [Google Scholar]
- Liu, Z.; Han, T.; Werner, H.; Rosen, C.J.; Schaffler, M.B.; Yakar, S. Reduced serum igf-1 associated with hepatic osteodystrophy is a main determinant of low cortical but not trabecular bone mass. J. Bone Min. Res. 2018, 33, 123–136. [Google Scholar] [CrossRef]
- Bachagol, D.; Joseph, G.S.; Ellur, G.; Patel, K.; Aruna, P.; Mittal, M.; China, S.P.; Singh, R.P.; Sharan, K. Stimulation of liver igf-1 expression promotes peak bone mass achievement in growing rats: A study with pomegranate seed oil. J. Nutr. Biochem. 2018, 52, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Marek, B.; Kajdaniuk, D.; Niedziolka, D.; Borgiel-Marek, H.; Nowak, M.; Sieminska, L.; Ostrowska, Z.; Glogowska-Szelag, J.; Piecha, T.; Otremba, L.; et al. Growth hormone/insulin-like growth factor-1 axis, calciotropic hormones and bone mineral density in young patients with chronic viral hepatitis. Endokrynol. Pol. 2015, 66, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. Tgf-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M.; Weiskirchen, R.; Breitkopf, K.; Dooley, S. Roles of tgf-beta in hepatic fibrosis. Front. Biosci. 2002, 7, d793–d807. [Google Scholar] [CrossRef]
- Breitkopf-Heinlein, K.; Meyer, C.; Konig, C.; Gaitantzi, H.; Addante, A.; Thomas, M.; Wiercinska, E.; Cai, C.; Li, Q.; Wan, F.; et al. Bmp-9 interferes with liver regeneration and promotes liver fibrosis. Gut 2017, 66, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Meurer, S.K.; Gressner, O.A.; Herrmann, J.; Borkham-Kamphorst, E.; Gressner, A.M. Bmp-7 as antagonist of organ fibrosis. Front. Biosci. 2009, 14, 4992–5012. [Google Scholar] [CrossRef]
- Diamond, T.; Pojer, R.; Stiel, D.; Alfrey, A.; Posen, S. Does iron affect osteoblast function? Studies in vitro and in patients with chronic liver disease. Calcif. Tissue Int. 1991, 48, 373–379. [Google Scholar] [CrossRef]
- Valenti, L.; Varenna, M.; Fracanzani, A.L.; Rossi, V.; Fargion, S.; Sinigaglia, L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos. Int. 2009, 20, 549–555. [Google Scholar] [CrossRef]
- Janes, C.H.; Dickson, E.R.; Okazaki, R.; Bonde, S.; McDonagh, A.F.; Riggs, B.L. Role of hyperbilirubinemia in the impairment of osteoblast proliferation associated with cholestatic jaundice. J. Clin. Investig. 1995, 95, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Shire, N.J.; Watts, N.B.; Schmitter, T.; Szabo, G.; Zucker, S.D. Hyperbilirubinemia is not a major contributing factor to altered bone mineral density in patients with chronic liver disease. J. Clin. Densitom. 2006, 9, 105–113. [Google Scholar] [CrossRef]
- Weinreb, M.; Pollak, R.D.; Ackerman, Z. Experimental cholestatic liver disease through bile-duct ligation in rats results in skeletal fragility and impaired osteoblastogenesis. J. Hepatol. 2004, 40, 385–390. [Google Scholar] [CrossRef]
- Christensen, M.H.; Apalset, E.M.; Nordbo, Y.; Varhaug, J.E.; Mellgren, G.; Lien, E.A. 1,25-dihydroxyvitamin d and the vitamin d receptor gene polymorphism apa1 influence bone mineral density in primary hyperparathyroidism. PLoS ONE 2013, 8, e56019. [Google Scholar] [CrossRef]
- Hay, J.E. Vitamin d receptor polymorphism and posttransplantation bone loss. Liver Transpl. 2001, 7, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Pares, A.; Guanabens, N.; Alvarez, L.; De Osaba, M.J.; Oriola, J.; Pons, F.; Caballeria, L.; Monegal, A.; Salvador, G.; Jo, J.; et al. Collagen type ialpha1 and vitamin d receptor gene polymorphisms and bone mass in primary biliary cirrhosis. Hepatology 2001, 33, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Pares, A.; Guanabens, N.; Rodes, J. Gene polymorphisms as predictors of decreased bone mineral density and osteoporosis in primary biliary cirrhosis. Eur. J. Gastroenterol. Hepatol. 2005, 17, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Springer, J.E.; Cole, D.E.; Rubin, L.A.; Cauch-Dudek, K.; Harewood, L.; Evrovski, J.; Peltekova, V.D.; Heathcote, E.J. Vitamin d-receptor genotypes as independent genetic predictors of decreased bone mineral density in primary biliary cirrhosis. Gastroenterology 2000, 118, 145–151. [Google Scholar] [CrossRef]
- Krishnamoorthy, T.L.; Miezynska-Kurtycz, J.; Hodson, J.; Gunson, B.K.; Neuberger, J.; Milkiewicz, P.; Oo, Y.H. Longterm corticosteroid use after liver transplantation for autoimmune hepatitis is safe and associated with a lower incidence of recurrent disease. Liver Transpl. 2016, 22, 34–41. [Google Scholar] [CrossRef]
- Bozkaya, G.; Nart, A.; Uslu, A.; Onman, T.; Aykas, A.; Dogan, M.; Karaca, B. Impact of calcineurin inhibitors on bone metabolism in primary kidney transplant patients. Transpl. Proc. 2008, 40, 151–155. [Google Scholar] [CrossRef]
- Lee, J.; Kim, J.H.; Kim, K.; Jin, H.M.; Lee, K.B.; Chung, D.J.; Kim, N. Ribavirin enhances osteoclast formation through osteoblasts via up-regulation of trance/rankl. Mol. Cell. Biochem. 2007, 296, 17–24. [Google Scholar] [CrossRef]
- Narayana, K.; D’Souza, U.J.; Seetharama Rao, K.P. The genotoxic and cytotoxic effects of ribavirin in rat bone marrow. Mutat. Res. 2002, 521, 179–185. [Google Scholar] [CrossRef]
- Solis-Herruzo, J.A.; Castellano, G.; Fernandez, I.; Munoz, R.; Hawkins, F. Decreased bone mineral density after therapy with alpha interferon in combination with ribavirin for chronic hepatitis c. J. Hepatol. 2000, 33, 812–817. [Google Scholar] [CrossRef]
- Gasser, R.W. Cholestasis and metabolic bone disease—A clinical review. Wien. Med. Wochenschr. 2008, 158, 553–557. [Google Scholar] [CrossRef]
- Wintermeyer, E.; Ihle, C.; Ehnert, S.; Stockle, U.; Ochs, G.; de Zwart, P.; Flesch, I.; Bahrs, C.; Nussler, A.K. Crucial role of vitamin d in the musculoskeletal system. Nutrients 2016, 8, 319. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin d: A d-lightful solution for health. J. Investig. Med. 2011, 59, 872–880. [Google Scholar] [CrossRef]
- Nussler, A.K.; Wildemann, B.; Freude, T.; Litzka, C.; Soldo, P.; Friess, H.; Hammad, S.; Hengstler, J.G.; Braun, K.F.; Trak-Smayra, V.; et al. Chronic ccl4 intoxication causes liver and bone damage similar to the human pathology of hepatic osteodystrophy: A mouse model to analyse the liver-bone axis. Arch. Toxicol. 2014, 88, 997–1006. [Google Scholar] [CrossRef]
- Hochrath, K.; Ehnert, S.; Ackert-Bicknell, C.L.; Lau, Y.; Schmid, A.; Krawczyk, M.; Hengstler, J.G.; Dunn, J.; Hiththetiya, K.; Rathkolb, B.; et al. Modeling hepatic osteodystrophy in abcb4 deficient mice. Bone 2013, 55, 501–511. [Google Scholar] [CrossRef]
- Arteh, J.; Narra, S.; Nair, S. Prevalence of vitamin d deficiency in chronic liver disease. Dig. Dis. Sci. 2010, 55, 2624–2628. [Google Scholar] [CrossRef] [PubMed]
- Deluca, H.F. History of the discovery of vitamin d and its active metabolites. BoneKEy Rep. 2014, 3, 479. [Google Scholar] [CrossRef] [PubMed]
- Shinchuk, L.; Holick, M.F. Vitamin d and rehabilitation: Improving functional outcomes. Nutr. Clin. Pr. 2007, 22, 297–304. [Google Scholar] [CrossRef]
- Booth, D.R.; Ding, N.; Parnell, G.P.; Shahijanian, F.; Coulter, S.; Schibeci, S.D.; Atkins, A.R.; Stewart, G.J.; Evans, R.M.; Downes, M.; et al. Cistromic and genetic evidence that the vitamin d receptor mediates susceptibility to latitude-dependent autoimmune diseases. Genes Immun. 2016, 17, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Kim, S.H.; Lee, N.; Lee, W.W.; Hwang, K.A.; Shin, M.S.; Lee, S.H.; Kim, W.U.; Kang, I. 1,25-dihyroxyvitamin d3 promotes foxp3 expression via binding to vitamin d response elements in its conserved noncoding sequence region. J. Immunol. 2012, 188, 5276–5282. [Google Scholar] [CrossRef]
- Zittermann, A.; Kuhn, J.; Ernst, J.B.; Becker, T.; Larisch, J.; Dreier, J.; Knabbe, C.; Borgermann, J.; Gummert, J.F. Circulating 25-hydroxyvitamin d and 1,25-dihydroxyvitamin d concentrations and postoperative infections in cardiac surgical patients: The calcitop-study. PLoS ONE 2016, 11, e0158532. [Google Scholar] [CrossRef]
- Shymanskyi, I.; Lisakovska, O.; Mazanova, A.; Labudzynskyi, D.; Veliky, M. Vitamin d3 modulates impaired crosstalk between rank and glucocorticoid receptor signaling in bone marrow cells after chronic prednisolone administration. Front. Endocrinol. 2018, 9, 303. [Google Scholar] [CrossRef]
- Wacker, M.; Holick, M.F. Vitamin d-effects on skeletal and extraskeletal health and the need for supplementation. Nutrients 2013, 5, 111–148. [Google Scholar] [CrossRef]
- Takahashi, N.; Udagawa, N.; Suda, T. Vitamin d endocrine system and osteoclasts. BoneKEy Rep. 2014, 3, 495. [Google Scholar] [CrossRef]
- Mercer, K.E.; Wynne, R.A.; Lazarenko, O.P.; Lumpkin, C.K.; Hogue, W.R.; Suva, L.J.; Chen, J.R.; Mason, A.Z.; Badger, T.M.; Ronis, M.J. Vitamin d supplementation protects against bone loss associated with chronic alcohol administration in female mice. J. Pharm. Exp. Ther. 2012, 343, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Rode, A.; Fourlanos, S.; Nicoll, A. Oral vitamin d replacement is effective in chronic liver disease. Gastroenterol. Clin. Biol. 2010, 34, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, N.M.; Sakhaee, K. Treatment of osteoporosis in patients with chronic liver disease and in liver transplant recipients. Curr. Treat. Opt. Gastroenterol. 2006, 9, 456–463. [Google Scholar] [CrossRef]
- Yurci, A.; Kalkan, A.O.; Ozbakir, O.; Karaman, A.; Torun, E.; Kula, M.; Baskol, M.; Gursoy, S.; Yucesoy, M.; Bayram, F. Efficacy of different therapeutic regimens on hepatic osteodystrophy in chronic viral liver disease. Eur. J. Gastroenterol. Hepatol. 2011, 23, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Gomez, J.M.; Bouillon, R. Is calcifediol better than cholecalciferol for vitamin d supplementation? Osteoporos. Int. 2018, 29, 1697–1711. [Google Scholar] [CrossRef]
- Cesareo, R.; Attanasio, R.; Caputo, M.; Castello, R.; Chiodini, I.; Falchetti, A.; Guglielmi, R.; Papini, E.; Santonati, A.; Scillitani, A.; et al. Italian association of clinical endocrinologists (ame) and italian chapter of the american association of clinical endocrinologists (aace) position statement: Clinical management of vitamin d deficiency in adults. Nutrients 2018, 10, 546. [Google Scholar] [CrossRef] [PubMed]
- Pilz, S.; Zittermann, A.; Trummer, C.; Theiler-Schwetz, V.; Lerchbaum, E.; Keppel, M.H.; Grubler, M.R.; Marz, W.; Pandis, M. Vitamin d testing and treatment: A narrative review of current evidence. Endocr. Connect. 2019, 8, R27–R43. [Google Scholar] [CrossRef]
- Guanabens, N.; Pares, A. Management of osteoporosis in liver disease. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 438–445. [Google Scholar] [CrossRef]
- Luxon, B.A. Bone disorders in chronic liver diseases. Curr. Gastroenterol. Rep. 2012, 13, 40–48. [Google Scholar] [CrossRef]
- Rudic, J.S.; Giljaca, V.; Krstic, M.N.; Bjelakovic, G.; Gluud, C. Bisphosphonates for osteoporosis in primary biliary cirrhosis. Cochrane Database Syst. Rev. 2011, 12, CD009144. [Google Scholar]
- Wariaghli, G.; Allali, F.; El Maghraoui, A.; Hajjaj-Hassouni, N. Osteoporosis in patients with primary biliary cirrhosis. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Danford, C.J.; Trivedi, H.D.; Bonder, A. Bone health in patients with liver diseases. J. Clin. Densitom. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.L.M.; MacDonald, P.N. Vitamin d: More than a “bone-a-fide” hormone. Mol. Endocrinol. 2003, 17, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Lavi-Moshayoff, V.; Wasserman, G.; Meir, T.; Silver, J.; Naveh-Many, T. Pth increases fgf23 gene expression and mediates the high-fgf23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am. J. Physiol. Ren. Physiol 2010, 299, F882–F889. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Naveh-Many, T. Fgf23 and the parathyroid glands. Pediatr. Nephrol. 2010, 25, 2241–2245. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.S.; Brown, A.J. Direct suppression of pth gene expression by the vitamin d prohormones doxercalciferol and calcidiol requires the vitamin d receptor. J. Mol. Endocrinol. 2011, 46, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin d metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Aslan, D.; Andersen, M.D.; Gede, L.B.; de Franca, T.K.; Jorgensen, S.R.; Schwarz, P.; Jorgensen, N.R. Mechanisms for the bone anabolic effect of parathyroid hormone treatment in humans. Scand. J. Clin. Lab. Investig. 2012, 72, 14–22. [Google Scholar] [CrossRef]
- Anagnostis, P.; Gkekas, N.K.; Potoupnis, M.; Kenanidis, E.; Tsiridis, E.; Goulis, D.G. New therapeutic targets for osteoporosis. Maturitas 2019, 120, 1–6. [Google Scholar] [CrossRef]
- Dresner-Pollak, R.; Gabet, Y.; Steimatzky, A.; Hamdani, G.; Bab, I.; Ackerman, Z.; Weinreb, M. Human parathyroid hormone 1-34 prevents bone loss in experimental biliary cirrhosis in rats. Gastroenterology 2008, 134, 259–267. [Google Scholar] [CrossRef]
- Prie, D.; Forand, A.; Francoz, C.; Elie, C.; Cohen, I.; Courbebaisse, M.; Eladari, D.; Lebrec, D.; Durand, F.; Friedlander, G. Plasma fibroblast growth factor 23 concentration is increased and predicts mortality in patients on the liver-transplant waiting list. PLoS ONE 2013, 8, e66182. [Google Scholar] [CrossRef]
- He, X.; Shen, Y.; Ma, X.; Ying, L.; Peng, J.; Pan, X.; Bao, Y.; Zhou, J. The association of serum fgf23 and non-alcoholic fatty liver disease is independent of vitamin d in type 2 diabetes patients. Clin. Exp. Pharm. Physiol. 2018, 45, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Bihari, C.; Lal, D.; Thakur, M.; Sukriti, S.; Mathur, D.; Patil, A.G.; Anand, L.; Kumar, G.; Sharma, S.; Thapar, S.; et al. Suboptimal level of bone-forming cells in advanced cirrhosis are associated with hepatic osteodystrophy. Hepatol. Commun. 2018, 2, 1095–1110. [Google Scholar] [CrossRef]
- Tejwani, V.; Qian, Q. Calcium regulation and bone mineral metabolism in elderly patients with chronic kidney disease. Nutrients 2013, 5, 1913–1936. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Lopez, N.; Roman-Garcia, P.; Rodriguez-Rebollar, A.; Fernandez-Martin, J.L.; Naves-Diaz, M.; Cannata-Andia, J.B. Indirect regulation of pth by estrogens may require fgf23. J. Am. Soc. Nephrol. 2009, 20, 2009–2017. [Google Scholar] [CrossRef] [PubMed]
- Carpinteri, R.; Porcelli, T.; Mejia, C.; Patelli, I.; Bilezikian, J.P.; Canalis, E.; Angeli, A.; Giustina, A.; Mazziotti, G. Glucocorticoid-induced osteoporosis and parathyroid hormone. J. Endocrinol. Investig. 2010, 33, 16–21. [Google Scholar]
- Felsenfeld, A.J.; Levine, B.S. Calcitonin, the forgotten hormone: Does it deserve to be forgotten? Clin. Kidney J. 2015, 8, 180–187. [Google Scholar] [CrossRef]
- Cochran, M.; Peacock, M.; Sachs, G.; Nordin, B.E. Renal effects of calcitonin. Br. Med. J. 1970, 1, 135–137. [Google Scholar] [CrossRef]
- Mirzaei, S.; Krotla, G.; Knoll, P.; Koriska, K.; Kohn, H. Possible effect of calcitonin deficiency on bone mass after subtotal thyroidectomy. Acta Med. Austriaca 1999, 26, 29–31. [Google Scholar] [PubMed]
- Giannini, S.; Nobile, M.; Sartori, L.; Binotto, P.; Ciuffreda, M.; Gemo, G.; Pelizzo, M.R.; D’Angelo, A.; Crepaldi, G. Bone density and mineral metabolism in thyroidectomized patients treated with long-term l-thyroxine. Clin. Sci. 1994, 87, 593–597. [Google Scholar] [CrossRef]
- Gonzalez, D.C.; Mautalen, C.A.; Correa, P.H.; el Tamer, E.; el Tamer, S. Bone mass in totally thyroidectomized patients. Role of calcitonin deficiency and exogenous thyroid treatment. Acta Endocrinol. 1991, 124, 521–525. [Google Scholar] [CrossRef][Green Version]
- Novince, C.M.; Michalski, M.N.; Koh, A.J.; Sinder, B.P.; Entezami, P.; Eber, M.R.; Pettway, G.J.; Rosol, T.J.; Wronski, T.J.; Kozloff, K.M.; et al. Proteoglycan 4: A dynamic regulator of skeletogenesis and parathyroid hormone skeletal anabolism. J. Bone Min. Res. 2013, 27, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Carey, R.M.; Siragy, H.M. The intrarenal renin-angiotensin system and diabetic nephropathy. Trends Endocrin. Met. 2003, 14, 274–281. [Google Scholar] [CrossRef]
- Williams, S.; Malatesta, K.; Norris, K. Vitamin d and chronic kidney disease. Ethn. Dis. 2009, 19, As8–As11. [Google Scholar]
- Putz-Bankuti, C.; Pilz, S.; Stojakovic, T.; Scharnagl, H.; Pieber, T.R.; Trauner, M.; Obermayer-Pietsch, B.; Stauber, R.E. Association of 25-hydroxyvitamin d levels with liver dysfunction and mortality in chronic liver disease. Liver Int. 2012, 32, 845–851. [Google Scholar] [CrossRef]
- Li, Y.C. Renoprotective effects of vitamin d analogs. Kidney Int. 2010, 78, 134–139. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; Consortium, I.-L. Tgf-beta signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef]
- Weng, H.L.; Liu, Y.; Chen, J.L.; Huang, T.; Xu, L.J.; Godoy, P.; Hu, J.H.; Zhou, C.; Stickel, F.; Marx, A.; et al. The etiology of liver damage imparts cytokines transforming growth factor beta1 or interleukin-13 as driving forces in fibrogenesis. Hepatology 2009, 50, 230–243. [Google Scholar] [CrossRef]
- Bonewald, L.F.; Dallas, S.L. Role of active and latent transforming growth factor beta in bone formation. J. Cell. Biochem. 1994, 55, 350–357. [Google Scholar] [CrossRef]
- Erlebacher, A.; Filvaroff, E.H.; Ye, J.Q.; Derynck, R. Osteoblastic responses to tgf-beta during bone remodeling. Mol. Biol. Cell 1998, 9, 1903–1918. [Google Scholar] [CrossRef]
- Robey, P.G.; Young, M.F.; Flanders, K.C.; Roche, N.S.; Kondaiah, P.; Reddi, A.H.; Termine, J.D.; Sporn, M.B.; Roberts, A.B. Osteoblasts synthesize and respond to transforming growth factor-type beta (tgf-beta) in vitro. J. Cell Biol. 1987, 105, 457–463. [Google Scholar] [CrossRef]
- Pfeilschifter, J.; Bonewald, L.; Mundy, G.R. Characterization of the latent transforming growth factor beta complex in bone. J. Bone Min. Res. 1990, 5, 49–58. [Google Scholar] [CrossRef]
- Oursler, M.J. Osteoclast synthesis and secretion and activation of latent transforming growth factor beta. J. Bone Min. Res. 1994, 9, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Sivakumar, P.; Jones, C.J.; Chen, Q.; Peters, D.M.; Mosher, D.F.; Humphries, M.J.; Kielty, C.M. Fibronectin regulates latent transforming growth factor-beta (tgf beta) by controlling matrix assembly of latent tgf beta-binding protein-1. J. Biol. Chem. 2005, 280, 18871–18880. [Google Scholar] [CrossRef]
- Harris, S.E.; Bonewald, L.F.; Harris, M.A.; Sabatini, M.; Dallas, S.; Feng, J.Q.; Ghosh-Choudhury, N.; Wozney, J.; Mundy, G.R. Effects of transforming growth factor beta on bone nodule formation and expression of bone morphogenetic protein 2, osteocalcin, osteopontin, alkaline phosphatase, and type i collagen mrna in long-term cultures of fetal rat calvarial osteoblasts. J. Bone Min. Res. 1994, 9, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Ignotz, R.A.; Massague, J. Cell adhesion protein receptors as targets for transforming growth factor-beta action. Cell 1987, 51, 189–197. [Google Scholar] [CrossRef]
- Noda, M.; Camilliere, J.J. In vivo stimulation of bone formation by transforming growth factor-beta. Endocrinology 1989, 124, 2991–2994. [Google Scholar] [CrossRef]
- Erlebacher, A.; Derynck, R. Increased expression of tgf-beta 2 in osteoblasts results in an osteoporosis-like phenotype. J. Cell Biol. 1996, 132, 195–210. [Google Scholar] [CrossRef]
- Hata, K.; Takahata, Y.; Murakami, T.; Nishimura, R. Transcriptional network controlling endochondral ossification. J. Bone Metab. 2017, 24, 75–82. [Google Scholar] [CrossRef]
- Zimmermann, G.; Henle, P.; Kusswetter, M.; Moghaddam, A.; Wentzensen, A.; Richter, W.; Weiss, S. Tgf-beta1 as a marker of delayed fracture healing. Bone 2005, 36, 779–785. [Google Scholar] [CrossRef]
- Lind, M. Growth factor stimulation of bone healing. Effects on osteoblasts, osteomies, and implants fixation. Acta Orthop. Scand. Suppl. 1998, 283, 2–37. [Google Scholar]
- Lian, N.; Lin, T.; Liu, W.; Wang, W.; Li, L.; Sun, S.; Nyman, J.S.; Yang, X. Transforming growth factor beta suppresses osteoblast differentiation via the vimentin activating transcription factor 4 (atf4) axis. J. Biol. Chem. 2012, 287, 35975–35984. [Google Scholar] [CrossRef]
- Lian, N.; Wang, W.; Li, L.; Elefteriou, F.; Yang, X. Vimentin inhibits atf4-mediated osteocalcin transcription and osteoblast differentiation. J. Biol. Chem. 2009, 284, 30518–30525. [Google Scholar] [CrossRef]
- Nishiguchi, M.; Yuasa, K.; Saito, K.; Fukumoto, E.; Yamada, A.; Hasegawa, T.; Yoshizaki, K.; Kamasaki, Y.; Nonaka, K.; Fujiwara, T.; et al. Amelogenin is a negative regulator of osteoclastogenesis via downregulation of rankl, m-csf and fibronectin expression in osteoblasts. Arch. Oral Biol. 2007, 52, 237–243. [Google Scholar] [CrossRef]
- Ventura, E.; Weller, M.; Macnair, W.; Eschbach, K.; Beisel, C.; Cordazzo, C.; Claassen, M.; Zardi, L.; Burghardt, I. Tgf-beta induces oncofetal fibronectin that, in turn, modulates tgf-beta superfamily signaling in endothelial cells. J. Cell Sci. 2018, 131, jcs209619. [Google Scholar] [CrossRef]
- Sens, C.; Altrock, E.; Rau, K.; Klemis, V.; von Au, A.; Pettera, S.; Uebel, S.; Damm, T.; Tiwari, S.; Moser, M.; et al. An o-glycosylation of fibronectin mediates hepatic osteodystrophy through alpha4beta1 integrin. J. Bone Min. Res. 2017, 32, 70–81. [Google Scholar] [CrossRef]
- Kawelke, N.; Bentmann, A.; Hackl, N.; Hager, H.D.; Feick, P.; Geursen, A.; Singer, M.V.; Nakchbandi, I.A. Isoform of fibronectin mediates bone loss in patients with primary biliary cirrhosis by suppressing bone formation. J. Bone Min. Res. 2008, 23, 1278–1286. [Google Scholar] [CrossRef]
- Ehnert, S.; Zhao, J.; Pscherer, S.; Freude, T.; Dooley, S.; Kolk, A.; Stockle, U.; Nussler, A.K.; Hube, R. Transforming growth factor beta1 inhibits bone morphogenic protein (bmp)-2 and bmp-7 signaling via upregulation of ski-related novel protein n (snon): Possible mechanism for the failure of bmp therapy? BMC Med. 2012, 10, 101. [Google Scholar] [CrossRef]
- Sosa, I.; Cvijanovic, O.; Celic, T.; Cuculic, D.; Crncevic-Orlic, Z.; Vukelic, L.; Zoricic Cvek, S.; Dudaric, L.; Bosnar, A.; Bobinac, D. Hepatoregenerative role of bone morphogenetic protein-9. Med. Sci. Monit. 2011, 17, HY33–HY35. [Google Scholar] [CrossRef][Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and smad-independent pathways in tgf-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of tgf-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Mueller, T.D.; Nickel, J. Promiscuity and specificity in bmp receptor activation. FEBS Lett. 2012, 586, 1846–1859. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Shinozaki, M.; Hara, T.; Furuya, T.; Miyazono, K. Two major smad pathways in tgf-beta superfamily signalling. Genes Cells 2002, 7, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.P. Tgf-beta and bmp signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef]
- Zhu, W.; Kim, J.; Cheng, C.; Rawlins, B.A.; Boachie-Adjei, O.; Crystal, R.G.; Hidaka, C. Noggin regulation of bone morphogenetic protein (bmp) 2/7 heterodimer activity in vitro. Bone 2006, 39, 61–71. [Google Scholar] [CrossRef]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Alliston, T.; Delston, R.; Derynck, R. Repression of runx2 function by tgf-beta through recruitment of class ii histone deacetylases by smad3. EMBO J. 2005, 24, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- Ehnert, S.; Sreekumar, V.; Aspera-Werz, R.H.; Sajadian, S.O.; Wintermeyer, E.; Sandmann, G.H.; Bahrs, C.; Hengstler, J.G.; Godoy, P.; Nussler, A.K. Tgf-beta1 impairs mechanosensation of human osteoblasts via hdac6-mediated shortening and distortion of primary cilia. J. Mol. Med. 2017, 95, 653–663. [Google Scholar] [CrossRef]
- Ehnert, S.; Heuberger, E.; Linnemann, C.; Nussler, A.K.; Pscherer, S. Tgf-β1-dependent downregulation of hdac9 inhibits maturation of human osteoblasts. J. Funct. Morphol. Kinesiol. 2017, 2, 41. [Google Scholar] [CrossRef]
- Choo, M.K.; Yeo, H.; Zayzafoon, M. Nfatc1 mediates hdac-dependent transcriptional repression of osteocalcin expression during osteoblast differentiation. Bone 2009, 45, 579–589. [Google Scholar] [CrossRef]
- Jensen, E.D.; Schroeder, T.M.; Bailey, J.; Gopalakrishnan, R.; Westendorf, J.J. Histone deacetylase 7 associates with runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J. Bone Min. Res. 2008, 23, 361–372. [Google Scholar] [CrossRef]
- Vrtacnik, P.; Zupan, J.; Mlakar, V.; Kranjc, T.; Marc, J.; Kern, B.; Ostanek, B. Epigenetic enzymes influenced by oxidative stress and hypoxia mimetic in osteoblasts are differentially expressed in patients with osteoporosis and osteoarthritis. Sci. Rep. 2018, 8, 16215. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; McKinsey, T.A.; Zhang, C.L.; Richardson, J.A.; Hill, J.A.; Olson, E.N. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol. Cell. Biol. 2004, 24, 8467–8476. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wei, X.; Wang, S.; Jiao, Q.; Zhang, Y.; Du, G.; Wang, X.; Wei, F.; Zhang, J.; Wei, L. Compression regulates gene expression of chondrocytes through hdac4 nuclear relocation via pp2a-dependent hdac4 dephosphorylation. Biochim. Biophys. Acta 2016, 1863, 1633–1642. [Google Scholar] [CrossRef]
- Guan, Y.; Chen, Q.; Yang, X.; Haines, P.; Pei, M.; Terek, R.; Wei, X.; Zhao, T.; Wei, L. Subcellular relocation of histone deacetylase 4 regulates growth plate chondrocyte differentiation through Ca2+/calmodulin-dependent kinase iv. Am. J. Physiol. Cell Physiol. 2012, 303, C33–C40. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, T.; Chen, T.; Johnson, J.; Westendorf, J.J.; Partridge, N.C. The deletion of hdac4 in mouse osteoblasts influences both catabolic and anabolic effects in bone. J. Bone Min. Res. 2018, 33, 1362–1375. [Google Scholar] [CrossRef]
- Feigenson, M.; Shull, L.C.; Taylor, E.L.; Camilleri, E.T.; Riester, S.M.; van Wijnen, A.J.; Bradley, E.W.; Westendorf, J.J. Histone deacetylase 3 deletion in mesenchymal progenitor cells hinders long bone development. J. Bone Min. Res. 2017, 32, 2453–2465. [Google Scholar] [CrossRef]
- Razidlo, D.F.; Whitney, T.J.; Casper, M.E.; McGee-Lawrence, M.E.; Stensgard, B.A.; Li, X.; Secreto, F.J.; Knutson, S.K.; Hiebert, S.W.; Westendorf, J.J. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PLoS ONE 2010, 5, e11492. [Google Scholar] [CrossRef] [PubMed]
- Knutson, S.K.; Chyla, B.J.; Amann, J.M.; Bhaskara, S.; Huppert, S.S.; Hiebert, S.W. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008, 27, 1017–1028. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef]
- McGee-Lawrence, M.E.; Carpio, L.R.; Schulze, R.J.; Pierce, J.L.; McNiven, M.A.; Farr, J.N.; Khosla, S.; Oursler, M.J.; Westendorf, J.J. Hdac3 deficiency increases marrow adiposity and induces lipid storage and glucocorticoid metabolism in osteochondroprogenitor cells. J. Bone Min. Res. 2016, 31, 116–128. [Google Scholar] [CrossRef]
- Zimmermann, S.; Kiefer, F.; Prudenziati, M.; Spiller, C.; Hansen, J.; Floss, T.; Wurst, W.; Minucci, S.; Gottlicher, M. Reduced body size and decreased intestinal tumor rates in hdac2-mutant mice. Cancer Res. 2007, 67, 9047–9054. [Google Scholar] [CrossRef]
- Stemig, M.; Astelford, K.; Emery, A.; Cho, J.J.; Allen, B.; Huang, T.H.; Gopalakrishnan, R.; Mansky, K.C.; Jensen, E.D. Deletion of histone deacetylase 7 in osteoclasts decreases bone mass in mice by interactions with mitf. PLoS ONE 2015, 10, e0123843. [Google Scholar] [CrossRef]
- Haberland, M.; Mokalled, M.H.; Montgomery, R.L.; Olson, E.N. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009, 23, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Rivadeneira, F.; Styrkarsdottir, U.; Estrada, K.; Halldorsson, B.V.; Hsu, Y.H.; Richards, J.B.; Zillikens, M.C.; Kavvoura, F.K.; Amin, N.; Aulchenko, Y.S.; et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat. Genet. 2009, 41, 1199–1206. [Google Scholar] [PubMed]
- Westendorf, J.J.; Zaidi, S.K.; Cascino, J.E.; Kahler, R.; van Wijnen, A.J.; Lian, J.B.; Yoshida, M.; Stein, G.S.; Li, X. Runx2 (cbfa1, aml-3) interacts with histone deacetylase 6 and represses the p21(cip1/waf1) promoter. Mol. Cell. Biol. 2002, 22, 7982–7992. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Spatz, J.; Nishimori, S.; Doench, J.; Root, D.; Babij, P.; Nagano, K.; Baron, R.; Brooks, D.; Bouxsein, M.; et al. Hdac5 controls mef2c-driven sclerostin expression in osteocytes. J. Bone Min. Res. 2015, 30, 400–411. [Google Scholar] [CrossRef]
- Chen, W.; Sheng, P.; Huang, Z.; Meng, F.; Kang, Y.; Huang, G.; Zhang, Z.; Liao, W.; Zhang, Z. Microrna-381 regulates chondrocyte hypertrophy by inhibiting histone deacetylase 4 expression. Int. J. Mol. Sci. 2016, 17, 1377. [Google Scholar] [CrossRef]
- Arnold, M.A.; Kim, Y.; Czubryt, M.P.; Phan, D.; McAnally, J.; Qi, X.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Mef2c transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell 2007, 12, 377–389. [Google Scholar] [CrossRef]
- Jeon, E.J.; Lee, K.Y.; Choi, N.S.; Lee, M.H.; Kim, H.N.; Jin, Y.H.; Ryoo, H.M.; Choi, J.Y.; Yoshida, M.; Nishino, N.; et al. Bone morphogenetic protein-2 stimulates runx2 acetylation. J. Biol. Chem. 2006, 281, 16502–16511. [Google Scholar] [CrossRef]
- Schroeder, T.M.; Kahler, R.A.; Li, X.; Westendorf, J.J. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J. Biol. Chem. 2004, 279, 41998–42007. [Google Scholar] [CrossRef] [PubMed]
- Lamour, V.; Detry, C.; Sanchez, C.; Henrotin, Y.; Castronovo, V.; Bellahcene, A. Runx2- and histone deacetylase 3-mediated repression is relieved in differentiating human osteoblast cells to allow high bone sialoprotein expression. J. Biol. Chem. 2007, 282, 36240–36249. [Google Scholar] [CrossRef] [PubMed]
- Hesse, E.; Saito, H.; Kiviranta, R.; Correa, D.; Yamana, K.; Neff, L.; Toben, D.; Duda, G.; Atfi, A.; Geoffroy, V.; et al. Zfp521 controls bone mass by hdac3-dependent attenuation of runx2 activity. J. Cell Biol. 2010, 191, 1271–1283. [Google Scholar] [CrossRef]
- Lee, H.W.; Suh, J.H.; Kim, A.Y.; Lee, Y.S.; Park, S.Y.; Kim, J.B. Histone deacetylase 1-mediated histone modification regulates osteoblast differentiation. Mol. Endocrinol. 2006, 20, 2432–2443. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, P.; Ge, J.; Cheng, J.; Dong, W.; Yuan, H.; Du, Y.; Yang, M.; Sun, R.; Jiang, H. Histone deacetylase 8 suppresses osteogenic differentiation of bone marrow stromal cells by inhibiting histone h3k9 acetylation and runx2 activity. Int. J. Biochem. Cell Biol. 2014, 54, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Pala, R.; Alomari, N.; Nauli, S.M. Primary cilium-dependent signaling mechanisms. Int. J. Mol. Sci. 2017, 18, 2272. [Google Scholar] [CrossRef] [PubMed]
- Veland, I.R.; Awan, A.; Pedersen, L.B.; Yoder, B.K.; Christensen, S.T. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol. 2009, 111, p39–p53. [Google Scholar] [CrossRef]
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class i and iia hdacs mediate hif-1alpha stability through phd2-dependent mechanism, while hdac6, a class iib member, promotes hif-1alpha transcriptional activity in nucleus pulposus cells of the intervertebral disc. J. Bone Min. Res. 2016, 31, 1287–1299. [Google Scholar] [CrossRef]
- Rimando, M.G.; Wu, H.H.; Liu, Y.A.; Lee, C.W.; Kuo, S.W.; Lo, Y.P.; Tseng, K.F.; Liu, Y.S.; Lee, O.K. Glucocorticoid receptor and histone deacetylase 6 mediate the differential effect of dexamethasone during osteogenesis of mesenchymal stromal cells (mscs). Sci. Rep. 2016, 6, 37371. [Google Scholar] [CrossRef]
- Paino, F.; La Noce, M.; Tirino, V.; Naddeo, P.; Desiderio, V.; Pirozzi, G.; De Rosa, A.; Laino, L.; Altucci, L.; Papaccio, G. Histone deacetylase inhibition with valproic acid downregulates osteocalcin gene expression in human dental pulp stem cells and osteoblasts: Evidence for hdac2 involvement. Stem Cells 2014, 32, 279–289. [Google Scholar] [CrossRef]
- La Noce, M.; Mele, L.; Laino, L.; Iolascon, G.; Pieretti, G.; Papaccio, G.; Desiderio, V.; Tirino, V.; Paino, F. Cytoplasmic interactions between the glucocorticoid receptor and hdac2 regulate osteocalcin expression in vpa-treated MSCs. Cells 2019, 8, 217. [Google Scholar] [CrossRef]
- Carpio, L.R.; Bradley, E.W.; Westendorf, J.J. Histone deacetylase 3 suppresses erk phosphorylation and matrix metalloproteinase (mmp)-13 activity in chondrocytes. Connect. Tissue Res. 2017, 58, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Carpio, L.R.; Bradley, E.W.; McGee-Lawrence, M.E.; Weivoda, M.M.; Poston, D.D.; Dudakovic, A.; Xu, M.; Tchkonia, T.; Kirkland, J.L.; van Wijnen, A.J.; et al. Histone deacetylase 3 supports endochondral bone formation by controlling cytokine signaling and matrix remodeling. Sci. Signal. 2016, 9, ra79. [Google Scholar] [CrossRef]
- Nakatani, T.; Chen, T.; Partridge, N.C. Mmp-13 is one of the critical mediators of the effect of hdac4 deletion on the skeleton. Bone 2016, 90, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, E.; Selvamurugan, N.; Westendorf, J.J.; Olson, E.N.; Partridge, N.C. Hdac4 represses matrix metalloproteinase-13 transcription in osteoblastic cells, and parathyroid hormone controls this repression. J. Biol. Chem. 2010, 285, 9616–9626. [Google Scholar] [CrossRef]
- Shimizu, E.; Selvamurugan, N.; Westendorf, J.J.; Partridge, N.C. Parathyroid hormone regulates histone deacetylases in osteoblasts. Ann. N. Y. Acad. Sci. 2007, 1116, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, C.D.; Zhang, N.; Tong, W.X.; Zhang, Y.F.; Shan, S.Z.; Zhang, X.L.; Li, Q.F. Mechanical stimulation orchestrates the osteogenic differentiation of human bone marrow stromal cells by regulating hdac1. Cell Death Dis. 2016, 7, e2221. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hassan, M.Q.; Jafferji, M.; Aqeilan, R.I.; Garzon, R.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Biological functions of mir-29b contribute to positive regulation of osteoblast differentiation. J. Biol. Chem. 2009, 284, 15676–15684. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, W.; Wang, H.; Yang, Q.; Zhang, L.; Jin, F.; Jin, Y. Mutual inhibition between hdac9 and mir-17 regulates osteogenesis of human periodontal ligament stem cells in inflammatory conditions. Cell Death Dis. 2018, 9, 480. [Google Scholar] [CrossRef]
- Li, C.J.; Cheng, P.; Liang, M.K.; Chen, Y.S.; Lu, Q.; Wang, J.Y.; Xia, Z.Y.; Zhou, H.D.; Cao, X.; Xie, H.; et al. Microrna-188 regulates age-related switch between osteoblast and adipocyte differentiation. J. Clin. Investig. 2015, 125, 1509–1522. [Google Scholar] [CrossRef]
- Ota, S.; Zhou, Z.Q.; Romero, M.P.; Yang, G.; Hurlin, P.J. Hdac6 deficiency or inhibition blocks fgfr3 accumulation and improves bone growth in a model of achondroplasia. Hum. Mol. Genet. 2017, 26, 3651. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Kwon, S.; Yamaguchi, T.; Cubizolles, F.; Rousseaux, S.; Kneissel, M.; Cao, C.; Li, N.; Cheng, H.L.; Chua, K.; et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 2008, 28, 1688–1701. [Google Scholar] [CrossRef] [PubMed]
- Cantley, M.D.; Fairlie, D.P.; Bartold, P.M.; Marino, V.; Gupta, P.K.; Haynes, D.R. Inhibiting histone deacetylase 1 suppresses both inflammation and bone loss in arthritis. Rheumatology 2015, 54, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Lee, J.H.; Bae, S.C.; Ryoo, H.M.; Kim, H.H.; Ha, H.; Lee, Z.H. Histone deacetylase inhibitor ms-275 stimulates bone formation in part by enhancing dhx36-mediated tnap transcription. J. Bone Min. Res. 2011, 26, 2161–2173. [Google Scholar] [CrossRef]
- McGee-Lawrence, M.E.; Pierce, J.L.; Yu, K.; Culpepper, N.R.; Bradley, E.W.; Westendorf, J.J. Loss of hdac3 in osteoprogenitors increases bone expression of osteoprotegerin, improving systemic insulin sensitivity. J. Cell. Physiol. 2018, 233, 2671–2680. [Google Scholar] [CrossRef] [PubMed]
- Blixt, N.C.; Faulkner, B.K.; Astleford, K.; Lelich, R.; Schering, J.; Spencer, E.; Gopalakrishnan, R.; Jensen, E.D.; Mansky, K.C. Class ii and iv hdacs function as inhibitors of osteoclast differentiation. PLoS ONE 2017, 12, e0185441. [Google Scholar] [CrossRef]
- Li, H.; Xie, H.; Liu, W.; Hu, R.; Huang, B.; Tan, Y.F.; Xu, K.; Sheng, Z.F.; Zhou, H.D.; Wu, X.P.; et al. A novel microrna targeting hdac5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J. Clin. Investig. 2009, 119, 3666–3677. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, W.; Jiao, G.; Li, C.; Liu, H. Mir-455-3p activates nrf2/are signaling via hdac2 and protects osteoblasts from oxidative stress. Int. J. Biol. Macromol. 2018, 107, 2094–2101. [Google Scholar] [CrossRef]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables nf-kappab suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef]
- Dou, C.; Li, N.; Ding, N.; Liu, C.; Yang, X.; Kang, F.; Cao, Z.; Quan, H.; Hou, T.; Xu, J.; et al. Hdac2 regulates foxo1 during rankl-induced osteoclastogenesis. Am. J. Physiol. Cell Physiol. 2016, 310, C780–C787. [Google Scholar] [CrossRef]
- Kim, D.S.; Kwon, J.E.; Lee, S.H.; Kim, E.K.; Ryu, J.G.; Jung, K.A.; Choi, J.W.; Park, M.J.; Moon, Y.M.; Park, S.H.; et al. Attenuation of rheumatoid inflammation by sodium butyrate through reciprocal targeting of hdac2 in osteoclasts and hdac8 in t cells. Front. Immunol. 2018, 9, 1525. [Google Scholar] [CrossRef] [PubMed]
- McGee-Lawrence, M.E.; Bradley, E.W.; Dudakovic, A.; Carlson, S.W.; Ryan, Z.C.; Kumar, R.; Dadsetan, M.; Yaszemski, M.J.; Chen, Q.; An, K.N.; et al. Histone deacetylase 3 is required for maintenance of bone mass during aging. Bone 2013, 52, 296–307. [Google Scholar] [CrossRef]
- Bradley, E.W.; Carpio, L.R.; Olson, E.N.; Westendorf, J.J. Histone deacetylase 7 (hdac7) suppresses chondrocyte proliferation and beta-catenin activity during endochondral ossification. J. Biol. Chem. 2015, 290, 118–126. [Google Scholar] [CrossRef]
- Jin, Z.; Wei, W.; Dechow, P.C.; Wan, Y. Hdac7 inhibits osteoclastogenesis by reversing rankl-triggered beta-catenin switch. Mol. Endocrinol. 2013, 27, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.; Kaiser, B.; Romsa, A.; Schwarz, T.; Gopalakrishnan, R.; Jensen, E.D.; Mansky, K.C. Hdac3 and hdac7 have opposite effects on osteoclast differentiation. J. Biol. Chem. 2011, 286, 12056–12065. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Wei, W.; Huynh, H.; Wan, Y. Hdac9 inhibits osteoclastogenesis via mutual suppression of ppargamma/rankl signaling. Mol. Endocrinol. 2015, 29, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Shakespear, M.R.; Hohenhaus, D.M.; Kelly, G.M.; Kamal, N.A.; Gupta, P.; Labzin, L.I.; Schroder, K.; Garceau, V.; Barbero, S.; Iyer, A.; et al. Histone deacetylase 7 promotes toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J. Biol. Chem. 2013, 288, 25362–25374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.X.; Wang, Y.S.; Cai, Q.Q.; Wang, J.Q.; Yao, W.T. Up-regulation of hdac9 promotes cell proliferation through suppressing p53 transcription in osteosarcoma. Int. J. Clin. Exp. Med. 2015, 8, 11818–11823. [Google Scholar]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. Hdacs and hdac inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- De Souza, C.; Chatterji, B.P. Hdac inhibitors as novel anti-cancer therapeutics. Recent Pat. Anticancer Drug Discov. 2015, 10, 145–162. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging hdac inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the sost gene in patients with van buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (sost). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Brunkow, M.E.; Gardner, J.C.; Van Ness, J.; Paeper, B.W.; Kovacevich, B.R.; Proll, S.; Skonier, J.E.; Zhao, L.; Sabo, P.J.; Fu, Y.; et al. Bone dysplasia sclerosteosis results from loss of the sost gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 2001, 68, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Ellies, D.L.; Viviano, B.; McCarthy, J.; Rey, J.P.; Itasaki, N.; Saunders, S.; Krumlauf, R. Bone density ligand, sclerostin, directly interacts with lrp5 but not lrp5g171v to modulate wnt activity. J. Bone Min. Res. 2006, 21, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xue, P.; Wu, X.; Ma, J.; Wang, Y.; Li, Y. Role of sclerostin and dkk1 in bone remodeling in type 2 diabetic patients. Endocr. Res. 2018, 43, 29–38. [Google Scholar] [CrossRef]
- Kamiya, N.; Ye, L.; Kobayashi, T.; Mochida, Y.; Yamauchi, M.; Kronenberg, H.M.; Feng, J.Q.; Mishina, Y. Bmp signaling negatively regulates bone mass through sclerostin by inhibiting the canonical wnt pathway. Development 2008, 135, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Li, P.; Wu, H.; Shafiquzzaman, M.; Murakami, S.; Schneider, M.D.; Mishina, Y.; Li, B.; Li, J. Bmpria is required for osteogenic differentiation and rankl expression in adult bone marrow mesenchymal stromal cells. Sci. Rep. 2018, 8, 8475. [Google Scholar] [CrossRef]
- Liu, J.; Xu, K.; Wen, G.; Guo, H.; Li, S.; Wu, X.; Dai, R.; Sheng, Z.; Liao, E. Comparison of the effects of genistein and zoledronic acid on the bone loss in opg-deficient mice. Bone 2008, 42, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Moschen, A.R.; Kaser, A.; Stadlmann, S.; Millonig, G.; Kaser, S.; Muhllechner, P.; Habior, A.; Graziadei, I.; Vogel, W.; Tilg, H. The rankl/opg system and bone mineral density in patients with chronic liver disease. J. Hepatol. 2005, 43, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Valdecasas-Campelo, E.; Gonzalez-Reimers, E.; Santolaria-Fernandez, F.; De la Vega-Prieto, M.J.; Milena-Abril, A.; Sanchez-Perez, M.J.; Martinez-Riera, A.; Gomez-Rodriguez Mde, L. Serum osteoprotegerin and rankl levels in chronic alcoholic liver disease. Alcohol Alcohol. 2006, 41, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Guanabens, N.; Enjuanes, A.; Alvarez, L.; Peris, P.; Caballeria, L.; Jesus Martinez de Osaba, M.; Cerda, D.; Monegal, A.; Pons, F.; Pares, A. High osteoprotegerin serum levels in primary biliary cirrhosis are associated with disease severity but not with the mrna gene expression in liver tissue. J. Bone Min. Metab. 2009, 27, 347–354. [Google Scholar] [CrossRef]
- Monegal, A.; Navasa, M.; Peris, P.; Alvarez, L.; Pons, F.; Rodes, J.; Guanabens, N. Serum osteoprotegerin and its ligand in cirrhotic patients referred for orthotopic liver transplantation: Relationship with metabolic bone disease. Liver Int. 2007, 27, 492–497. [Google Scholar] [CrossRef]
- Fabrega, E.; Orive, A.; Garcia-Unzueta, M.; Amado, J.A.; Casafont, F.; Pons-Romero, F. Osteoprotegerin and receptor activator of nuclear factor-kappab ligand system in the early post-operative period of liver transplantation. Clin. Transpl. 2006, 20, 383–388. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R. Denosumab for the treatment of osteoporosis. Osteoporos. Sarcopenia 2017, 3, 8–17. [Google Scholar] [CrossRef]
- Zaheer, S.; LeBoff, M.; Lewiecki, E.M. Denosumab for the treatment of osteoporosis. Expert Opin. Drug Metab. Toxicol. 2015, 11, 461–470. [Google Scholar] [CrossRef]
- Takeno, A.; Yamamoto, M.; Notsu, M.; Sugimoto, T. Administration of anti-receptor activator of nuclear factor-kappa b ligand (rankl) antibody for the treatment of osteoporosis was associated with amelioration of hepatitis in a female patient with growth hormone deficiency: A case report. BMC Endocr. Disord. 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reimers, E.; Martin-Gonzalez, C.; de la Vega-Prieto, M.J.; Pelazas-Gonzalez, R.; Fernandez-Rodriguez, C.; Lopez-Prieto, J.; Alvisa-Negrin, J.; Santolaria-Fernandez, F. Serum sclerostin in alcoholics: A pilot study. Alcohol Alcohol. 2013, 48, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Anastasilakis, A.D.; Kountouras, J.; Makras, P.; Papatheodorou, A.; Kokkoris, P.; Sakellariou, G.T.; Terpos, E. Circulating sclerostin and dickkopf-1 levels in patients with nonalcoholic fatty liver disease. J. Bone Min. Metab. 2016, 34, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reimers, E.; Lopez-Prieto, J.; Pelazas-Gonzalez, R.; Aleman-Valls, M.R.; Jose de la Vega-Prieto, M.; Jorge-Ripper, C.; Duran-Castellon, M.C.; Santolaria-Fernandez, F. Serum sclerostin in hepatitis c virus infected patients. J. Bone Metab. 2014, 21, 69–75. [Google Scholar] [CrossRef]
- Guanabens, N.; Ruiz-Gaspa, S.; Gifre, L.; Miquel, R.; Peris, P.; Monegal, A.; Dubrueil, M.; Arias, A.; Pares, A. Sclerostin expression in bile ducts of patients with chronic cholestasis may influence the bone disease in primary biliary cirrhosis. J. Bone Min. Res. 2016, 31, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: A randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- McClung, M.R. Sclerostin antibodies in osteoporosis: Latest evidence and therapeutic potential. Adv. Musculoskelet. Dis. 2017, 9, 263–270. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year | Patients (N) | Rate (%) | 95% CI | Forest Plot | ||
|---|---|---|---|---|---|---|
| HOD | Total | |||||
| Studies on patients with primary biliary cirrhosis (PBC) | ||||||
| 1 | Guañabens et al., 1990 [17] | 7 | 20 | 35.00 | 9.07–60.93 |  |
| 2 | Lindor et al., 1995 [16] | 31 | 88 | 35.00 | 22.64–47.36 | |
| 3 | Menon et al., 2001 [14] | 35 | 176 | 20.00 | 13.39–26.61 | |
| 4 | Mounach et al., 2008 [15] | 17 | 33 | 51.50 | 27.01–75.99 | |
| Summary | 90 | 317 | 32.35 | 18.90–45.80 | ||
| Studies on patients with primary sclerosing cholangitis (PSC) | ||||||
| 1 | Angulo et al., 1998 [32] | 14 | 81 | 17.00 | 8.02–25.98 |  |
| 2 | Angulo et al., 2011 [1] | 178 | 237 | 75.00 | 63.97–86.03 | |
| 3 | Keller et al., 2016 [13] | 15 | 40 | 37.50 | 18.52–56.48 | |
| Summary | 207 | 358 | 43.18 | 3.44–82.92 | ||
| Studies on patients with viral hepatitis | ||||||
| 1 | Duarte et al., 2001 [33] | 25 | 100 | 25.00 | 15.20–34.80 |  |
| 2 | Auletta et al., 2005 [34] | 19 | 30 | 64.00 | 35.37–92.63 | |
| 3 | Hofmann et al., 2008 [35] | 17 | 30 | 56.00 | 29.22–82.78 | |
| 4 | Lin et al., 2012 [36] | 32 | 69 | 46.30 | 30.24–62.36 | |
| 5 | El-Husseini et al., 2013 [11] | 23 | 33 | 69.70 | 41.22–98.18 | |
| 6 | Orsini et al., 2013 [10] | 8 | 60 | 13.30 | 4.07–22-53 | |
| 7 | Lai et al., 2015 [37] | 25 | 60 | 42.00 | 25.60–58.40 | |
| 8 | Huang et al., 2017 [38] | 54 | 148 | 36.30 | 26.59–46.01 | |
| 9 | Bering et al., 2018 [39] | 28 | 104 | 27.10 | 17.09–37.11 | |
| Summary | 231 | 634 | 37.87 | 27.61–48.13 | ||
| Studies on patients with viral cirrhosis | ||||||
| 1 | Gallegy-Rojo et al., 1998 [40] | 17 | 32 | 53.00 | 27.78–78.22 |  |
| 2 | George et al., 2009 [41] | 49 | 72 | 68.00 | 48.95–87.05 | |
| 3 | Choudhary et al., 2011 [12] | 112 | 115 | 97.00 | 79.00–115.00 | |
| 4 | Goubraim et al., 2013 [42] | 37 | 46 | 80.50 | 54.57–106.43 | |
| 5 | Karoli et al., 2016 [43] | 72 | 72 | 100.00 | 76.90–123.10 | |
| Summary | 287 | 337 | 80.27 | 63.19–97.36 | ||
| Studies on patients with alcoholic liver disease | ||||||
| 1 | Spencer et al., 1986 [44] | 45 | 96 | 46.88 | 33.18–60.57 |  |
| 2 | Diamond et al., 1989 [21] | 18 | 28 | 64.29 | 34.59–93-98 | |
| 3 | Gonzalez-Calvin et al., 1993 [45] | 9 | 39 | 23.08 | 8.00–38.15 | |
| 4 | Kim et al., 2003 [46] | 14 | 19 | 73.68 | 35.09–112.28 | |
| 5 | Malik et al., 2009 [20] | 10 | 57 | 17.54 | 6.67–28.42 | |
| 6 | Savic et al., 2014 [19] | 6 | 30 | 20.00 | 4.00–36.00 | |
| Summary | 102 | 269 | 37.87 | 20.61–51.17 | ||
| Studies on patients with non-alcoholic fatty liver disease (NAFDL) or non-alcoholic steatohepatitis (NASH) | ||||||
| 1 | Pardee et al., 2012 [22] | 17 | 38 | 45.00 | 23.67–66.33 |  |
| 2 | Kim et al., 2017 [23] | 80 | 129 | 62.00 | 48.41–75.59 | |
| 3 | Chen et al., 2018 [24] | 116 | 365 | 31.80 | 26.01–37.59 | |
| Summary | 213 | 532 | 45.71 | 24.50–66.92 | ||
| Studies on patients with hemochromatosis | ||||||
| 1 | Diamond et al., 1989 [25] | 10 | 22 | 45.00 | 16.97–73.03 |  |
| 2 | Sinigaglia et al., 1997 [26] | 9 | 32 | 28.00 | 9.67–46.33 | |
| 3 | Guggenbuhl et al., 2005 [27] | 30 | 38 | 78.90 | 50.66–107.14 | |
| Summary | 49 | 92 | 49.26 | 19.41–79.10 | ||
| Studies on patients with Wilson disease | ||||||
| 1 | Hegedus et al., 2002 [31] | 9 | 21 | 43.00 | 14.95–71.05 |  |
| 2 | Selimoglu et al., 2008 [30] | 28 | 31 | 90.30 | 56.85–123.75 | |
| 3 | Quemeneur et al., 2014 [29] | 11 | 85 | 13.00 | 5.33–20.67 | |
| 4 | Weiss et al., 2015 [28] | 87 | 148 | 58.80 | 46.45–71.15 | |
| Summary | 135 | 285 | 49.31 | 16.10–82.53 | ||
| Risk Factors | Proposed Mechanisms | Ref. |
|---|---|---|
| Age | Independent of CLD, age may cause disbalances in osteoclast and osteoblast function. This is often associated with altered hormonal status or epigenetic changes. | [62] |
| Severity of liver damage | HOD is correlated with severity of the liver disease; HOD is more common in patients with end-stage liver disease and cirrhosis than in patients with fibrosis or hepatitis. | [62] |
| Low body mass index | A low body mass index (BMI) often correlates with low BMD both in healthy subjects and patients with CLD. A cut-off is usually set at a BMI below 19 kg/m2. | [63,64,65] |
| Dietary deficiencies | Malnutrition or dietary deficiencies frequently occur in patients with CLD (12% of OLT patients), due to altered nutritional requirements during ascites or other complications. | [66,67] |
| Alcohol consumption | Ethanol affects bone directly via a toxic effect on osteoblasts and indirectly by altering PTH, vitamin D, testosterone, IGF-1, cytokines (e.g., TNF or IL-6) and cortisol levels. | [68,69,70,71] |
| Cigarette consumption | Independent of CLD, smoking affects osteoblast and osteoclast function, favoring the development of severe osteoporosis and increasing the risk for fragility fractures. | [62] |
| Physical exercise | In patients with CLD, exercise levels are often reduced compared to healthy individuals; thus, the bone receives less mechanical stimulation. | [72,73] |
| Muscle wasting | Muscle wasting is very common in patients with CLD. When it occurs independent of malnutrition, it may be an indicator for the manifestation of HOD. | [74] |
| Hormonal status | Early menopause and post-menopausal status additionally favors bone loss in women. | [64] |
| Hypogonadism may cause osteoporosis independent of CLD. Parenchymal damage during CLD may cause hypogonadism due to an altered hypothalamic–pituitary–thyroid function with reduced release of gonadotrophins and primary gonadal failure. | [75] | |
| Anomalies of vitamin D and calcium metabolism | CLD patients may have reduced vitamin D (VitD) absorption in the gut. | [19,76,77,78,79] |
| Enterohepatic circulation of VitD might be disturbed in patients with CLD. | ||
| CLD patients frequently show impaired hepatic hydroxylation of VitD. | ||
| CLD patients may have increased urinary VitD excretion. | ||
| Reduced tissue sensitivity to VitD may contribute to the development of HOD. | ||
| VitD deficiency may cause hyperparathyroidism which increases bone turnover. | [80,81] | |
| Vitamin K deficiency | Vitamin K (VitK) is required for the formation of osteocalcin and osteonectin. VitK inhibits osteoclast viability, maturation, and function. | [82,83,84] |
| Growth hormones | IGF-1 levels, which decrease during CLD, were linked to HOD. | [85,86] |
| CLD is associated with a progressive increase in growth hormone (GH) resistance. | [87] | |
| Active transforming growth factor β (TGF-β) is produced in inflamed liver tissue. | [88,89] | |
| In response to damage, liver may produce bone morphogenetic proteins (BMPs). | [90,91] | |
| Iron and copper | Iron may directly affect osteoblast function. An excessive pituitary iron deposition may favor the development of hypogonadism independent of the CLD. | [27,92,93] |
| Increased bilirubin | Increased levels of unconjugated bilirubin (hyper-bilirubinemia) were associated with a decreased osteoblast function, mediated possibly via regulation of IGF-1. | [66,94,95,96] |
| Genetic factors | Genetic polymorphisms were described which may favor the development of HOD, including genes encoding vitamin D receptors or collagen type 1A1. | [97,98,99,100,101] |
| Medication | Corticosteroids affect bone structure by increasing osteoclasts activity and by decreasing differentiation, recruitment, and lifespan of osteoblasts. | [102] |
| Calcineurin inhibitors are used in conjunction with corticosteroids; thus, the independent effect of these agents on bone metabolism in humans is uncertain. | [103] | |
| Antiviral agents, e.g., ribavirin, may directly affect osteoclast and osteoblast function. | [104,105,106] | |
| Cholestyramine, a bile-acid sequestrant used to treat pruritus or itching during CLD, was reported to adversely affect the intestinal absorption of VitD. | [107] | |
| The effect of medication, e.g., diuretics, anticoagulants, and chemotherapy, used in the treatment of advanced liver disease, on bone metabolism in humans is uncertain. |
| HDACs | Proposed Mechanisms | References | |
|---|---|---|---|
| Deletion/Inhibition of | 1, 9 | Expression is decreased during osteogenic differentiation. | [188,189,190,214,228] |
| 9 | Expression is blocked by TGF-β signaling. | [188,189] | |
| 4, 9 | Expression is regulated by microRNAs (miR-17, miR-29b, miR-188). | [229,230,231] | |
| 1, 6 | Associated with improved skeletal phenotypes. | [228,232,233,234] | |
| 2–5, 7–9 | Associated with impaired skeletal phenotypes. | [192,193,194,195,196,197,198,199,200,201,202,203,204] | |
| 1, 7 | Induce expression of osteoblastic genes, e.g., TNAP. | [191,235] | |
| 3 | Drives MSC differentiation towards adipogenic lineage. | [198,199,200,201] | |
| 3, 4 | Favor expression of OPG, FGF-21, MMP3, MMP10, and MMP13. | [197,208,223,224,236] | |
| 4, 5, 9–11 | Increase osteoclast size and demineralization activity, together with increased expression of c-Fos, NFATc1, and Cathepsin K. | [237] | |
| Increased Expression of | 2, 4–7 | Increased during osteogenic differentiation. | [186,187,188,189,190,191] |
| 1–3, 6, 11 | Induced by TGF-β signaling. | [188,189,190,214,228] | |
| 1 | Induced by mechanical stimulation. | [228] | |
| 6 | Induced by hypoxia and oxidative stress. | [192] | |
| 4, 5 | Associated with impaired skeletal phenotypes. | [186,238] | |
| 5 | Polymorphisms are associated with decreased BMD. | [205] | |
| 5, 6 | Suppress expression of transcription factors, e.g., Runx2 or osterix. | [210,220] | |
| 1–8 | Repress transcriptional activity of, e.g., Runx2, p300, Mef2, Mef2c, NFATc1, Zfp521, or TCF by direct interaction/binding. | [187,190,191,206,207,208,209,210,211,212,213,214,215] | |
| 4, 5 | Deacetylate Runx2, affecting its transcriptional activity. | [187,210] | |
| 4, 5 | Deacetylate Runx2, promoting its degradation. | [187,210] | |
| 4, 9 | Expression is regulated by miRNAs (miR-17, miR-29b, miR-188). | [229,230,231] | |
| 4 | Its cytoplasmic–nuclear shuttling is regulated by mechanical load. | [194] | |
| 2 | Regulates proliferation, oxidative stress, and apoptosis by (binding) regulating transcriptional activity of Nrf2/ARE. | [239,240] | |
| 4 | Interaction with PTH regulates expression of genes, e.g., MMP13. | [225,226,227] | |
| 6 | Promotes Hif-1α transcriptional activity. | [219] | |
| 2 | Favors osteoclastogenesis via Akt-mediated suppression of FoxO1. | [241] | |
| 2, 6 | Interact with glucocorticoid receptor to regulate inflammation and expression of genes, e.g., osteocalcin or collagen during osteogenesis. | [220,221,222,242] | |
| 3 | Required for bone maintenance during aging. | [243] | |
| 3 | Represses activity of MMP13, proposed regulation via ERK1/2. | [223,224] | |
| 5 | Regulates PTH-driven sclerostin expression in osteocytes via Mef2. | [207] | |
| 6 | Affects structural integrity of primary cilia, the mechanosensory organelle on osteoblasts, which regulates signaling pathways. | [188] | |
| 3, 7, 9 | Regulate osteoclastogenesis via RANKL, Wnt, and PPARγ. | [244,245,246,247] | |
| 1, 3, 7 | Regulate inflammation, proposedly involving STAT and NF-κB. | [224,234,248] | |
| 9 | Promotes proliferation of osteogenic cells, interacting with p53. | [189,249] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ehnert, S.; Aspera-Werz, R.H.; Ruoß, M.; Dooley, S.; Hengstler, J.G.; Nadalin, S.; Relja, B.; Badke, A.; Nussler, A.K. Hepatic Osteodystrophy—Molecular Mechanisms Proposed to Favor Its Development. Int. J. Mol. Sci. 2019, 20, 2555. https://doi.org/10.3390/ijms20102555
Ehnert S, Aspera-Werz RH, Ruoß M, Dooley S, Hengstler JG, Nadalin S, Relja B, Badke A, Nussler AK. Hepatic Osteodystrophy—Molecular Mechanisms Proposed to Favor Its Development. International Journal of Molecular Sciences. 2019; 20(10):2555. https://doi.org/10.3390/ijms20102555
Chicago/Turabian StyleEhnert, Sabrina, Romina H. Aspera-Werz, Marc Ruoß, Steven Dooley, Jan G. Hengstler, Silvio Nadalin, Borna Relja, Andreas Badke, and Andreas K. Nussler. 2019. "Hepatic Osteodystrophy—Molecular Mechanisms Proposed to Favor Its Development" International Journal of Molecular Sciences 20, no. 10: 2555. https://doi.org/10.3390/ijms20102555
APA StyleEhnert, S., Aspera-Werz, R. H., Ruoß, M., Dooley, S., Hengstler, J. G., Nadalin, S., Relja, B., Badke, A., & Nussler, A. K. (2019). Hepatic Osteodystrophy—Molecular Mechanisms Proposed to Favor Its Development. International Journal of Molecular Sciences, 20(10), 2555. https://doi.org/10.3390/ijms20102555