Three-Dimensional Quantitative Structure-Activity Relationships (3D-QSAR) on a Series of Piperazine-Carboxamides Fatty Acid Amide Hydrolase (FAAH) Inhibitors as a Useful Tool for the Design of New Cannabinoid Ligands


Abstract
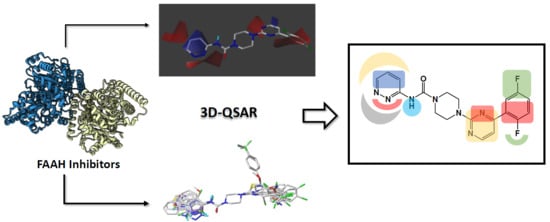
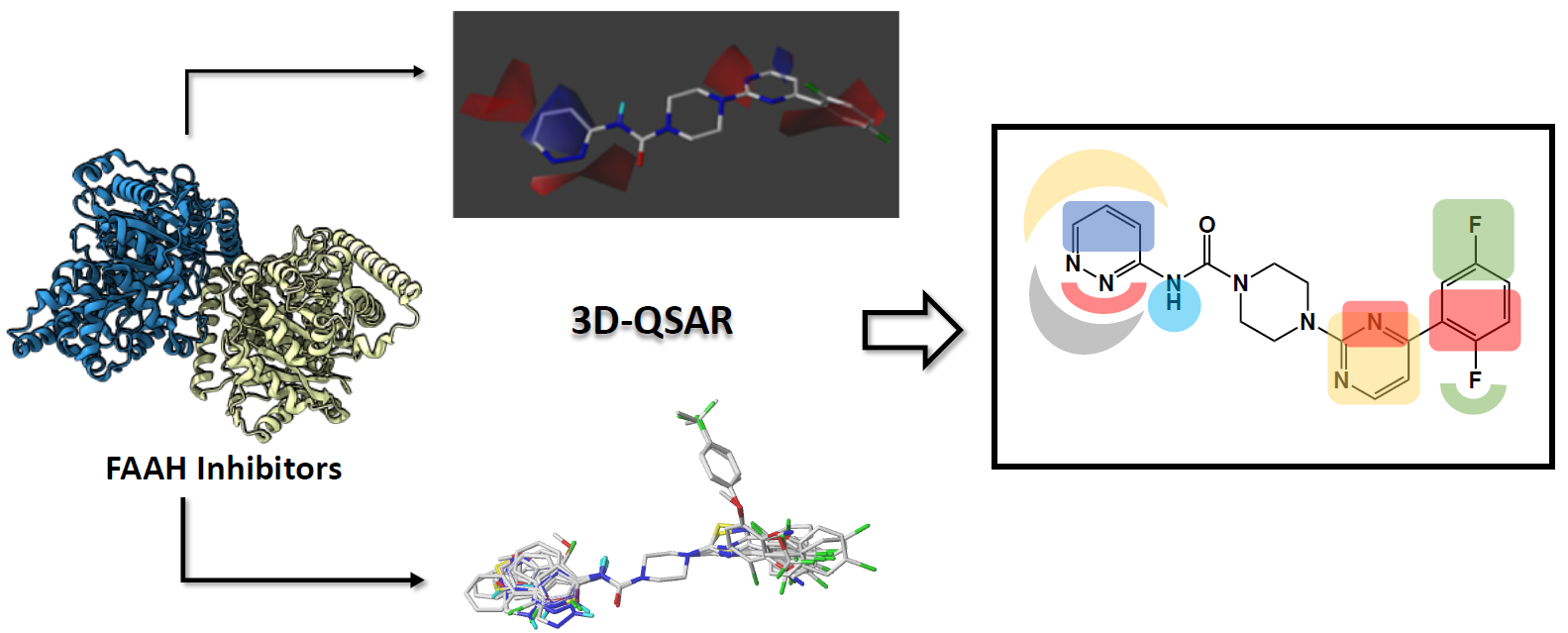
1. Introduction
2. Results and Discussion
2.1. Statistical Results
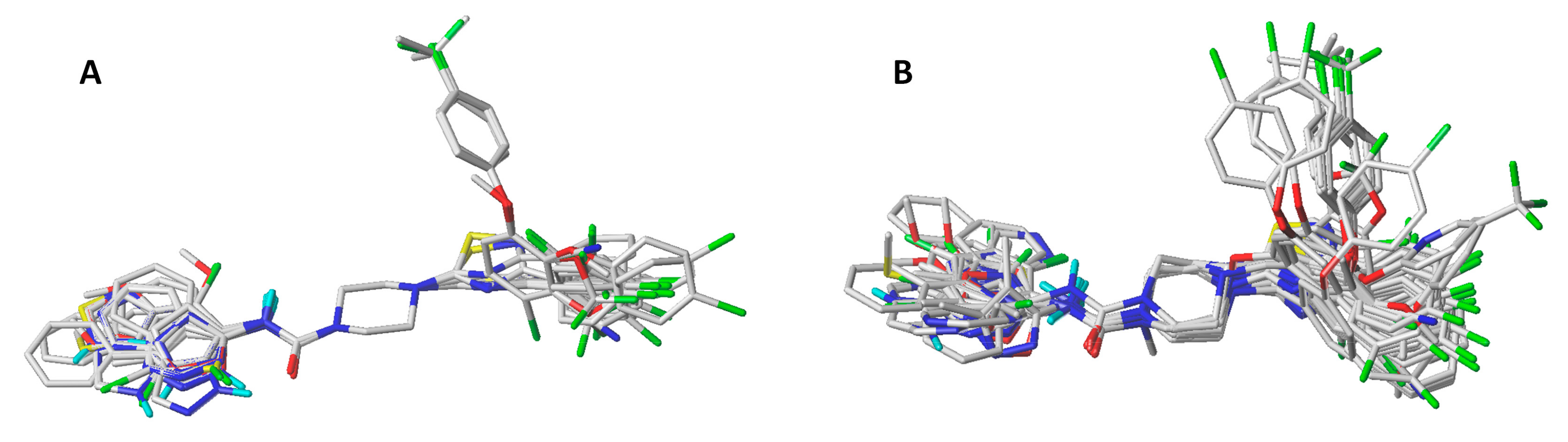
2.2. Applicability Domain
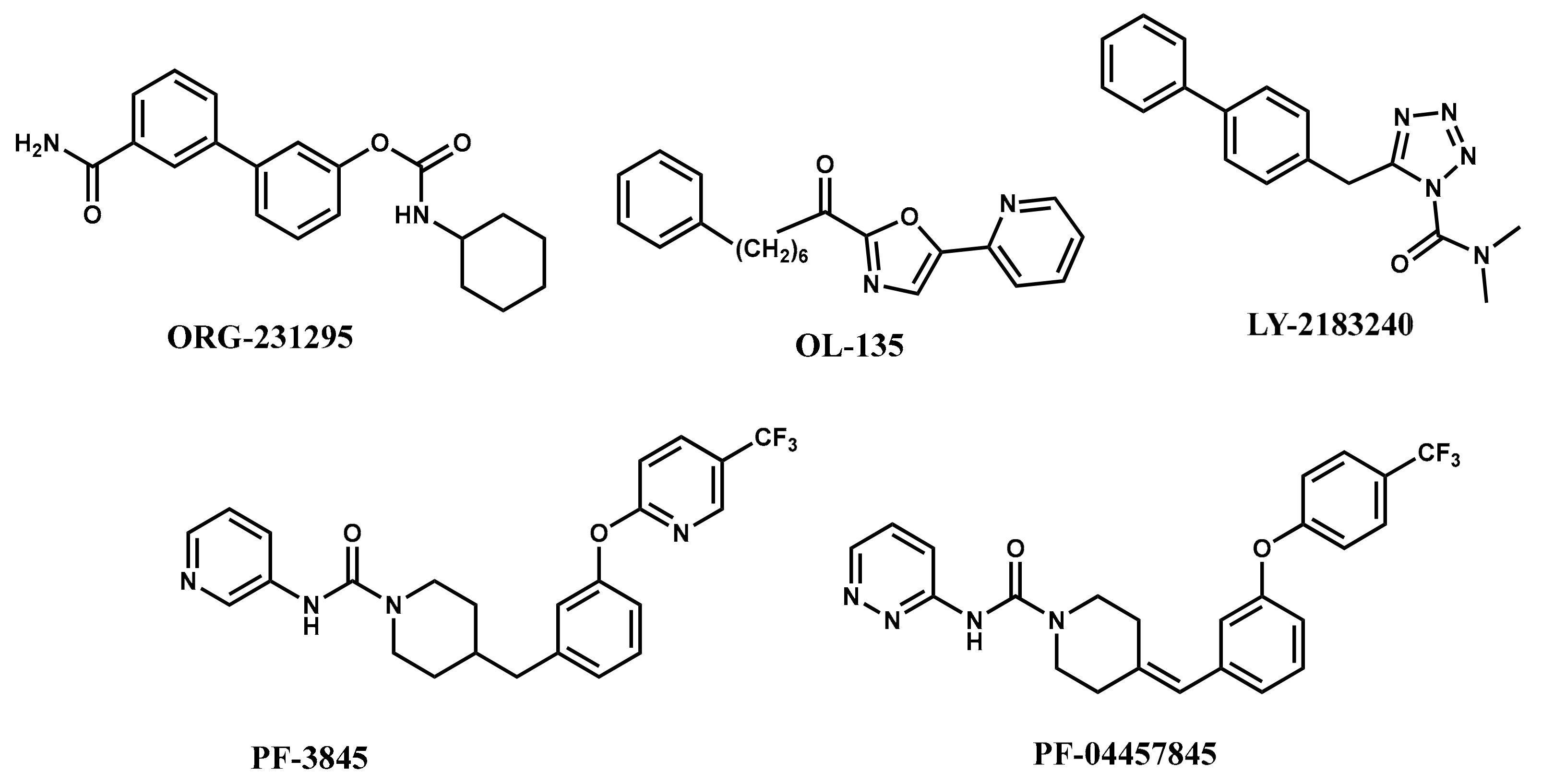
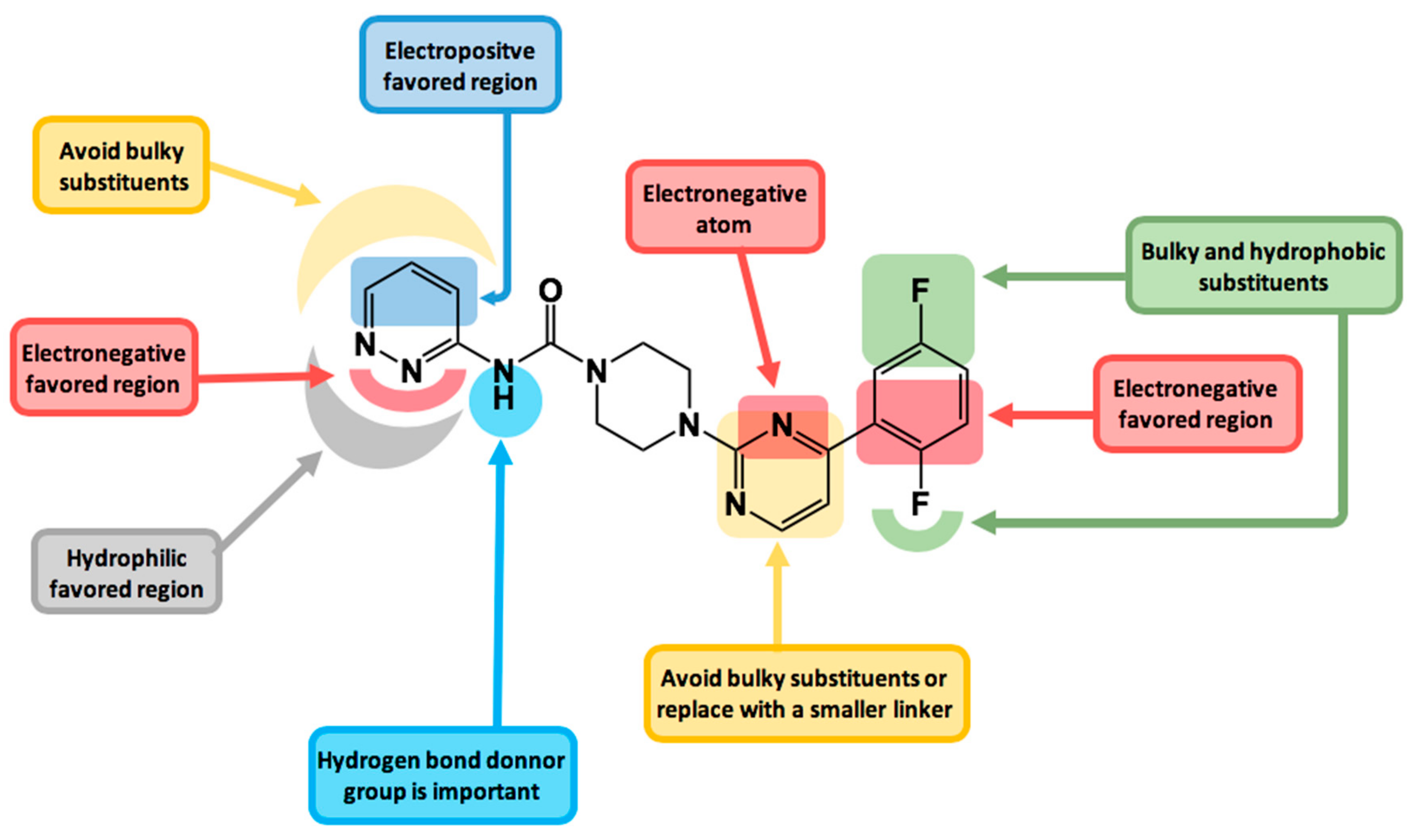
2.3. Contour Maps Analysis
2.3.1. Steric Contour Map
2.3.2. Electrostatic Contour Map
2.3.3. Hydrophobic Contour Map
2.3.4. Donor and Acceptor Contour Maps
2.4. Design of New FAAH Inhibitors
3. Materials and Methods
3.1. Molecular Alignment
3.2. CoMSIA Field Calculation
3.3. Data Set Selection and Inhibitory Activity
3.4. Internal Validation and Partial Least Squares (PLS) Analysis
3.5. External Validation
3.6. Applicability Domain (AD) Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D-QSAR | Three-dimensional Quantitative Structure-Activity Relationship |
| CoMSIA | Comparative Molecular Similarity Indices Analysis |
| FAAH | Fatty Acid Amide Hydrolase |
| CB | Cannabinoid |
References
- Pagotto, U.; Marsicano, G.; Cota, D.; Lutz, B.; Pasquali, R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr. Rev. 2006, 27, 73–100. [Google Scholar] [CrossRef] [PubMed]
- Manzanares, J.; Julian, M.D.; Carrascosa, A. Role of the cannabinoid system in pain control and therapeutic implications for the management of acute and chronic pain episodes. Curr. Neuropharmacol. 2006, 4, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. FAAH and MAGL inhibitors: Therapeutic opportunities from regulating endocannabinoid levels. Curr. Opin. Investig. Drugs 2010, 11, 51–62. [Google Scholar] [PubMed]
- Long, J.Z.; Cravatt, B.F. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev. 2011, 111, 6022–6063. [Google Scholar] [CrossRef] [PubMed]
- Paylor, B.; Holt, S.; Fowler, C.J. The potency of the fatty acid amide hydrolase inhibitor URB597 is dependent upon the assay pH. Pharmacol. Res. 2006, 54, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Mor, M.; Lodola, A.; Rivara, S.; Vacondio, F.; Duranti, A.; Tontini, A.; Sanchini, S.; Piersanti, G.; Clapper, J.R.; King, A.R.; et al. Synthesis and quantitative structure-activity relationship of fatty acid amide hydrolase inhibitors: Modulation at the N-portion of biphenyl-3-yl alkylcarbamates. J. Med. Chem. 2008, 51, 3487–3498. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saario, S.M.; Poso, A.; Juvonen, R.O.; Jarvinen, T.; Salo-Ahen, O.M. Fatty acid amide hydrolase inhibitors from virtual screening of the endocannabinoid system. J. Med. Chem. 2006, 49, 4650–4656. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Ligresti, A.; Allara, M.; di Marzo, V.; Lazzati, Z.; D’Ursi, P.; Marabotti, A.; Milanesi, L.; Spallarossa, A.; Ranise, A.; et al. Homology modeling in tandem with 3D-QSAR analyses: A computational approach to depict the agonist binding site of the human CB2 receptor. Eur. J. Med. Chem. 2011, 46, 4489–4505. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Cesarini, S.; Mosti, L.; Fossa, P. CoMFA and CoMSIA analyses on 1,2,3,4-tetrahydropyrrolo[3,4-b]indole and benzimidazole derivatives as selective CB2 receptor agonists. J. Mol. Model. 2010, 16, 1481–1498. [Google Scholar] [CrossRef]
- Cichero, E.; Cesarini, S.; Mosti, L.; Fossa, P. CoMFA and CoMSIA analyses on 4-oxo-1,4-dihydroquinoline and 4-oxo-1,4-dihydro-1,5-, -1,6- and -1,8-naphthyridine derivatives as selective CB2 receptor agonists. J. Mol. Model. 2010, 16, 677–691. [Google Scholar] [CrossRef]
- Romero-Parra, J.; Chung, H.; Tapia, R.A.; Faundez, M.; Morales-Verdejo, C.; Lorca, M.; Lagos, C.F.; Di Marzo, V.; David Pessoa-Mahana, C.; Mella, J. Combined CoMFA and CoMSIA 3D-QSAR study of benzimidazole and benzothiophene derivatives with selective affinity for the CB2 cannabinoid receptor. Eur. J. Pharm. Sci. 2017, 101, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mella-Raipan, J.A.; Lagos, C.F.; Recabarren-Gajardo, G.; Espinosa-Bustos, C.; Romero-Parra, J.; Pessoa-Mahana, H.; Iturriaga-Vasquez, P.; Pessoa-Mahana, C.D. Design, synthesis, binding and docking-based 3D-QSAR studies of 2-pyridylbenzimidazoles-a new family of high affinity CB1 cannabinoid ligands. Molecules 2013, 18, 3972–4001. [Google Scholar] [CrossRef] [PubMed]
- Mella-Raipan, J.; Hernandez-Pino, S.; Morales-Verdejo, C.; Pessoa-Mahana, D. 3D-QSAR/CoMFA-based structure-affinity/selectivity relationships of aminoalkylindoles in the cannabinoid CB1 and CB2 receptors. Molecules 2014, 19, 2842–2861. [Google Scholar] [CrossRef] [PubMed]
- Dainese, E.; Gasperi, V.; Maccarrone, M. Partial QSAR analysis of some selected natural inhibitors of FAAH suggests a working hypothesis for the development of endocannabinoid-based drugs. Curr. Drug Targets CNS Neurol. Disord. 2005, 4, 709–714. [Google Scholar] [CrossRef]
- Kasnanen, H.; Myllymaki, M.J.; Minkkila, A.; Kataja, A.O.; Saario, S.M.; Nevalainen, T.; Koskinen, A.M.; Poso, A. 3-Heterocycle-phenyl N-alkylcarbamates as FAAH inhibitors: Design, synthesis and 3D-QSAR studies. ChemMedChem 2010, 5, 213–231. [Google Scholar] [CrossRef] [PubMed]
- Vacondio, F.; Silva, C.; Lodola, A.; Fioni, A.; Rivara, S.; Duranti, A.; Tontini, A.; Sanchini, S.; Clapper, J.R.; Piomelli, D.; et al. Structure-property relationships of a class of carbamate-based fatty acid amide hydrolase (FAAH) inhibitors: Chemical and biological stability. ChemMedChem 2009, 4, 1495–1504. [Google Scholar] [CrossRef]
- Han, D.; Wang, B.; Jin, H.; Wang, H.; Chen, M. Design, synthesis and CoMFA studies of OEA derivatives as FAAH inhibitors. Med. Chem. Res. 2017, 26, 2951–2966. [Google Scholar] [CrossRef]
- Keith, J.M.; Apodaca, R.; Tichenor, M.; Xiao, W.; Jones, W.; Pierce, J.; Seierstad, M.; Palmer, J.; Webb, M.; Karbarz, M.; et al. Aryl piperazinyl ureas as inhibitors of fatty acid amide hydrolase (FAAH) in rat, dog, and primate. ACS Med. Chem. Lett. 2012, 3, 823–827. [Google Scholar] [CrossRef]
- Durdagi, S.; Papadopoulos, M.G.; Papahatjis, D.P.; Mavromoustakos, T. Combined 3D QSAR and molecular docking studies to reveal novel cannabinoid ligands with optimum binding activity. Bioorg. Med. Chem. Lett. 2007, 17, 6754–6763. [Google Scholar] [CrossRef]
- Golbraikh, A.; Shen, M.; Xiao, Z.; Xiao, Y.D.; Lee, K.H.; Tropsha, A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003, 17, 241–253. [Google Scholar] [CrossRef]
- Roy, K.; Mitra, I.; Kar, S.; Ojha, P.K.; Das, R.N.; Kabir, H. Comparative studies on some metrics for external validation of QSPR models. J. Chem. Inf. Model. 2012, 52, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Chirico, N.; Gramatica, P. Real external predictivity of QSAR models. Part 2. New intercomparable thresholds for different validation criteria and the need for scatter plot inspection. J. Chem. Inf. Model. 2012, 52, 2044–2058. [Google Scholar] [CrossRef] [PubMed]
- Rucker, C.; Rucker, G.; Meringer, M. y-Randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Lorca, M.; Morales-Verdejo, C.; Vásquez-Velásquez, D.; Andrades-Lagos, A.; Campanini-Salinas, J.; Soto-Delgado, J.; Recabarren-Gajardo, G.; Mella, J. Structure-activity relationships based on 3D-QSAR CoMFA/CoMSIA and design of aryloxypropanol-amine agonists with selectivity for the human β3-adrenergic receptor and anti-obesity and anti-diabetic profiles. Molecules 2018, 23, 1191. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemometr. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Aronov, A.M. Ligand structural aspects of hERG channel blockade. Curr. Top. Med. Chem. 2008, 8, 1113–1127. [Google Scholar] [CrossRef] [PubMed]
- Certara ENHANCES SYBYL-X Drug Design and Discovery Software Suite. Available online: https://www.certara.com/pressreleases/certara-enhances-sybyl-x-drug-design-and-discovery-software-suite/ (accessed on 9 May 2019).
- Xue, C.X.; Cui, S.Y.; Liu, M.C.; Hu, Z.D.; Fan, B.T. 3D QSAR studies on antimalarial alkoxylated and hydroxylated chalcones by CoMFA and CoMSIA. Eur. J. Med. Chem. 2004, 39, 745–753. [Google Scholar] [CrossRef]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef]
- Gustin, D.J.; Ma, Z.; Min, X.; Li, Y.; Hedberg, C.; Guimaraes, C.; Porter, A.C.; Lindstrom, M.; Lester-Zeiner, D.; Xu, G.; et al. Identification of potent, noncovalent fatty acid amide hydrolase (FAAH) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2492–2496. [Google Scholar] [CrossRef]
- Kono, M.; Matsumoto, T.; Kawamura, T.; Nishimura, A.; Kiyota, Y.; Oki, H.; Miyazaki, J.; Igaki, S.; Behnke, C.A.; Shimojo, M.; et al. Synthesis, SAR study, and biological evaluation of a series of piperazine ureas as fatty acid amide hydrolase (FAAH) inhibitors. Bioorg. Med. Chem. 2013, 21, 28–41. [Google Scholar] [CrossRef]
- Keith, J.M.; Apodaca, R.; Xiao, W.; Seierstad, M.; Pattabiraman, K.; Wu, J.; Webb, M.; Karbarz, M.J.; Brown, S.; Wilson, S.; et al. Thiadiazolopiperazinyl ureas as inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2008, 18, 4838–4843. [Google Scholar] [CrossRef]
- Kono, M.; Matsumoto, T.; Imaeda, T.; Kawamura, T.; Fujimoto, S.; Kosugi, Y.; Odani, T.; Shimizu, Y.; Matsui, H.; Shimojo, M.; et al. Design, synthesis, and biological evaluation of a series of piperazine ureas as fatty acid amide hydrolase inhibitors. Bioorg. Med. Chem. 2014, 22, 1468–1478. [Google Scholar] [CrossRef]
- Tichenor, M.S.; Keith, J.M.; Jones, W.M.; Pierce, J.M.; Merit, J.; Hawryluk, N.; Seierstad, M.; Palmer, J.A.; Webb, M.; Karbarz, M.J.; et al. Heteroaryl urea inhibitors of fatty acid amide hydrolase: Structure-mutagenicity relationships for arylamine metabolites. Bioorg. Med. Chem. Lett. 2012, 22, 7357–7362. [Google Scholar] [CrossRef]
- Keith, J.M.; Tichenor, M.S.; Apodaca, R.L.; Xiao, W.; Jones, W.M.; Seierstad, M.; Pierce, J.M.; Palmer, J.A.; Webb, M.; Karbarz, M.J.; et al. The SAR of brain penetration for a series of heteroaryl urea FAAH inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3109–3114. [Google Scholar] [CrossRef]
- Keith, J.M.; Jones, W.M.; Pierce, J.M.; Seierstad, M.; Palmer, J.A.; Webb, M.; Karbarz, M.J.; Scott, B.P.; Wilson, S.J.; Luo, L.; et al. Heteroarylureas with spirocyclic diamine cores as inhibitors of fatty acid amide hydrolase. Bioorg. Med. Chem. Lett. 2014, 24, 737–741. [Google Scholar] [CrossRef]
- Clark, M.; Cramer, R.D.; Van Opdenbosch, N. Validation of the general purpose Tripos 5.2 force field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q 2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Tropsha, A. Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Chakraborty, P.; Mitra, I.; Ojha, P.K.; Kar, S.; Das, R.N. Some case studies on application of “r(m)2” metrics for judging quality of quantitative structure-activity relationship predictions: Emphasis on scaling of response data. J. Comput. Chem. 2013, 34, 1071–1082. [Google Scholar] [CrossRef]
- Mauri, A.; Consonni, V.; Pavan, M.; Todeschini, R. Dragon software: An easy approach to molecular descriptor calculations MATCH Commun. Math. Comput. Chem. 2006, 56, 237–248. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | q2 | N | SEP | SEE | r2ncv | F | Field Contributions | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S | E | H | D | A | |||||||
| CoMSIA-S | 0.293 | 3 | 1.042 | 0.903 | 0.470 | 20.383 | 1 | ||||
| CoMSIA-E | 0.534 | 10 | 0.893 | 0.476 | 0.867 | 40.592 | 1 | ||||
| CoMSIA-H | 0.317 | 4 | 1.032 | 0.788 | 0.602 | 25.722 | 1 | ||||
| CoMSIA-D | 0.253 | 8 | 1.112 | 1.031 | 0.359 | 4.482 | 1 | ||||
| CoMSIA-A | 0.520 | 5 | 0.871 | 0.638 | 0.743 | 38.785 | 1 | ||||
| CoMSIA-SE | 0.519 | 10 | 0.907 | 0.417 | 0.898 | 54.799 | 0.314 | 0.686 | |||
| CoMSIA-SEH | 0.534 | 8 | 0.879 | 0.216 | 0.977 | 110.701 | 0.183 | 0.492 | 0.324 | ||
| CoMSIA-SEHD | 0.628 | 7 | 0.779 | 0.382 | 0.910 | 94.448 | 0.159 | 0.458 | 0.260 | 0.123 | |
| CoMSIA-SEHA | 0.688 | 7 | 0.713 | 0.332 | 0.933 | 128.486 | 0.131 | 0.346 | 0.177 | 0.347 | |
| CoMSIA-SED | 0.626 | 9 | 0.793 | 0.382 | 0.913 | 73.600 | 0.245 | 0.639 | 0.116 | ||
| CoMSIA-SEA | 0.725 | 7 | 0.670 | 0.350 | 0.925 | 114.665 | 0.182 | 0.387 | 0.421 | ||
| CoMSIA-SEDA | 0.765 | 7 | 0.620 | 0.327 | 0.934 | 132.475 | 0.154 | 0.357 | 0.099 | 0.389 | |
| CoMSIA-SH | 0.316 | 4 | 1.033 | 0.772 | 0.618 | 27.502 | 0.408 | 0.592 | |||
| CoMSIA-SD | 0.364 | 19 | 1.128 | 0.525 | 0.862 | 17.433 | 0.814 | 0.186 | |||
| CoMSIA-SA | 0.572 | 7 | 0.836 | 0.484 | 0.857 | 55.453 | 0.344 | 0.656 | |||
| CoMSIA-SHD | 0.426 | 3 | 0.939 | 0.799 | 0.585 | 32.396 | 0.219 | 0.479 | 0.303 | ||
| CoMSIA-SHA | 0.529 | 6 | 0.870 | 0.483 | 0.855 | 64.909 | 0.201 | 0.303 | 0.496 | ||
| CoMSIA-SDA | 0.719 | 7 | 0.678 | 0.404 | 0.900 | 83.485 | 0.235 | 0.210 | 0.555 | ||
| CoMSIA-SHDA | 0.673 | 7 | 0.731 | 0.366 | 0.918 | 103.744 | 0.156 | 0.240 | 0.164 | 0.440 | |
| CoMSIA-EH | 0.550 | 10 | 0.877 | 0.391 | 0.911 | 63.307 | 0.537 | 0.427 | |||
| CoMSIA-ED | 0.616 | 9 | 0.804 | 0.407 | 0.902 | 64.163 | 0.856 | 0.144 | |||
| CoMSIA-EA | 0.701 | 6 | 0.693 | 0.408 | 0.896 | 95.020 | 0.498 | 0.502 | |||
| CoMSIA-EHD | 0.641 | 8 | 0.771 | 0.376 | 0.915 | 85.695 | 0.525 | 0.343 | 0.132 | ||
| CoMSIA-EHA | 0.691 | 7 | 0.710 | 0.390 | 0.925 | 115.138 | 0.390 | 0.234 | 0.376 | ||
| CoMSIA-EDA | 0.752 | 7 | 0.636 | 0.366 | 0.918 | 103.539 | 0.453 | 0.111 | 0.437 | ||
| CoMSIA-EHDA | 0.742 | 8 | 0.654 | 0.311 | 0.942 | 128.936 | 0.341 | 0.211 | 0.093 | 0.355 | |
| CoMSIA-HD | 0.428 | 9 | 0.981 | 0.528 | 0.834 | 35.186 | 0.804 | 0.196 | |||
| CoMSIA-HA | 0.537 | 6 | 0.862 | 0.493 | 0.849 | 61.685 | 0.426 | 0.574 | |||
| CoMSIA-HDA | 0.682 | 10 | 0.738 | 0.331 | 0.936 | 90.828 | 0.356 | 0.155 | 0.490 | ||
| CoMSIA-DA | 0.705 | 11 | 0.716 | 0.465 | 0.876 | 39.132 | 0.240 | 0.760 | |||
| CoMSIA-ALL | 0.734 | 7 | 0.659 | 0.320 | 0.937 | 138.360 | 0.110 | 0.304 | 0.156 | 0.100 | 0.330 |
| Condition | Parameters | Threshold Value | CoMSIA |
|---|---|---|---|
| 1 | q2 | >0.5 | 0.734 |
| 2 | r2 | >0.6 | 0.966 |
| 3a | r02 | Close to value of r2 | 0.920 |
| 3b | r′02 | Close to value of r2 | 0.944 |
| 4a | k | 0.85 < k < 1.15 | 1.004 |
| 4b | k′ | 0.85 < k′ < 1.15 | 0.995 |
| 5a | (r2−r20)/r2 | <0.1 | 0.048 |
| 5b | (r2−r’20)/r2 | <0.1 | 0.023 |
| 06 | |r20−r′20| | <0.3 | 0.024 |
| 7 | >0.5 | 0.723 | |
| 8 | Q2F1 | >0.7 | 0.944 |
| 9 | Q2F2 | >0.7 | 0.943 |
| 10 | Q2F3 | >0.7 | 0.951 |
| 11 | CCC | >0.85 | 0.967 |
| 12 | ∆r2m | <0.2 | 0.056 |
| Iteration | q2 | r2ncv | Iteration | q2 | r2ncv |
|---|---|---|---|---|---|
| Random 1 | −0.013 | 0.107 | Random 6 | 0.006 | 0.119 |
| Random 2 | −0.030 | 0.087 | Random 7 | −0.093 | 0.183 |
| Random 3 | −0.052 | 0.082 | Random 8 | 0.085 | 0.188 |
| Random 4 | −0.198 | 0.108 | Random 9 | −0.034 | 0.086 |
| Random 5 | −0.202 | 0.179 | Random 10 | −0.100 | 0.073 |
| Mol | Exp. pIC50 | Pred. pIC50 | Residual | Mol | Exp. pIC50 | Pred. pIC50 | Residual |
|---|---|---|---|---|---|---|---|
| 1 | 5.331 | 5.620 | −0.29 | 46 t | 8.009 | 7.774 | 0.23 |
| 2 t | 6.076 | 6.034 | 0.04 | 47 | 6.575 | 6.286 | 0.29 |
| 3 | 6.893 | 6.317 | 0.58 | 48 | 7.987 | 7.699 | 0.29 |
| 4 t | 5.607 | 5.961 | −0.35 | 49 | 7.886 | 7.915 | −0.03 |
| 5 | 5.558 | 6.107 | −0.55 | 50 | 7.301 | 7.231 | 0.07 |
| 6 | 5.176 | 5.295 | −0.12 | 51 | 5.574 | 6.018 | −0.44 |
| 7 | 5.331 | 5.438 | −0.11 | 52 t | 7.638 | 7.624 | 0.01 |
| 8 | 6.456 | 6.032 | 0.42 | 53 | 5.933 | 6.164 | −0.23 |
| 9 t | 5.815 | 6.141 | −0.33 | 54 | 5.984 | 5.876 | 0.11 |
| 10 | 6.310 | 6.291 | 0.02 | 55 | 6.495 | 6.381 | 0.11 |
| 11 | 7.131 | 6.816 | 0.31 | 56 | 7.155 | 7.619 | −0.46 |
| 12 | 7.208 | 7.689 | −0.48 | 57 | 6.495 | 6.436 | 0.06 |
| 13 | 7.921 | 7.810 | 0.11 | 58 t | 7.155 | 7.268 | −0.11 |
| 14 t | 7.337 | 7.386 | −0.05 | 59 | 7.638 | 7.512 | 0.13 |
| 15 | 7.824 | 7.497 | 0.33 | 60 | 7.444 | 7.467 | −0.02 |
| 16 | 8.854 | 8.718 | 0.14 | 61 | 7.553 | 7.408 | 0.14 |
| 17 | 7.921 | 8.062 | −0.14 | 62 | 6.854 | 6.741 | 0.11 |
| 18 | 8.319 | 7.972 | 0.35 | 63 | 8.658 | 8.820 | −0.16 |
| 19 | 8.432 | 8.206 | 0.23 | 64 | 8.824 | 8.740 | 0.08 |
| 20 t | 6.959 | 7.170 | −0.21 | 65 | 10.143 | 10.287 | −0.14 |
| 21 | 8.770 | 8.758 | 0.01 | 66 | 10.602 | 10.685 | −0.08 |
| 22 | 8.538 | 8.559 | −0.02 | 67 t | 10.143 | 9.759 | 0.38 |
| 23 t | 6.921 | 7.421 | −0.50 | 68 | 10.097 | 10.144 | −0.05 |
| 24 | 8.620 | 8.273 | 0.35 | 69 | 8.061 | 8.023 | 0.04 |
| 25 | 9.036 | 9.126 | −0.09 | 70 | 8.114 | 8.106 | 0.01 |
| 26 | 8.678 | 8.415 | 0.26 | 71 | 7.495 | 7.397 | 0.10 |
| 27 | 9.174 | 9.057 | 0.12 | 72 | 6.987 | 6.787 | 0.20 |
| 28 | 8.495 | 8.427 | 0.07 | 73 t | 7.553 | 7.707 | −0.15 |
| 29 | 8.921 | 9.236 | −0.32 | 74 | 7.553 | 7.757 | −0.20 |
| 30 t | 9.066 | 8.814 | 0.25 | 75 | 8.237 | 7.347 | 0.89 |
| 31 | 7.482 | 6.743 | 0.74 | 76 | 5.886 | 5.739 | 0.15 |
| 32 | 6.818 | 6.490 | 0.33 | 77 | 8.174 | 8.153 | 0.02 |
| 33 | 5.731 | 6.422 | −0.69 | 78 t | 8.469 | 8.155 | 0.31 |
| 34 | 6.762 | 6.654 | 0.11 | 79 | 7.161 | 7.463 | −0.30 |
| 35 | 5.886 | 6.710 | −0.82 | 80 | 8.000 | 8.225 | −0.23 |
| 36 | 6.714 | 6.734 | −0.02 | 81 | 7.658 | 8.237 | −0.58 |
| 37 t | 6.460 | 6.848 | −0.39 | 82 | 9.301 | 9.394 | −0.09 |
| 38 | 6.590 | 6.753 | −0.16 | 83 | 7.337 | 7.216 | 0.12 |
| 39 | 7.499 | 7.072 | 0.43 | 84 t | 7.482 | 7.278 | 0.20 |
| 40 | 7.018 | 6.901 | 0.12 | 85 | 7.000 | 7.134 | −0.13 |
| 41 t | 6.845 | 6.878 | −0.03 | 86 | 7.770 | 8.110 | −0.34 |
| 42 | 5.972 | 6.412 | −0.44 | 87 | 6.854 | 6.957 | −0.10 |
| 43 | 6.079 | 6.471 | −0.39 | 88 | 9.000 | 8.926 | 0.07 |
| 44 | 7.161 | 7.148 | 0.01 | 89 | 8.886 | 8.686 | 0.20 |
| 45 t | 7.574 | 7.855 | −0.28 | 90 | 9.000 | 8.960 | 0.04 |
| N° | Structure | Pred. pIC50 | N° | Structure | Pred. pIC50 |
|---|---|---|---|---|---|
| 1x |  | 10.889 | 6x |  | 11.744 |
| 2x |  | 11.388 | 7x |  | 11.599 |
| 3x |  | 11.490 | 8x |  | 11.822 |
| 4x |  | 10.990 | 9x |  | 12.416 |
| 5x |  | 11.253 | 10x |  | 12.196 |
 |
 |
 |
 |
 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorca, M.; Valdes, Y.; Chung, H.; Romero-Parra, J.; Pessoa-Mahana, C.D.; Mella, J. Three-Dimensional Quantitative Structure-Activity Relationships (3D-QSAR) on a Series of Piperazine-Carboxamides Fatty Acid Amide Hydrolase (FAAH) Inhibitors as a Useful Tool for the Design of New Cannabinoid Ligands. Int. J. Mol. Sci. 2019, 20, 2510. https://doi.org/10.3390/ijms20102510
Lorca M, Valdes Y, Chung H, Romero-Parra J, Pessoa-Mahana CD, Mella J. Three-Dimensional Quantitative Structure-Activity Relationships (3D-QSAR) on a Series of Piperazine-Carboxamides Fatty Acid Amide Hydrolase (FAAH) Inhibitors as a Useful Tool for the Design of New Cannabinoid Ligands. International Journal of Molecular Sciences. 2019; 20(10):2510. https://doi.org/10.3390/ijms20102510
Chicago/Turabian StyleLorca, Marcos, Yudisladys Valdes, Hery Chung, Javier Romero-Parra, C. David Pessoa-Mahana, and Jaime Mella. 2019. "Three-Dimensional Quantitative Structure-Activity Relationships (3D-QSAR) on a Series of Piperazine-Carboxamides Fatty Acid Amide Hydrolase (FAAH) Inhibitors as a Useful Tool for the Design of New Cannabinoid Ligands" International Journal of Molecular Sciences 20, no. 10: 2510. https://doi.org/10.3390/ijms20102510
APA StyleLorca, M., Valdes, Y., Chung, H., Romero-Parra, J., Pessoa-Mahana, C. D., & Mella, J. (2019). Three-Dimensional Quantitative Structure-Activity Relationships (3D-QSAR) on a Series of Piperazine-Carboxamides Fatty Acid Amide Hydrolase (FAAH) Inhibitors as a Useful Tool for the Design of New Cannabinoid Ligands. International Journal of Molecular Sciences, 20(10), 2510. https://doi.org/10.3390/ijms20102510