Epigenetic Mechanisms in Hepatic Stellate Cell Activation During Liver Fibrosis and Carcinogenesis



Abstract
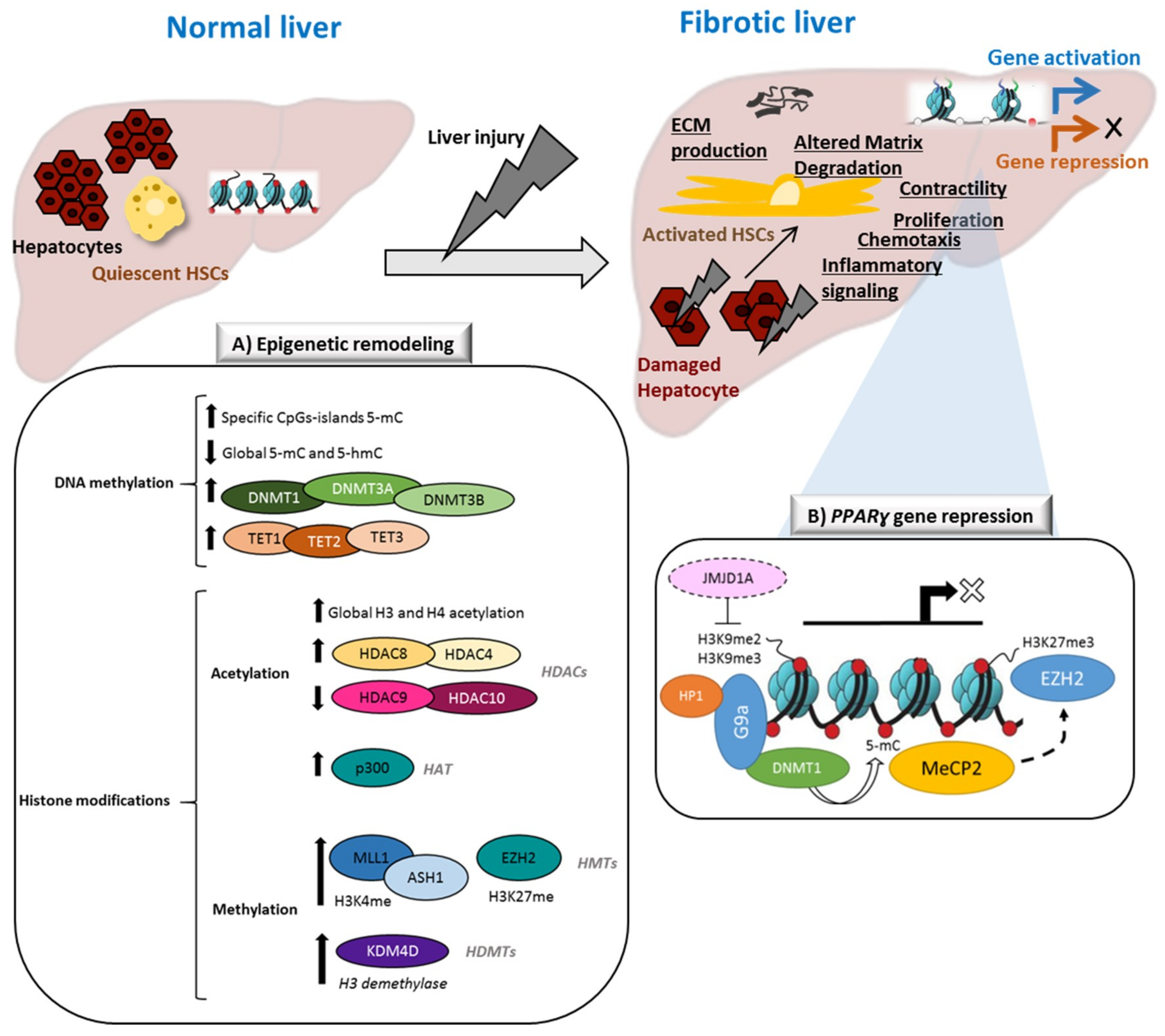
1. Introduction
2. Changes in DNA Methylation during HSCs Activation
3. Reprogramming of the Histone Code in HSCs Activation
3.1. Histone Acetylation in HSCs Activation
3.2. Histone Methylation in HSCs Activation
4. Crosstalk between DNA Methylation and the Histone Code: Another Layer of Complexity in Fibrogenic Activation
5. Translational Perspectives
5.1. Epigenetic Marks as Biomarkers for Liver Fibrosis
5.2. Epigenetic Drugs Targeting the Fibrotic Stroma
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CLD | Chronic Liver Disease |
| ECM | Extracellular Matrix |
| HSCs | Hepatic Stellate Cells |
| DNA | Deoxyribonucleic Acid |
| PTM | Postranslational Modifications |
| HCC | Hepatocellular Carcinoma |
| CpG | Cytosine Guanine Dinucleotide |
| MeCP2 | Methyl-CpG-Binding Domain Protein 2 |
| MBD1-4 | Methyl-CpG-Binding Domain Protein 1-4 |
| DNMTs | DNA-Methyltransferases |
| 5mC | 5-Methylcytosine |
| 5hmC | 5-Hydroxymethylcytosine |
| TETs | Ten Eleven Translocation Enzymes |
| APC | Adenomatous Polyposis Coli Protein |
| Wnt5a | Wnt Family Member 5A |
| ACTG | Actin, Gamma-Enteric Smooth Muscle |
| LOXL | Lysyl Oxidase Like |
| Col4a | Collagen Type IV Alpha |
| ADAMTs | ADAM Metallopeptidase with Thrombospondin Type 1 Motif |
| MMP | Matrix Metalloproteinase |
| TGFβR | Transforming Growth Factor Beta Receptor |
| PTEN | Fosfatidilinositol-3,4,5-Trisfosfato 3-Fosfatasa |
| HATs | Histone Acetyltransferases |
| HDACs | Hisone Deacetylases |
| HMTs | Histone Methyltransferases |
| HDMTs | Hisone Demethylases |
| α-SMA | Alpha Smooth Muscle Actin |
| CTGF | Connective Tissue Growth Factor |
| CXCL12 | C-X-C Motif Chemokine 12 |
| IL | Interleukin |
| PDGFA | Platelet Derived Growth Factor Subunit A |
| VEGFA | Vascular Endothelial Growth Factor A |
| SIRT | Seven Sirtuin Proteins |
| mRNA | Messenger Ribonucleic Acid |
| HGF | Hepatocyte Growth Factor |
| TSA | Trichostatin A |
| ERK | Extracellular Signal–Regulated Kinases |
| PI3K | Phosphatidylinositol-3- Kinase |
| MLL | Mixed Lineage Leukemia Protein |
| MEF | Mouse Embryonic Fibroblast |
| COMPASS | Complex Proteins Associated with Set1 |
| ASH2 | Set1/Ash2 Histone Methyltransferase complex subunit ASH2 |
| SET1 | Histone-lysine N-methyltransferase SETD1A |
| TIMP | Tissue Inhibitors of Metalloproteinases |
| EZH2 | Enhancer of Zeste 2 Polycomb Repressive Complex 2 Subunit |
| PCR2 | Polycomb Repressive Complex 2 |
| PDGF-BB | Platelet-Derived Growth Factor-BB |
| PPARɣ | Peroxisome Proliferator-Activated Receptor Gamma |
| JMJD | Jumonji Domain-Containing Protein |
| KDM4D | Lysine Demethylase 4D |
| HP1 | Heterochromatin Protein 1 |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| ALD | Alcoholic Liver Disease |
| NASH | Non-Alcoholic Steatohepatitis |
| SAHA | Suberoy- Lanilide Hydroxamic Acid |
| DZNep | 3-Deazaneplanocin A hydrochloride |
| ScAb | Single-Chain Antibody |
References
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Schoepp, M.; Ströse, A.; Haier, J. Dysregulation of miRNA expression in cancer associated fibroblasts (CAFs) and its consequences on the tumor microenvironment. Cancers 2017, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- El–Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Berasain, C.; Avila, M.A. Regulation of hepatocyte identity and quiescence. Cell. Mol. Life Sci. 2015, 72, 3831–3851. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Page, A.; Mann, D.A.; Mann, J. The mechanisms of HSC activation and epigenetic regulation of HSCs phenotypes. Curr. Pathobiol. Rep. 2014, 2, 163–170. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Böhm, F.; Köhler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.; Schwabe, R. Origin and function of myofibroblasts in the liver. Semin. Liver Dis. 2015, 35, e1. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainier, D.Y.R. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Investig. 2013, 123, 1902–1910. [Google Scholar] [CrossRef]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef]
- Troeger, J.S.; Mederacke, I.; Gwak, G.-Y.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.-P.; Friedman, R.A.; Schwabe, R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012, 143, 1073–1083. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Mann, J.; Mann, D.A. Epigenetic regulation of wound healing and fibrosis. Curr. Opin. Rheumatol. 2013, 25, 101–107. [Google Scholar] [CrossRef]
- Mann, J.; Mann, D.A. Transcriptional regulation of hepatic stellate cells. Adv. Drug Deliv. Rev. 2009, 61, 497–512. [Google Scholar] [CrossRef]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; de Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Shenker, N.; Flanagan, J.M. Intragenic DNA methylation: Implications of this epigenetic mechanism for cancer research. Br. J. Cancer 2012, 106, 248–253. [Google Scholar] [CrossRef]
- Gujar, H.; Weisenberger, D.; Liang, G. The roles of human DNA methyltransferases and their isoforms in shaping the epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef]
- Edwards, J.R.; Yarychkivska, O.; Boulard, M.; Bestor, T.H. DNA methylation and DNA methyltransferases. Epigenet. Chromatin. 2017, 10, 23. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef]
- Delatte, B.; Deplus, R.; Fuks, F. Playing TETris with DNA modifications. EMBO J. 2014, 33, 1198–1211. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Wilson, C.L.; Mann, D.A.; Borthwick, L.A. Epigenetic reprogramming in liver fibrosis and cancer. Adv. Drug Deliv. Rev. 2017, 121, 124–132. [Google Scholar] [CrossRef]
- Dowson, C.; O’Reilly, S. DNA methylation in fibrosis. Eur. J. Cell Biol. 2016, 95, 323–330. [Google Scholar] [CrossRef]
- Mann, J.; Oakley, F.; Akiboye, F.; Elsharkawy, A.; Thorne, A.W.; Mann, D.A. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: Implications for wound healing and fibrogenesis. Cell Death Differ. 2007, 14, 275–285. [Google Scholar] [CrossRef]
- Komatsu, Y.; Waku, T.; Iwasaki, N.; Ono, W.; Yamaguchi, C.; Yanagisawa, J. Global analysis of DNA methylation in early-stage liver fibrosis. BMC Med. Genom. 2012, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Götze, S.; Schumacher, E.C.; Kordes, C.; Häussinger, D. Epigenetic changes during hepatic stellate cell activation. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- El Taghdouini, A.; Sørensen, A.L.; Reiner, A.H.; Coll, M.; Verhulst, S.; Mannaerts, I.; Øie, C.I.; Smedsrød, B.; Najimi, M.; Sokal, E.; et al. Genome-wide analysis of DNA methylation and gene expression patterns in purified, uncultured human liver cells and activated hepatic stellate cells. Oncotarget 2015, 6, 26729–26745. [Google Scholar] [CrossRef]
- Xiong, W.-J.; Hu, L.-J.; Jian, Y.-C.; Wang, L.-J.; Jiang, M.; Li, W.; He, Y. Wnt5a participates in hepatic stellate cell activation observed by gene expression profile and functional assays. World J. Gastroenterol. 2012, 18, 1745. [Google Scholar] [CrossRef]
- Miao, C.; Yang, Y.; He, X.; Huang, C.; Huang, Y.; Zhang, L.; Lv, X.-W.; Jin, Y.; Li, J. Wnt signaling in liver fibrosis: Progress, challenges and potential directions. Biochimie 2013, 95, 2326–2335. [Google Scholar] [CrossRef]
- Jiang, F.; Parsons, C.J.; Stefanovic, B. Gene expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J. Hepatol. 2006, 45, 401–409. [Google Scholar] [CrossRef]
- Bian, E.-B.; Huang, C.; Wang, H.; Chen, X.-X.; Zhang, L.; Lv, X.-W.; Li, J. Repression of Smad7 mediated by DNMT1 determines hepatic stellate cell activation and liver fibrosis in rats. Toxicol. Lett. 2014, 224, 175–185. [Google Scholar] [CrossRef]
- Bian, E.-B.; Huang, C.; Ma, T.-T.; Tao, H.; Zhang, H.; Cheng, C.; Lv, X.-W.; Li, J. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol. Appl. Pharmacol. 2012, 264, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Paoli, P.; Salvador, E.M.; White, S.; French, J.; Mann, J. Hepatic stellate cell transdifferentiation involves genome-wide remodeling of the DNA methylation landscape. J. Hepatol. 2016, 64, 661–673. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—Writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, M.S.; Wolberger, C. How does the histone code work? Biochem. Cell Biol. 2005, 83, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Liu, Z.; Tu, K.; Zhang, H.; Chen, C.; Yaqoob, U.; Wang, Y.; Wen, J.; van Deursen, J.; Sicard, D.; et al. P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology 2018, 154, 2209–2221. [Google Scholar] [CrossRef]
- Hammond, C.M.; Strømme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 2017, 18, 141–158. [Google Scholar] [CrossRef]
- Mannaerts, I.; Nuytten, N.R.; Rogiers, V.; Vanderkerken, K.; van Grunsven, L.A.; Geerts, A. Chronic administration of valproic acid inhibits activation of mouse hepatic stellate cells in vitro and in vivo. Hepatology 2010, 51, 603–614. [Google Scholar] [CrossRef]
- Qin, L.; Han, Y.-P. Epigenetic repression of matrix metalloproteinases in myofibroblastic hepatic stellate cells through histone deacetylases 4. Am. J. Pathol. 2010, 177, 1915–1928. [Google Scholar] [CrossRef]
- Mannaerts, I.; Eysackers, N.; Onyema, O.O.; van Beneden, K.; Valente, S.; Mai, A.; Odenthal, M.; van Grunsven, L.A. Class II HDAC inhibition hampers hepatic stellate cell activation by induction of MicroRNA-29. PLoS ONE 2013, 8, e55786. [Google Scholar] [CrossRef]
- Shaker, M.E.; Ghani, A.; Shiha, G.E.; Ibrahim, T.M.; Mehal, W.Z. Nilotinib induces apoptosis and autophagic cell death of activated hepatic stellate cells via inhibition of histone deacetylases. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 1992–2003. [Google Scholar] [CrossRef]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors: Insights into mechanisms of lethality. Expert Opin. Ther. Targets 2005, 9, 809–824. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix metalloproteinases in liver injury, repair and fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef]
- Yan, C.; Zhou, L.; Han, Y.-P. Contribution of hepatic stellate cells and matrix metalloproteinase 9 in acute liver failure. Liver Int. 2008, 28, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ruehl, M.; Somasundaram, R.; Hahn, E.G. Matrix as a modulator of hepatic fibrogenesis. Semin. Liver Dis. 2001, 21, 351–372. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-P.; Zhou, L.; Wang, J.; Xiong, S.; Garner, W.L.; French, S.W.; Tsukamoto, H. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J. Biol. Chem. 2004, 279, 4820–4828. [Google Scholar] [CrossRef] [PubMed]
- Pannem, R.R.; Dorn, C.; Hellerbrand, C.; Massoumi, R. Cylindromatosis gene CYLD regulates hepatocyte growth factor expression in hepatic stellate cells through interaction with histone deacetylase 7. Hepatology 2014, 60, 1066–1081. [Google Scholar] [CrossRef] [PubMed]
- Barter, M.J.; Pybus, L.; Litherland, G.J.; Rowan, A.D.; Clark, I.M.; Edwards, D.R.; Cawston, T.E.; Young, D.A. HDAC-mediated control of ERK- and PI3K-dependent TGF-β-induced extracellular matrix-regulating genes. Matrix Biol. 2010, 29, 602–612. [Google Scholar] [CrossRef]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim. Biophys. Acta 2007, 1773, 1572–1582. [Google Scholar] [CrossRef]
- Rombouts, K.; Niki, T.; Greenwel, P.; Vandermonde, A.; Wielant, A.; Hellemans, K.; de Bleser, P.; Yoshida, M.; Schuppan, D.; Rojkind, M.; et al. Trichostatin A, a histone deacetylase inhibitor, suppresses collagen synthesis and prevents TGF-beta(1)-induced fibrogenesis in skin fibroblasts. Exp. Cell Res. 2002, 278, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, M.; Hishikawa, K.; Marumo, T.; Fujita, T. Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-beta1 in human renal epithelial cells. J. Am. Soc. Nephrol. 2007, 18, 58–65. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, L.; Jiao, F.-Z.; Zhang, W.-B.; Chen, Q.; Gong, Z.-J. Histone deacetylase inhibitor suberoylanilide hydroxamic acid alleviates liver fibrosis by suppressing the transforming growth factor-β1 signal pathway. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Paoli, P.P.; Hill, S.J.; Howarth, R.; Wu, R.; Kweon, S.-M.; French, J.; White, S.; Tsukamoto, H.; Mann, D.A.; et al. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J. Hepatol. 2015, 62, 388–397. [Google Scholar] [CrossRef]
- Tian, W.; Fan, Z.; Li, J.; Hao, C.; Li, M.; Xu, H.; Wu, X.; Zhou, B.; Zhang, L.; Fang, M.; et al. Myocardin-related transcription factor A (MRTF-A) plays an essential role in hepatic stellate cell activation by epigenetically modulating TGF-β signaling. Int. J. Biochem. Cell Biol. 2016, 71, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.D.; Vakoc, C.R.; Rozovskaia, T.; Zheng, X.; Patel, S.; Nakamura, T.; Canaani, E.; Blobel, G.A. Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol. Cell. Biol. 2007, 27, 8466–8479. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Wilson, C.L.; Zeybel, M.; Walsh, M.; Amin, S.; Robinson, S.; White, S.A.; Burt, A.D.; Oakley, F.; Tsukamoto, H.; et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012, 56, 1129–1139. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef]
- Mann, J.; Chu, D.C.K.; Maxwell, A.; Oakley, F.; Zhu, N.-L.; Tsukamoto, H.; Mann, D.A. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010, 138, 705–714. [Google Scholar] [CrossRef]
- Martin-Mateos, R.; de Assuncao, T.M.; Arab, J.P.; Jalan-Sakrikar, N.; Yaqoob, U.; Greuter, T.; Verma, V.K.; Mathison, A.J.; Cao, S.; Lomberk, G.; et al. Enhancer of zeste homologue 2 inhibition attenuates TGF-β dependent hepatic stellate cell activation and liver fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 197–209. [Google Scholar] [CrossRef]
- Panebianco, C.; Oben, J.A.; Vinciguerra, M.; Pazienza, V. Senescence in hepatic stellate cells as a mechanism of liver fibrosis reversal: A putative synergy between retinoic acid and PPAR-gamma signalings. Clin. Exp. Med. 2017, 17, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Hazra, S.; Miyahara, T.; Rippe, R.A.; Tsukamoto, H. PPAR gamma and hepatic stellate cells. Comp. Hepatol. 2004, 3, S7. [Google Scholar] [CrossRef]
- Miyahara, T.; Schrum, L.; Rippe, R.; Xiong, S.; Yee, H.F.; Motomura, K.; Anania, F.A.; Willson, T.M.; Tsukamoto, H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 2000, 275, 35715–35722. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, S.; Zhao, Y.; Lin, C.; Zhong, F.; Jin, L.; He, F.; Wang, H. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015, 29, 1830–1841. [Google Scholar] [CrossRef]
- Dong, F.; Jiang, S.; Li, J.; Wang, Y.; Zhu, L.; Huang, Y.; Jiang, X.; Hu, X.; Zhou, Q.; Zhang, Z.; et al. The histone demethylase KDM4D promotes hepatic fibrogenesis by modulating Toll-like receptor 4 signaling pathway. EBioMedicine 2019, 39, 472–483. [Google Scholar] [CrossRef]
- Smith, E.; Shilatifard, A. The chromatin signaling pathway: Diverse mechanisms of recruitment of histone-modifying enzymes and varied biological outcomes. Mol. Cell 2010, 40, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Yadav, T.; Quivy, J.-P.; Almouzni, G. Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science 2018, 361, 1332–1336. [Google Scholar] [CrossRef]
- Hu, B.; Gharaee-Kermani, M.; Wu, Z.; Phan, S.H. Essential role of MeCP2 in the regulation of myofibroblast differentiation during pulmonary fibrosis. Am. J. Pathol. 2011, 178, 1500–1508. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, W.; Wani, M.; Yu, X.; Ashraf, M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS ONE 2014, 9, e88685. [Google Scholar] [CrossRef]
- Zhou, P.; Lu, Y.; Sun, X.-H. Zebularine suppresses TGF-beta-induced lens epithelial cell-myofibroblast transdifferentiation by inhibiting MeCP2. Mol. Vis. 2011, 17, 2717–2723. [Google Scholar]
- Bian, E.-B.; Huang, C.; Wang, H.; Chen, X.-X.; Tao, H.; Zhang, L.; Lv, X.; Li, J. The role of methyl-CpG binding protein 2 in liver fibrosis. Toxicology 2013, 309, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Mellén, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef]
- Bárcena-Varela, M.; Caruso, S.; Llerena, S.; Álvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Rebouissou, S.; Alvarez, L.; Jimenez, M.; et al. Dual Targeting of histone methyltransferase G9a and DNA-methyltransferase 1 for the treatment of experimental hepatocellular carcinoma. Hepatology 2019, 69, 587–603. [Google Scholar] [CrossRef]
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenet. 2015, 7, 25. [Google Scholar] [CrossRef]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef]
- Hardy, T.; Mann, D.A. Epigenetics in liver disease: From biology to therapeutics. Gut 2016, 65, 1895–1905. [Google Scholar] [CrossRef]
- Mann, J.; Reeves, H.L.; Feldstein, A.E. Liquid biopsy for liver diseases. Gut 2018, 67, 2204–2212. [Google Scholar] [CrossRef]
- Kuykendall, J.R. 5-azacytidine and decitabine monotherapies of myelodysplastic disorders. Ann. Pharmacother. 2005, 39, 1700–1709. [Google Scholar] [CrossRef]
- Issa, J.-P.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015, 16, 1099–1110. [Google Scholar] [CrossRef]
- Jansen, Y.J.L.; Verset, G.; Schats, K.; van Dam, P.-J.; Seremet, T.; Kockx, M.; van Laethem, J.-L.B.; Neyns, B. Phase I clinical trial of decitabine (5-aza-2′-deoxycytidine) administered by hepatic arterial infusion in patients with unresectable liver-predominant metastases. ESMO Open 2019, 4, e000464. [Google Scholar] [CrossRef]
- Kuang, Y.; El-Khoueiry, A.; Taverna, P.; Ljungman, M.; Neamati, N. Guadecitabine (SGI-110) priming sensitizes hepatocellular carcinoma cells to oxaliplatin. Mol. Oncol. 2015, 9, 1799–1814. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Park, K.C.; Park, J.H.; Jeon, J.Y.; Kim, S.Y.; Kim, J.M.; Lim, C.Y.; Lee, T.H.; Kim, H.K.; Lee, H.G.; Kim, S.M.; et al. A new histone deacetylase inhibitor improves liver fibrosis in BDL rats through suppression of hepatic stellate cells. Br. J. Pharmacol. 2014, 171, 4820–4830. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Wang, J.; Lam, W.; Kwong, S.; Li, F.; Friedman, S.L.; Zhou, S.; Ren, Q.; Xu, Z.; et al. A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-β and vascular endothelial growth factor signalling. Liver Int. 2013, 33, 504–515. [Google Scholar] [CrossRef]
- Pang, M.; Kothapally, J.; Mao, H.; Tolbert, E.; Ponnusamy, M.; Chin, Y.E.; Zhuang, S. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. Physiol. 2009, 297, F996–F1005. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.-S.; Xia, W.-S.; Yi, C.-G.; Wang, Y.-M.; Li, B.; Xia, W.; Liu, B.; Guo, S.-Z.; Sun, X.-D. Trichostatin A inhibits collagen synthesis and induces apoptosis in keloid fibroblasts. Arch. Dermatol. Res. 2011, 303, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.R.; Haitchi, H.M.; Thatcher, T.H.; Sime, P.J.; Kottmann, R.M.; Ganesan, A.; Packham, G.; O’Reilly, K.M.A.; Davies, D.E. Spiruchostatin A inhibits proliferation and differentiation of fibroblasts from patients with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 687–694. [Google Scholar] [CrossRef]
- Kirpich, I.; Zhang, J.; Gobejishvili, L.; Kharebava, G.; Barker, D.; Ghare, S.; Joshi-Barve, S.; McClain, C.J.; Barve, S. Binge ethanol-induced HDAC3 down-regulates Cpt1α expression leading to hepatic steatosis and injury. Alcohol. Clin. Exp. Res. 2013, 37, 1920–1929. [Google Scholar] [CrossRef]
- Watanabe, T.; Tajima, H.; Hironori, H.; Nakagawara, H.; Ohnishi, I.; Takamura, H.; Ninomiya, I.; Kitagawa, H.; Fushida, S.; Tani, T.; et al. Sodium valproate blocks the transforming growth factor (TGF)-β1 autocrine loop and attenuates the TGF-β1-induced collagen synthesis in a human hepatic stellate cell line. Int. J. Mol. Med. 2011, 28, 919–925. [Google Scholar] [CrossRef]
- Niki, T.; Rombouts, K.; de Bleser, P.; de Smet, K.; Rogiers, V.; Schuppan, D.; Yoshida, M.; Gabbiani, G.; Geerts, A. A histone deacetylase inhibitor, trichostatin A, suppresses myofibroblastic differentiation of rat hepatic stellate cells in primary culture. Hepatology 1999, 29, 858–867. [Google Scholar] [CrossRef]
- Rombouts, K.; Knittel, T.; Machesky, L.; Braet, F.; Wielant, A.; Hellemans, K.; de Bleser, P.; Gelman, I.; Ramadori, G.; Geerts, A. Actin filament formation, reorganization and migration are impaired in hepatic stellate cells under influence of trichostatin A, a histone deacetylase inhibitor. J. Hepatol. 2002, 37, 788–796. [Google Scholar] [CrossRef]
- Ding, D.; Chen, L.-L.; Zhai, Y.-Z.; Hou, C.-J.; Tao, L.-L.; Lu, S.-H.; Wu, J.; Liu, X.-P. Trichostatin A inhibits the activation of Hepatic stellate cells by Increasing C/EBP-α Acetylation in vivo and in vitro. Sci. Rep. 2018, 8, 4395. [Google Scholar] [CrossRef]
- Zeybel, M.; Luli, S.; Sabater, L.; Hardy, T.; Oakley, F.; Leslie, J.; Page, A.; Salvador, E.M.; Sharkey, V.; Tsukamoto, H.; et al. A proof-of-concept for epigenetic therapy of tissue fibrosis: Inhibition of liver fibrosis progression by 3-deazaneplanocin A. Mol. Ther. 2017, 25, 218–231. [Google Scholar] [CrossRef] [PubMed]
- José-Enériz, E.S.; Agirre, X.; Rabal, O.; Vilas-Zornoza, A.; Sanchez-Arias, J.A.; Miranda, E.; Ugarte, A.; Roa, S.; Paiva, B.; de Mendoza, A.E.; et al. Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 2017, 8, 15424. [Google Scholar] [CrossRef]


{kind=link}
| Expression | Specific Genes | Regulatory Mechanism |
|---|---|---|
| Increased expression | Apc2 [35] | Promoter-DNA hypomethylation |
| Wnt5a [39] | Promoter-DNA hypomethylation | |
| Actg2 [36] | Promoter-DNA hypomethylation | |
| Loxl1 [36] | Promoter-DNA hypomethylation | |
| Loxl2 [36] | Promoter-DNA hypomethylation | |
| Col4a1/2 [36] | Promoter-DNA hypomethylation | |
| α-SMA [45,65] | p300-dependent transcription | |
| Elastin [62] | MLL1-mediated H3K4 methylation | |
| Col1a1/2 [64,65] | COMPASS-mediated H3K4 methylation | |
| TIMP-1 [65] | ASH1-mediated H3K4 methylation | |
| ASH1-mediated H3K4 methylation | ||
| TGFβ1 [65] | ASH1-mediated H3K4 methylation | |
| Decreased expression | Adamts9 [36] | Promoter-DNA hypermethylation |
| Mmp15 [36] | Promoter-DNA hypermethylation | |
| Smad7 [40] | Promoter-DNA hypermethylation | |
| Pten [41] | Promoter-DNA hypermethylation | |
| Mmp9 [48] | HDAC4-mediated histone deacetylation | |
| HGF [56] | HDAC7-mediated histone deacteylation | |
| PPARγ [33,65,67] | Promoter DNA-hypermethylation | |
| EZH2-mediated H3K27 methylation | ||
| G9a-mediated H3K9 methylation | ||
| MeCp2-mediated repressive complex recruitment |
| Activated | Inactivated | Associated Epigenetic Marks |
|---|---|---|
| DNMTs [35] | TETs [42] | Global DNA hypomethylation Promoter-specific hypermethylation |
| p300 [45] | Histone and nonhistone targets acetylation | |
| HDAC1* [48]; HDAC4 [48]; HDAC5* [49]; HDAC6* [49]; HDAC8 [47] | HDAC9 [49]; HDAC10 [49] | Changes in H3 and H4 acetylation patterns |
| MLL1 [62]; ASH2 [63]; SET1 [63]; ASH1 [64] | Increased gene-specific H3K4me2/3 | |
| EZH2 [67,68,69] | Increased gene-specific H3K27me3 | |
| G9a [81] | Increased gene-specific H3K9me2 | |
| JMJD1A [72] | Reduced gene-specific H3K4me2/3 | |
| JMJD2D [73] | H3K9 demethylation |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barcena-Varela, M.; Colyn, L.; Fernandez-Barrena, M.G. Epigenetic Mechanisms in Hepatic Stellate Cell Activation During Liver Fibrosis and Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 2507. https://doi.org/10.3390/ijms20102507
Barcena-Varela M, Colyn L, Fernandez-Barrena MG. Epigenetic Mechanisms in Hepatic Stellate Cell Activation During Liver Fibrosis and Carcinogenesis. International Journal of Molecular Sciences. 2019; 20(10):2507. https://doi.org/10.3390/ijms20102507
Chicago/Turabian StyleBarcena-Varela, Marina, Leticia Colyn, and Maite G. Fernandez-Barrena. 2019. "Epigenetic Mechanisms in Hepatic Stellate Cell Activation During Liver Fibrosis and Carcinogenesis" International Journal of Molecular Sciences 20, no. 10: 2507. https://doi.org/10.3390/ijms20102507
APA StyleBarcena-Varela, M., Colyn, L., & Fernandez-Barrena, M. G. (2019). Epigenetic Mechanisms in Hepatic Stellate Cell Activation During Liver Fibrosis and Carcinogenesis. International Journal of Molecular Sciences, 20(10), 2507. https://doi.org/10.3390/ijms20102507