Differential Effects of a Full and Biased Ghrelin Receptor Agonist in a Mouse Kindling Model

 , ,
, ,
Abstract

1. Introduction
2. Results
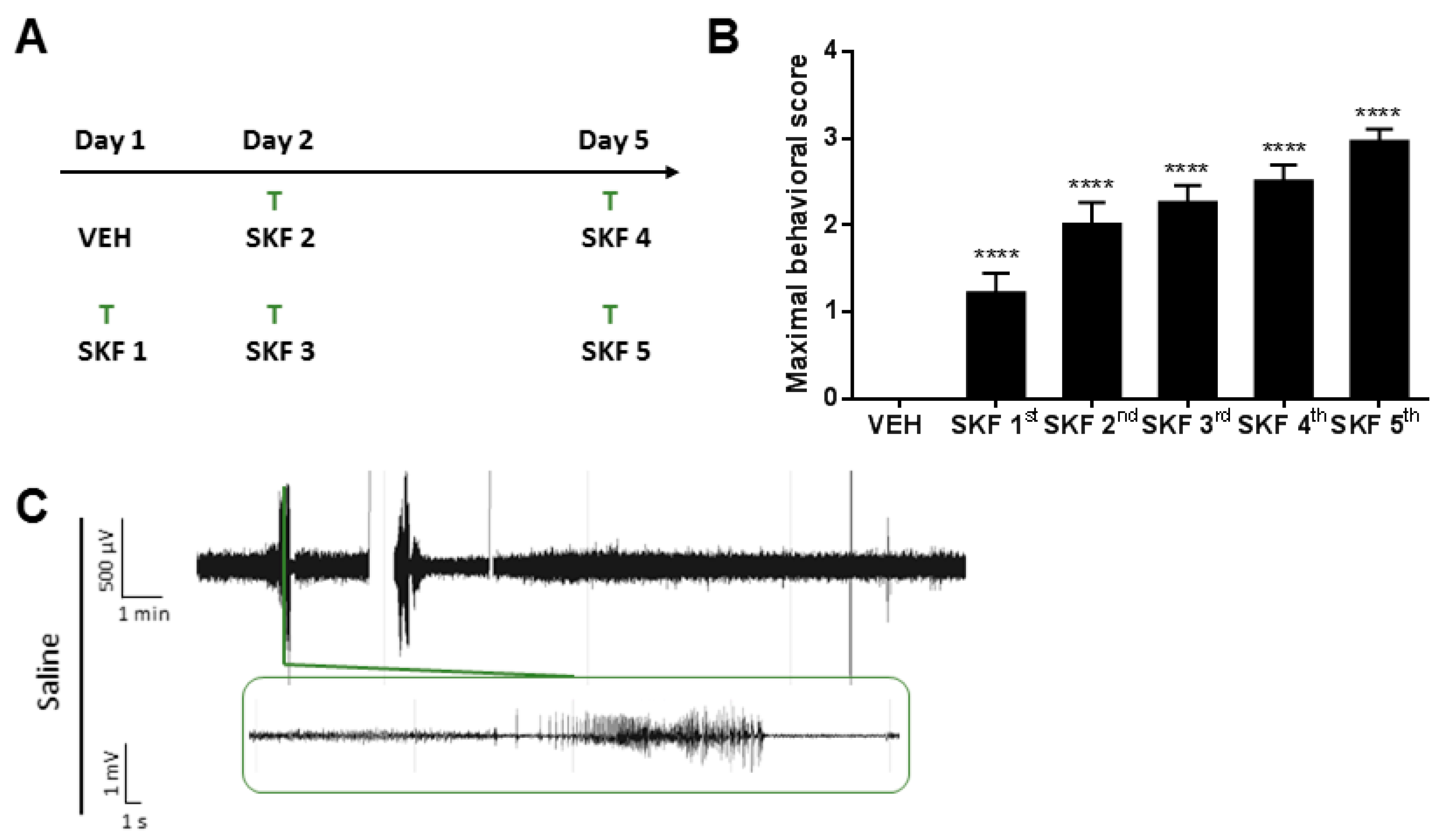
2.1. SKF81297 Induces Hippocampal Discharges Corresponding to Seizure-Like Behavior
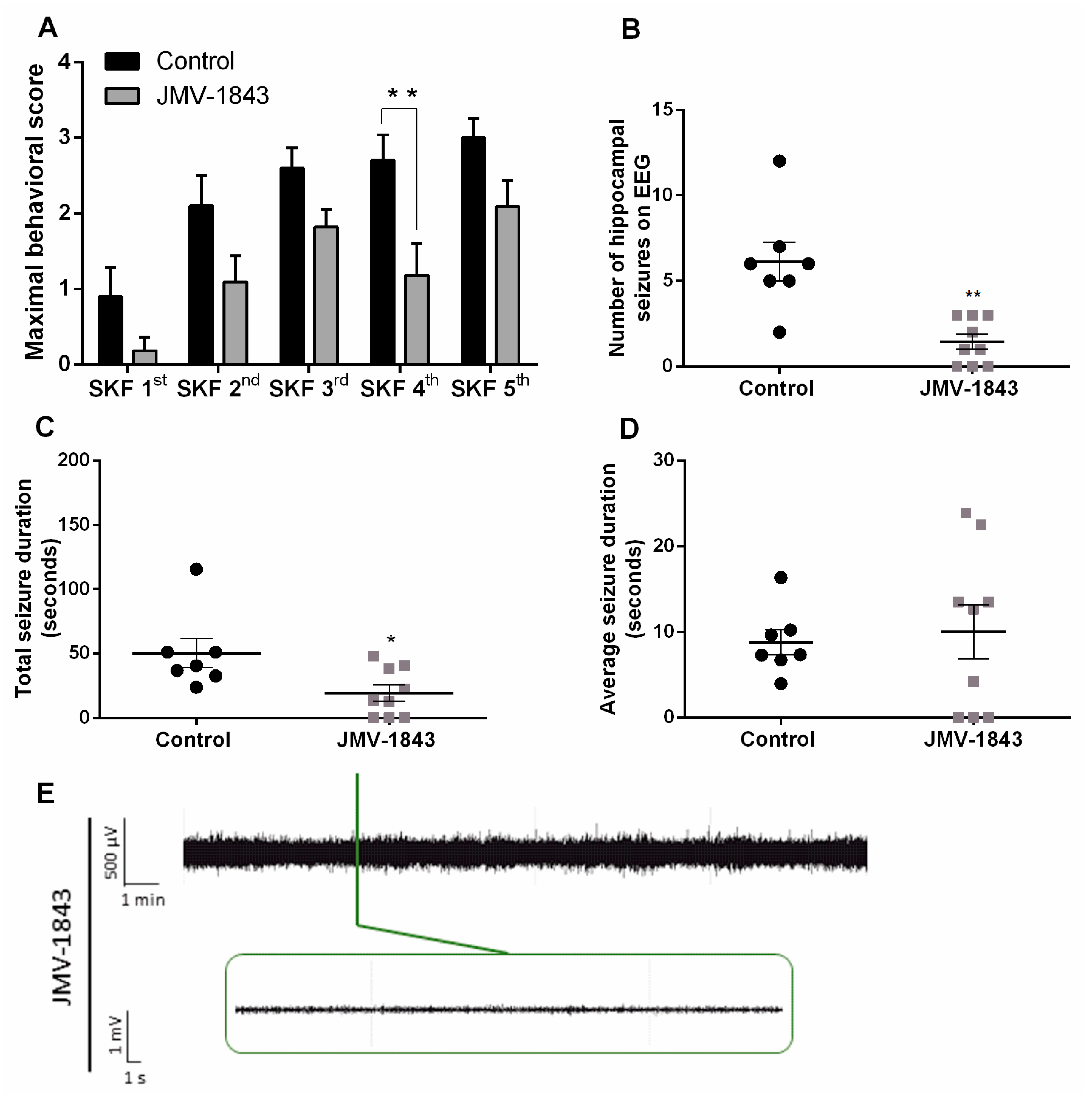
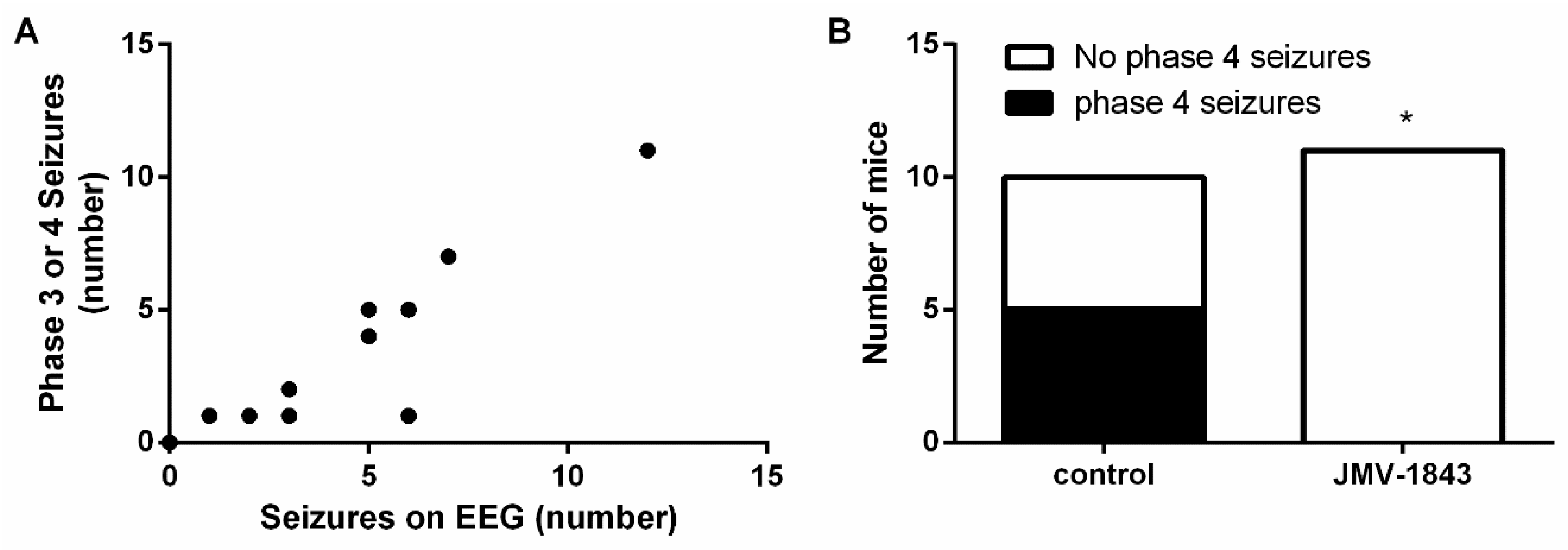
2.2. The Ghrelin-R Full agonist, JMV-1843, Is Anticonvulsive in the D1R-Mediated Kindling Model
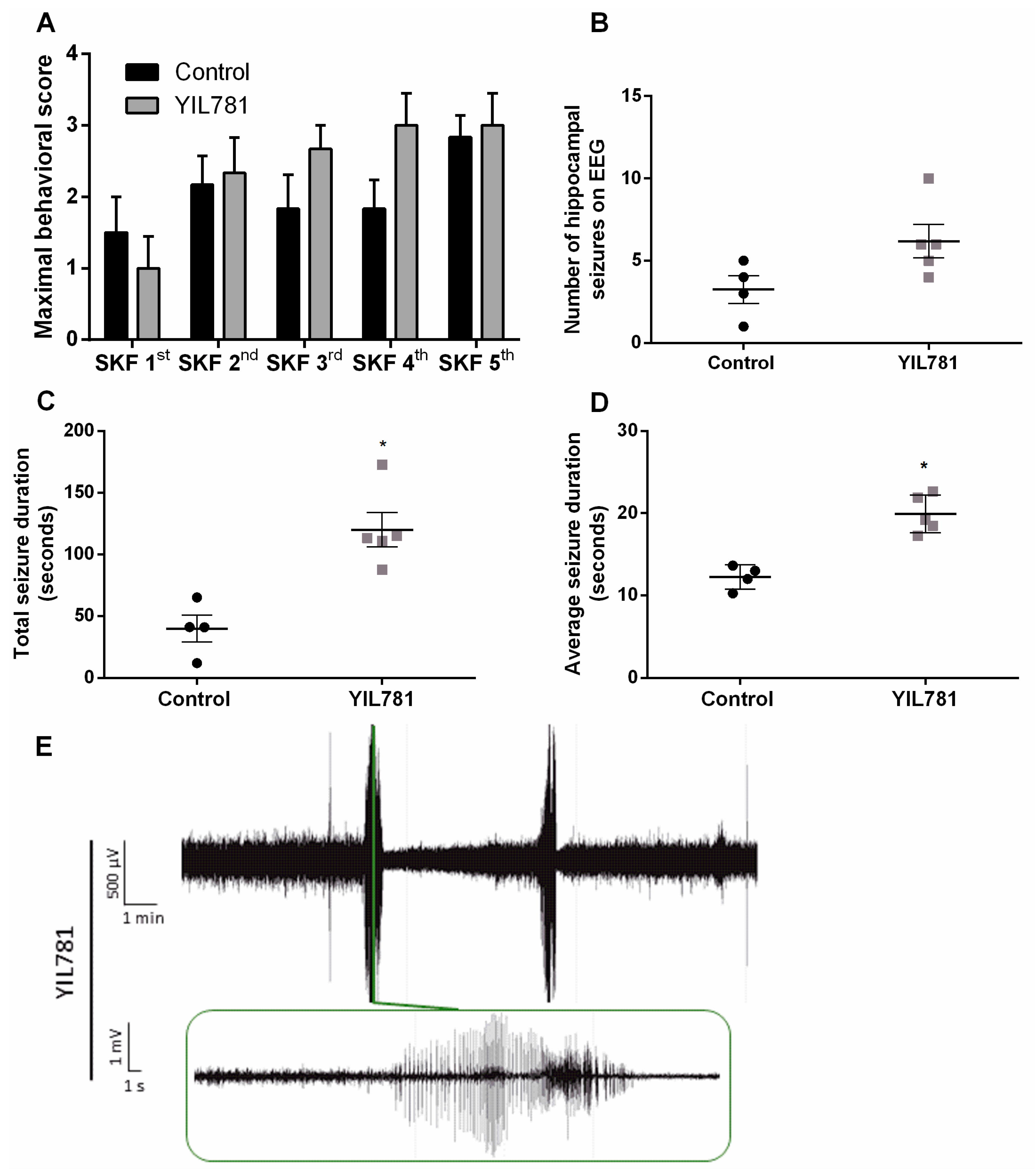
2.3. The Ghrelin-R Gαq Partial agonist, YIL781, Increases Seizure Burden in the D1R-Mediated Kindling Model
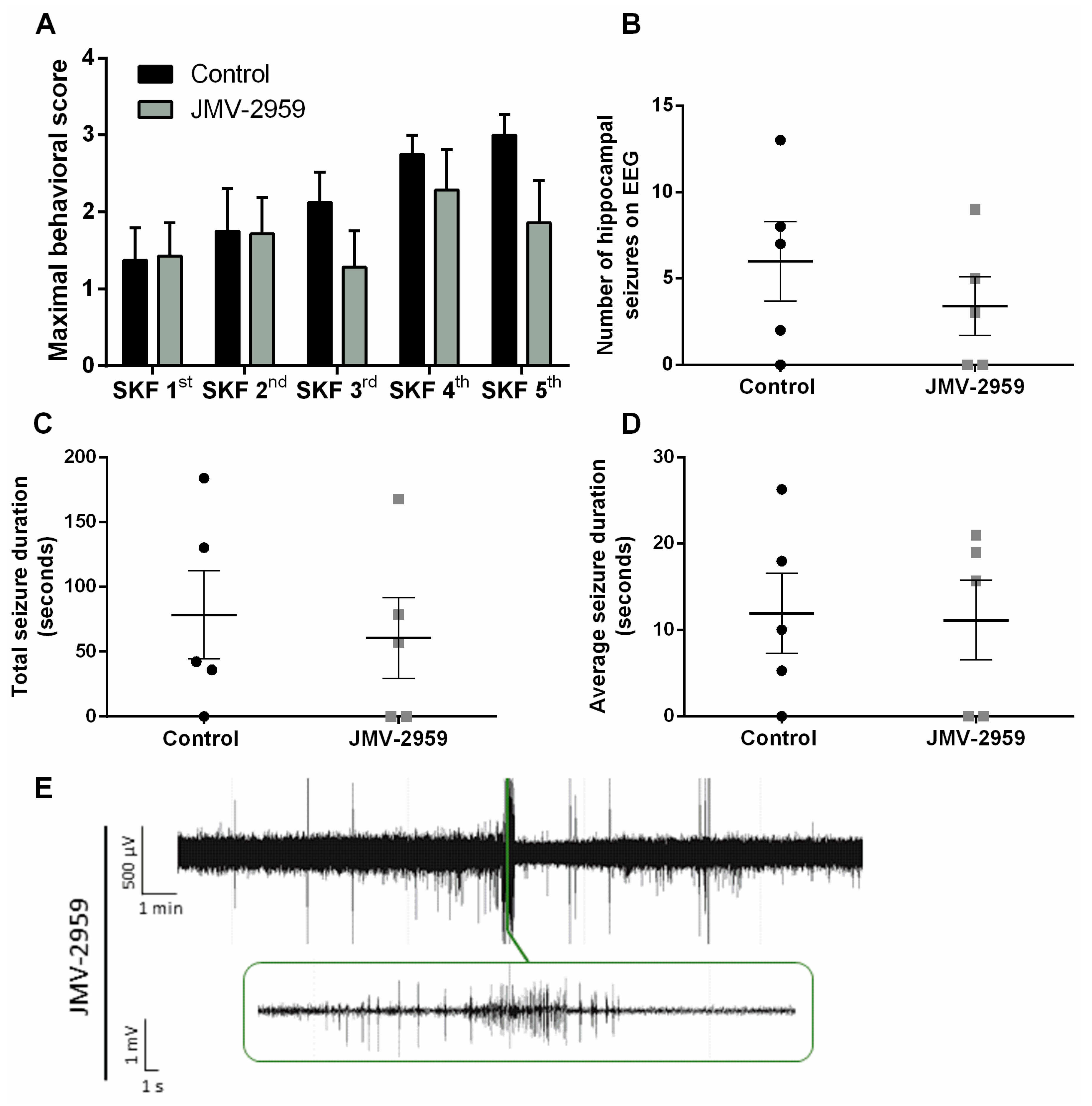
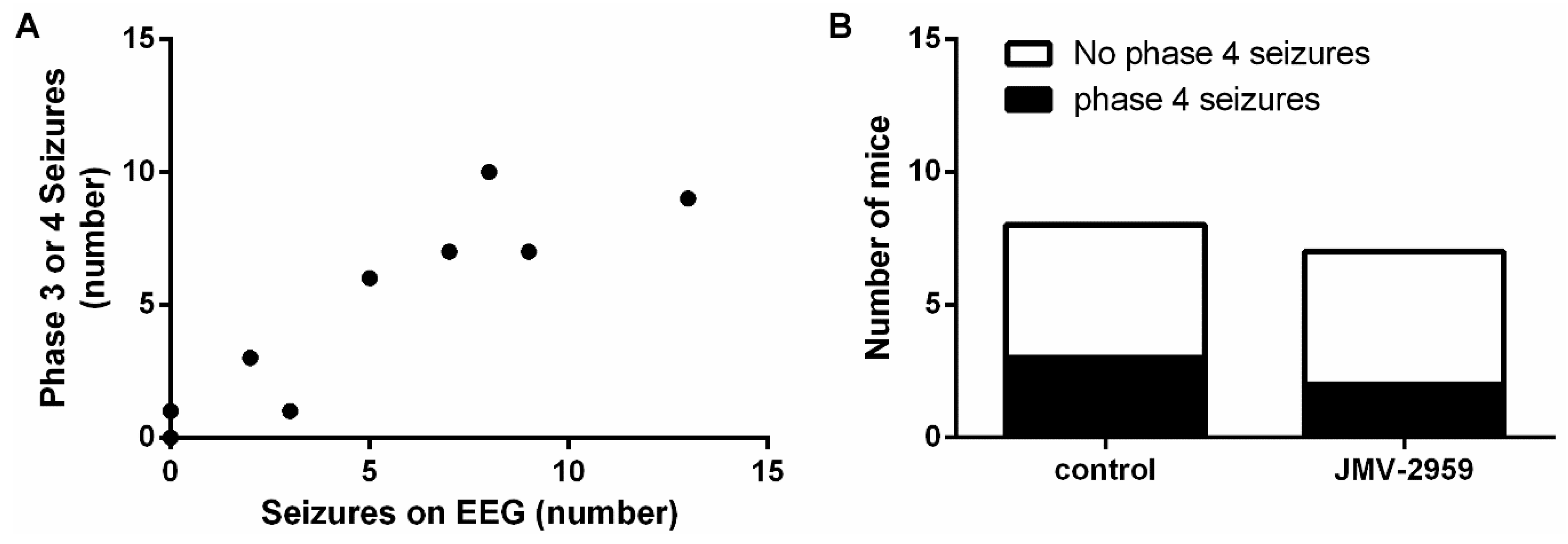
2.4. The Ghrelin-R Antagonist, JMV-2959, Does not Affect Seizures in the D1R-Mediated Kindling Model
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Electrode Placement and Transmitter Implantation
4.3. Treatment and D1R-Kindling Model
4.4. EEG Recordings
4.5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Adenylyl Cyclase |
| BBB | Blood–Brain Barrier |
| cAMP | Cyclic Adenosine Monophosphate |
| D1R | Dopamine 1 Receptor |
| EEG | Electro-Encephalography |
| ERK1/2 | Extracellular Signal-Regulated Kinases 1 and 2 |
| GH | Growth Hormone |
| Ghrelin-R | Ghrelin Receptor |
| GPCR | G-Protein Coupled Receptor |
| i.p. | Intraperitoneal |
| IP3 | Inositol 1,4,5-trisphosphate |
| MAPK | Mitogen-Activated Protein Kinase |
| min | Minute |
| µV | Microvolt |
| mV | Millivolt |
| PLC | Phospholipase C |
| PTZ | Pentylenetetrazol |
| RM | Repeated Measures |
| s | Second |
| SEM | Standard Error of the Mean |
| SKF | SKF81297 |
| SRE | Serum Response Element |
References
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the international league against epilepsy: Position paper of the ilae commission for classification and terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef]
- Dingledine, R.; Varvel, N.H.; Dudek, F.E. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv. Exp. Med. Biol. 2014, 813, 109–122. [Google Scholar] [PubMed]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef]
- Kwan, P.; Brodie, M.J. Refractory epilepsy: Mechanisms and solutions. Expert Rev. Neurother 2006, 6, 397–406. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, M.; Murakami, N.; Date, Y.; Kojima, M.; Matsuo, H.; Kangawa, K.; Matsukura, S. A role for ghrelin in the central regulation of feeding. Nature 2001, 409, 194–198. [Google Scholar] [CrossRef]
- Diano, S.; Farr, S.A.; Benoit, S.C.; McNay, E.C.; da Silva, I.; Horvath, B.; Gaskin, F.S.; Nonaka, N.; Jaeger, L.B.; Banks, W.A.; et al. Ghrelin controls hippocampal spine synapse density and memory performance. Nat. Neurosci. 2006, 9, 381–388. [Google Scholar] [CrossRef]
- Obay, B.D.; Tasdemir, E.; Tümer, C.; Bilgin, H.M.; Sermet, A. Antiepileptic effects of ghrelin on pentylenetetrazole-induced seizures in rats. Peptides 2007, 28, 1214–1219. [Google Scholar] [CrossRef]
- Ghahramanian Golzar, M.; Babri, S.; Ataie, Z.; Ebrahimi, H.; Mirzaie, F.; Mohaddes, G. Npy receptors blockade prevents anticonvulsant action of ghrelin in the hippocampus of rat. Adv. Pharm. Bull. 2013, 3, 265–271. [Google Scholar]
- Lee, J.; Lim, E.; Kim, Y.; Li, E.; Park, S. Ghrelin attenuates kainic acid-induced neuronal cell death in the mouse hippocampus. J. Endocrinol. 2010, 205, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Aslan, A.; Yildirim, M.; Ayyildiz, M.; Güven, A.; Agar, E. The role of nitric oxide in the inhibitory effect of ghrelin against penicillin-induced epileptiform activity in rat. Neuropeptides 2009, 43, 295–302. [Google Scholar] [CrossRef]
- Portelli, J.; Thielemans, L.; Ver Donck, L.; Loyens, E.; Coppens, J.; Aourz, N.; Aerssens, J.; Vermoesen, K.; Clinckers, R.; Schallier, A.; et al. Inactivation of the constitutively active ghrelin receptor attenuates limbic seizure activity in rodents. Neurotherapeutics 2012, 9, 658–672. [Google Scholar] [CrossRef]
- Lucchi, C.; Curia, G.; Vinet, J.; Gualtieri, F.; Bresciani, E.; Locatelli, V.; Torsello, A.; Biagini, G. Protective but not anticonvulsant effects of ghrelin and jmv-1843 in the pilocarpine model of status epilepticus. PLoS ONE 2013, 8, e72716. [Google Scholar] [CrossRef]
- Biagini, G.; Torsello, A.; Marinelli, C.; Gualtieri, F.; Vezzali, R.; Coco, S.; Bresciani, E.; Locatelli, V. Beneficial effects of desacyl-ghrelin, hexarelin and ep-80317 in models of status epilepticus. Eur. J. Pharmacol. 2011, 670, 130–136. [Google Scholar] [CrossRef]
- Obay, B.D.; Taşdemir, E.; Tümer, C.; Bilgin, H.; Atmaca, M. Dose dependent effects of ghrelin on pentylenetetrazole-induced oxidative stress in a rat seizure model. Peptides 2008, 29, 448–455. [Google Scholar] [CrossRef]
- Zhang, R.; Yang, G.; Wang, Q.; Guo, F.; Wang, H. Acylated ghrelin protects hippocampal neurons in pilocarpine-induced seizures of immature rats by inhibiting cell apoptosis. Mol. Biol. Rep. 2013, 40, 51–58. [Google Scholar] [CrossRef]
- Xu, J.; Wang, S.; Lin, Y.; Cao, L.; Wang, R.; Chi, Z. Ghrelin protects against cell death of hippocampal neurons in pilocarpine-induced seizures in rats. Neurosci. Lett. 2009, 453, 58–61. [Google Scholar] [CrossRef]
- Oztas, B.; Sahin, D.; Kir, H.; Eraldemir, F.C.; Musul, M.; Kuskay, S.; Ates, N. The effect of leptin, ghrelin, and neuropeptide-y on serum tnf-α, il-1β, il-6, fgf-2, galanin levels and oxidative stress in an experimental generalized convulsive seizure model. Neuropeptides 2017, 61, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Babri, S.; Amani, M.; Mohaddes, G.; Mirzaei, F.; Mahmoudi, F. Effects of intrahippocampal injection of ghrelin on spatial memory in ptz-induced seizures in male rats. Neuropeptides 2013, 47, 355–360. [Google Scholar] [CrossRef] [PubMed]
- M’Kadmi, C.; Leyris, J.P.; Onfroy, L.; Galés, C.; Saulière, A.; Gagne, D.; Damian, M.; Mary, S.; Maingot, M.; Denoyelle, S.; et al. Agonism, antagonism, and inverse agonism bias at the ghrelin receptor signaling. J. Biol. Chem. 2015, 290, 27021–27039. [Google Scholar] [CrossRef] [PubMed]
- Damian, M.; Marie, J.; Leyris, J.P.; Fehrentz, J.A.; Verdié, P.; Martinez, J.; Banères, J.L.; Mary, S. High constitutive activity is an intrinsic feature of ghrelin receptor protein: A study with a functional monomeric ghs-r1a receptor reconstituted in lipid discs. J. Biol. Chem. 2012, 287, 3630–3641. [Google Scholar] [CrossRef]
- Sivertsen, B.; Lang, M.; Frimurer, T.M.; Holliday, N.D.; Bach, A.; Els, S.; Engelstoft, M.S.; Petersen, P.S.; Madsen, A.N.; Schwartz, T.W.; et al. Unique interaction pattern for a functionally biased ghrelin receptor agonist. J. Biol. Chem. 2011, 286, 20845–20860. [Google Scholar] [CrossRef]
- Holliday, N.D.; Holst, B.; Rodionova, E.A.; Schwartz, T.W.; Cox, H.M. Importance of constitutive activity and arrestin-independent mechanisms for intracellular trafficking of the ghrelin receptor. Mol. Endocrinol. 2007, 21, 3100–3112. [Google Scholar] [CrossRef]
- Fernandez, G.; Cabral, A.; Andreoli, M.F.; Labarthe, A.; M’Kadmi, C.; Ramos, J.G.; Marie, J.; Fehrentz, J.A.; Epelbaum, J.; Tolle, V.; et al. Evidence supporting a role for constitutive ghrelin receptor signaling in fasting-induced hyperphagia in male mice. Endocrinology 2018, 159, 1021–1034. [Google Scholar] [CrossRef]
- Guerlavais, V.; Boeglin, D.; Mousseaux, D.; Oiry, C.; Heitz, A.; Deghenghi, R.; Locatelli, V.; Torsello, A.; Ghé, C.; Catapano, F.; et al. New active series of growth hormone secretagogues. J. Med. Chem. 2003, 46, 1191–1203. [Google Scholar] [CrossRef]
- Garcia, J.M.; Swerdloff, R.; Wang, C.; Kyle, M.; Kipnes, M.; Biller, B.M.K.; Cook, D.; Yuen, K.C.J.; Bonert, V.; Dobs, A.; et al. Macimorelin (aezs-130)-stimulated growth hormone (gh) test: Validation of a novel oral stimulation test for the diagnosis of adult gh deficiency. J. Clin. Endocrinol. Metab. 2013, 98, 2422–2429. [Google Scholar] [CrossRef]
- Evron, T.; Peterson, S.M.; Urs, N.M.; Bai, Y.; Rochelle, L.K.; Caron, M.G.; Barak, L.S. G protein and β-arrestin signaling bias at the ghrelin receptor. J Biol. Chem. 2014, 289, 33442–33455. [Google Scholar] [CrossRef]
- Mende, F.; Hundahl, C.; Plouffe, B.; Skov, L.J.; Sivertsen, B.; Madsen, A.N.; Lückmann, M.; Diep, T.A.; Offermanns, S.; Frimurer, T.M.; et al. Translating biased signaling in the ghrelin receptor system into differential in vivo functions. Proc. Natl. Acad. Sci. USA 2018, 115, E10255–E10264. [Google Scholar] [CrossRef]
- Holst, B.; Holliday, N.D.; Bach, A.; Elling, C.E.; Cox, H.M.; Schwartz, T.W. Common structural basis for constitutive activity of the ghrelin receptor family. J. Biol. Chem. 2004, 279, 53806–53817. [Google Scholar] [CrossRef]
- Moulin, A.; Demange, L.; Bergé, G.; Gagne, D.; Ryan, J.; Mousseaux, D.; Heitz, A.; Perrissoud, D.; Locatelli, V.; Torsello, A.; et al. Toward potent ghrelin receptor ligands based on trisubstituted 1,2,4-triazole structure. 2. Synthesis and pharmacological in vitro and in vivo evaluations. J. Med. Chem. 2007, 50, 5790–5806. [Google Scholar] [CrossRef]
- Coppens, J.; Aourz, N.; Walrave, L.; Fehrentz, J.A.; Martinez, J.; De Bundel, D.; Portelli, J.; Smolders, I. Anticonvulsant effect of a ghrelin receptor agonist in 6hz corneally kindled mice. Epilepsia 2016, 57, e195–e199. [Google Scholar] [CrossRef]
- Gangarossa, G.; Ceolin, L.; Paucard, A.; Lerner-Natoli, M.; Perroy, J.; Fagni, L.; Valjent, E. Repeated stimulation of dopamine d1-like receptor and hyperactivation of mtor signaling lead to generalized seizures, altered dentate gyrus plasticity, and memory deficits. Hippocampus 2014, 24, 1466–1481. [Google Scholar] [CrossRef]
- Kern, A.; Mavrikaki, M.; Ullrich, C.; Albarran-Zeckler, R.; Brantley, A.F.; Smith, R.G. Hippocampal dopamine/drd1 signaling dependent on the ghrelin receptor. Cell 2015, 163, 1176–1190. [Google Scholar] [CrossRef]
- Camiña, J.P.; Carreira, M.C.; El Messari, S.; Llorens-Cortes, C.; Smith, R.G.; Casanueva, F.F. Desensitization and endocytosis mechanisms of ghrelin-activated growth hormone secretagogue receptor 1a. Endocrinology 2004, 145, 930–940. [Google Scholar] [CrossRef]
- Esler, W.P.; Rudolph, J.; Claus, T.H.; Tang, W.; Barucci, N.; Brown, S.E.; Bullock, W.; Daly, M.; Decarr, L.; Li, Y.; et al. Small-molecule ghrelin receptor antagonists improve glucose tolerance, suppress appetite, and promote weight loss. Endocrinology 2007, 148, 5175–5185. [Google Scholar] [CrossRef]
- Gomez, J.L.; Ryabinin, A.E. The effects of ghrelin antagonists [d-lys(3) ]-ghrp-6 or jmv2959 on ethanol, water, and food intake in c57bl/6j mice. Alcohol. Clin. Exp. Res. 2014, 38, 2436–2444. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The arrive guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| JMV-1843 | YIL781 | JMV-2959 | |
|---|---|---|---|
| Gαq/11 | ↑ | ↑ | – |
| Gαi/o | ↑ | – | – |
| Gα12 | ↑ | ↑ | – |
| Gα13 | ↑ | – | – |
| β-arrestin | ↑ | – | – |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buckinx, A.; Van Den Herrewegen, Y.; Pierre, A.; Cottone, E.; Ben Haj Salah, K.; Fehrentz, J.-A.; Kooijman, R.; De Bundel, D.; Smolders, I. Differential Effects of a Full and Biased Ghrelin Receptor Agonist in a Mouse Kindling Model. Int. J. Mol. Sci. 2019, 20, 2480. https://doi.org/10.3390/ijms20102480
Buckinx A, Van Den Herrewegen Y, Pierre A, Cottone E, Ben Haj Salah K, Fehrentz J-A, Kooijman R, De Bundel D, Smolders I. Differential Effects of a Full and Biased Ghrelin Receptor Agonist in a Mouse Kindling Model. International Journal of Molecular Sciences. 2019; 20(10):2480. https://doi.org/10.3390/ijms20102480
Chicago/Turabian StyleBuckinx, An, Yana Van Den Herrewegen, Anouk Pierre, Eleonora Cottone, Khoubaib Ben Haj Salah, Jean-Alain Fehrentz, Ron Kooijman, Dimitri De Bundel, and Ilse Smolders. 2019. "Differential Effects of a Full and Biased Ghrelin Receptor Agonist in a Mouse Kindling Model" International Journal of Molecular Sciences 20, no. 10: 2480. https://doi.org/10.3390/ijms20102480
APA StyleBuckinx, A., Van Den Herrewegen, Y., Pierre, A., Cottone, E., Ben Haj Salah, K., Fehrentz, J.-A., Kooijman, R., De Bundel, D., & Smolders, I. (2019). Differential Effects of a Full and Biased Ghrelin Receptor Agonist in a Mouse Kindling Model. International Journal of Molecular Sciences, 20(10), 2480. https://doi.org/10.3390/ijms20102480