Role of Macrophages in Cardioprotection


 ,
,
Abstract
{kind=link}
{kind=link}
1. Cardiovascular Disease and Global Burden
2. Overview of Atherosclerosis
3. Macrophage Roles in Thrombus Formation
4. Macrophages: Inflammation and Activation
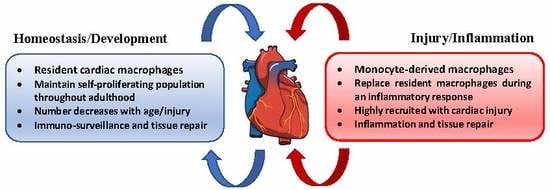
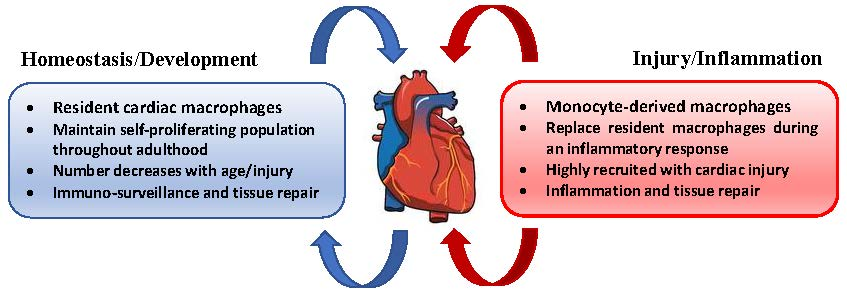
5. Cardiac Macrophages
6. Macrophages in Homeostasis
7. Macrophages in the Aging Heart
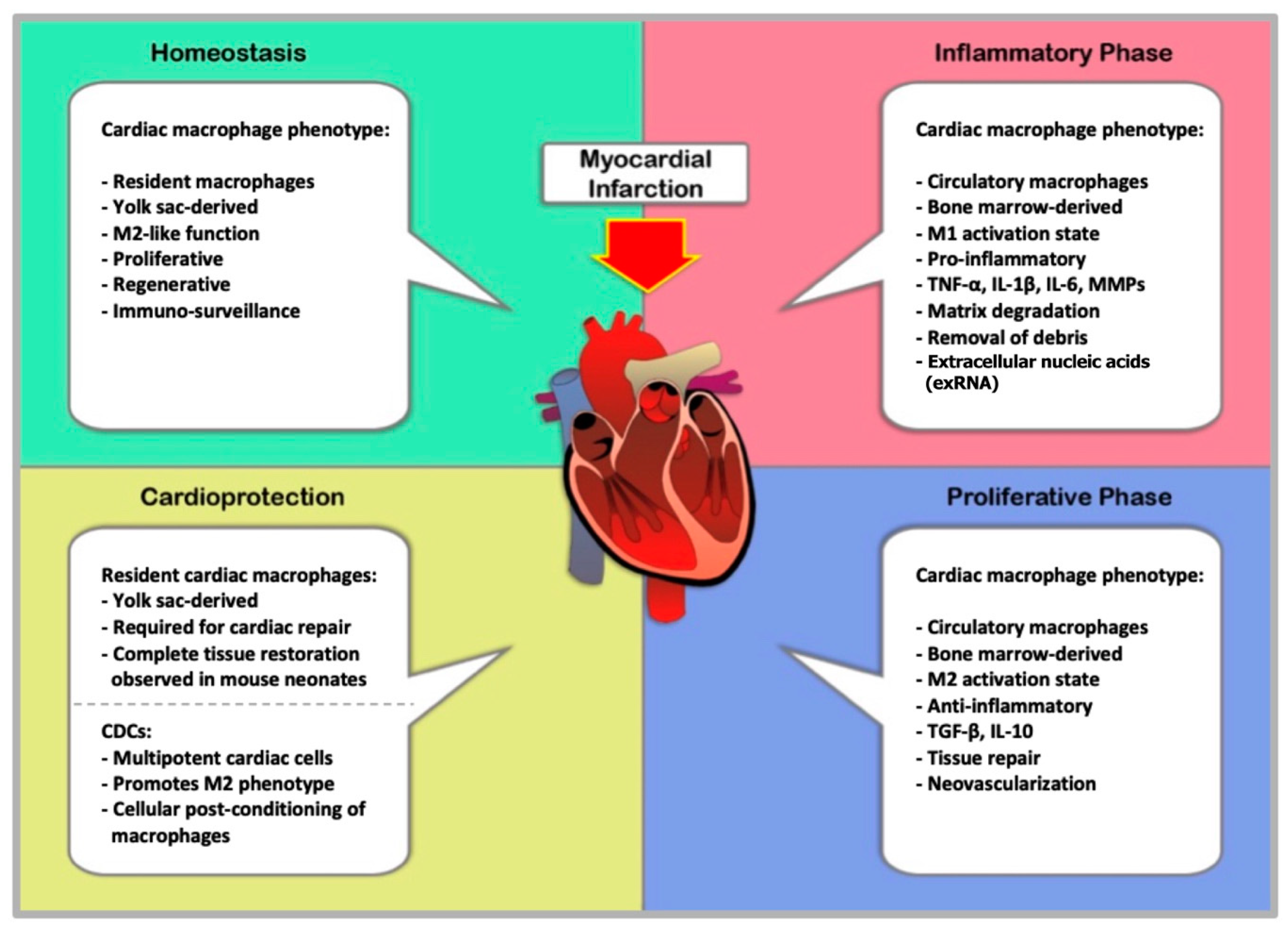
8. Pathology and Resolution of Myocardial Infarction
9. Macrophages in Myocardial Infarction
10. Macrophages and Tissue Repair after MI
11. Macrophage-Targeted Pharmaceutical Interventions
12. Cell-Mediated Cardioprotection in MI
13. Cardioprotection through Cellular Post-Conditioning
14. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| CVD | Cardiovascular diseases |
| DALYs | Disability adjusted life-years |
| MI | Myocardial infarction |
| PE | Pulmonary embolism |
| LDL | Low-density lipoprotein |
| ROS | Reactive oxygen species |
| ER | Endoplasmic reticulum |
| MMPs | Matrix metalloproteinases |
| ECM | Extracellular matrix |
| CXCLs | Chemokine (C-X-C motif) ligands |
| CCLs | Chemokine ligands |
| MIP | Macrophage Inflammatory Proteins |
| RANTES | Regulated on Activation, Normal T Cell Expressed and Secreted – CCL5 |
| MCP | Monocyte chemoattractant protein |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Damage-associated molecular patterns |
| PRRs | Pattern recognition receptors |
| TLRs | Toll-like receptors |
| MYD88 | Myeloid differentiation primary response 88 |
| IRAK-4 | Interleukin-1 receptor-associated kinase 4 |
| TRAF | TNF receptor associated factor |
| IKKβ | IκB kinase β |
| NF-κB | Nuclear factor-kappa B |
| ILs | Interleukins |
| IFN | Interferon |
| NK | Natural-killer cells |
| NO | Nitric oxide |
| SR | Scavenger receptors |
| MR | Mannose receptors |
| TGF-β | Transforming growth factor-β |
| MHC-II | Major histocompatibility complex class II molecules |
| CCR | Chemokine receptors |
| ARG 1 | Arginase-1 |
| GCR | Glucocorticoid receptor |
| FN-1 | Fibronectin-1 |
| TRIB1 | Tribbles homolog 1 is a protein kinase |
| VEGF | Vascular endothelial growth factor |
| BMDM | Bone marrow derived macrophages |
| CDC | Cardiosphere-derived cells |
| GFP | Green fluorescent protein |
References
- Mendis, S.; Puska, P.; Norrving, B.; World Health Organization; World Heart Federation; World Stroke Organization. Global Atlas on Cardiovascular Disease Prevention and Control; World Health Organization in collaboration with the World Heart Federation and the World Stroke Organization: Geneva, Switzerland, 2011; p online resource (vi, 155p.). [Google Scholar]
- Roth, G.A.; Johnson, C.; Abajobir, A.; Abd-Allah, F.; Abera, S.F.; Abyu, G.; Ahmed, M.; Aksut, B.; Alam, T.; Alam, K.; et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 2017, 70, 1–25. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart disease and stroke statistics-2018 update: A report from the american heart association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.; Diamond, J.; Vieco, A.; Chaudhuri, S.; Shinnar, E.; Cromer, S.; Perel, P.; Mensah, G.A.; Narula, J.; Johnson, C.O.; et al. Global atlas of cardiovascular disease 2000–2016: The path to prevention and control. Glob. Heart 2018, 13, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Simsekyilmaz, S.; Cabrera-Fuentes, H.A.; Meiler, S.; Kostin, S.; Baumer, Y.; Liehn, E.A.; Weber, C.; Boisvert, W.A.; Preissner, K.T.; Zernecke, A. Role of extracellular rna in atherosclerotic plaque formation in mice. Circulation 2014, 129, 598–606. [Google Scholar] [CrossRef]
- Simsekyilmaz, S.; Cabrera-Fuentes, H.A.; Meiler, S.; Kostin, S.; Baumer, Y.; Liehn, E.A.; Weber, C.; Boisvert, W.A.; Preissner, K.T.; Zernecke, A. Response to letter regarding article “role of extracellular rna in atherosclerotic plaque formation in mice”. Circulation 2014, 130, e144–e145. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. New Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Zaman, A.G.; Helft, G.; Worthley, S.G.; Badimon, J.J. The role of plaque rupture and thrombosis in coronary artery disease. Atherosclerosis 2000, 149, 251–266. [Google Scholar] [CrossRef]
- Munoz-Vega, M.; Masso, F.; Paez, A.; Carreon-Torres, E.; Cabrera-Fuentes, H.A.; Fragoso, J.M.; Perez-Hernandez, N.; Martinez, L.O.; Najib, S.; Vargas-Alarcon, G.; et al. Characterization of immortalized human dermal microvascular endothelial cells (hmec-1) for the study of hdl functionality. Lipids Health Dis. 2018, 17, 44. [Google Scholar] [CrossRef]
- Liehn, E.A.; Ponomariov, V.; Diaconu, R.; Streata, I.; Ioana, M.; Crespo-Avilan, G.E.; Hernandez-Resendiz, S.; Cabrera-Fuentes, H.A. Apolipoprotein e in cardiovascular diseases: Novel aspects of an old-fashioned enigma. Arch. Med. Res. 2018, 49, 522–529. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Yakala, G.K.; Cabrera-Fuentes, H.A.; Crespo-Avilan, G.E.; Rattanasopa, C.; Burlacu, A.; George, B.L.; Anand, K.; Mayan, D.C.; Corliano, M.; Hernandez-Resendiz, S.; et al. Furin inhibition reduces vascular remodeling and atherosclerotic lesion progression in mice. Arter. Thromb. Vasc. Biol. 2019, 39, 387–401. [Google Scholar] [CrossRef]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Liehn, E.A.; Cabrera-Fuentes, H.A. Inflammation between defense and disease: Impact on tissue repair and chronic sickness. Discoveries 2015, 3, e42. [Google Scholar] [CrossRef]
- Cabrera-Fuentes, H.A.; Aragones, J.; Bernhagen, J.; Boening, A.; Boisvert, W.A.; Botker, H.E.; Bulluck, H.; Cook, S.; Di Lisa, F.; Engel, F.B.; et al. From basic mechanisms to clinical applications in heart protection, new players in cardiovascular diseases and cardiac theranostics: Meeting report from the third international symposium on “new frontiers in cardiovascular research”. Basic Res. Cardiol. 2016, 111, 69. [Google Scholar] [CrossRef][Green Version]
- Hernandez-Resendiz, S.; Munoz-Vega, M.; Contreras, W.E.; Crespo-Avilan, G.E.; Rodriguez-Montesinos, J.; Arias-Carrion, O.; Perez-Mendez, O.; Boisvert, W.A.; Preissner, K.T.; Cabrera-Fuentes, H.A. Responses of endothelial cells towards ischemic conditioning following acute myocardial infarction. Cond. Med. 2018, 1, 247–258. [Google Scholar] [PubMed]
- Cabrera-Fuentes, H.A.; Alba-Alba, C.; Aragones, J.; Bernhagen, J.; Boisvert, W.A.; Botker, H.E.; Cesarman-Maus, G.; Fleming, I.; Garcia-Dorado, D.; Lecour, S.; et al. Meeting report from the 2nd international symposium on new frontiers in cardiovascular research. Protecting the cardiovascular system from ischemia: Between bench and bedside. Basic Res. Cardiol. 2016, 111, 7. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Padro, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc Care 2012, 1, 60–74. [Google Scholar] [CrossRef]
- Rothwell, P.M. Atherothrombosis and ischaemic stroke. BMJ 2007, 334, 379–380. [Google Scholar] [CrossRef]
- Oklu, R. Thrombosis. Cardiovasc. Diagn 2017, 7, S131–S133. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. Canakinumab for residual inflammatory risk. Eur. Heart J. 2017, 38, 3545–3548. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arter. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef]
- Wouters, K.; Shiri-Sverdlov, R.; van Gorp, P.J.; van Bilsen, M.; Hofker, M.H. Understanding hyperlipidemia and atherosclerosis: Lessons from genetically modified apoe and ldlr mice. Clin. Chem Lab. Med. 2005, 43, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mehta, J.L. Oxidized ldl, a critical factor in atherogenesis. Cardiovasc. Res. 2005, 68, 353–354. [Google Scholar] [CrossRef]
- Moore, K.J.; Koplev, S.; Fisher, E.A.; Tabas, I.; Björkegren, J.L.M.; Doran, A.C.; Kovacic, J.C. Macrophage trafficking, inflammatory resolution, and genomics in atherosclerosis: Jacc macrophage in cvd series (part 2). J. Am. Coll. Cardiol. 2018, 72, 2181–2197. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and atherosclerosis. Nat. Med. 2010, 16, 396–399. [Google Scholar] [CrossRef]
- Oh, J.; Riek, A.E.; Weng, S.; Petty, M.; Kim, D.; Colonna, M.; Cella, M.; Bernal-Mizrachi, C. Endoplasmic reticulum stress controls m2 macrophage differentiation and foam cell formation. J. Biol. Chem. 2012, 287, 11629–11641. [Google Scholar] [CrossRef]
- Yu, X.H.; Fu, Y.C.; Zhang, D.W.; Yin, K.; Tang, C.K. Foam cells in atherosclerosis. Clin. Chim. Acta 2013, 424, 245–252. [Google Scholar] [CrossRef]
- Kzhyshkowska, J.; Neyen, C.; Gordon, S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology 2012, 217, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Ivanova, E.A.; Chistiakov, D.A.; Nikiforov, N.G.; Orekhov, A.N. Macrophages and their role in atherosclerosis: Pathophysiology and transcriptome analysis. Biomed. Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef]
- Newby, A.C.; Zaltsman, A.B. Fibrous cap formation or destruction—The critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc. Res. 1999, 41, 345–360. [Google Scholar] [CrossRef]
- Ley, K.; Miller, Y.I.; Hedrick, C.C. Monocyte and macrophage dynamics during atherogenesis. Arter. Thromb. Vasc. Biol. 2011, 31, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.; Tabas, I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J. Lipid Res. 2009, 50, S382–S387. [Google Scholar] [CrossRef] [PubMed]
- Finn, A.V.; Nakano, M.; Narula, J.; Kolodgie, F.D.; Virmani, R. Concept of vulnerable/unstable plaque. Arter. Thromb. Vasc. Biol. 2010, 30, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Schrijvers, D.M.; De Meyer, G.R. Necrotic cell death in atherosclerosis. Basic Res. Cardiol. 2011, 106, 749–760. [Google Scholar] [CrossRef]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef]
- Alpuche, J.; Quirino, L.; Sanchez-Vega, J.T.; Yap, J.; Perez-Campos, E.; Cabrera-Fuentes, H.A. The role of platelets in ischemic conditioning. Cond. Med. 2018, 1, 313–318. [Google Scholar]
- Insull, W., Jr. The pathology of atherosclerosis: Plaque development and plaque responses to medical treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef]
- Brandt, E.; Ludwig, A.; Petersen, F.; Flad, H.D. Platelet-derived cxc chemokines: Old players in new games. Immunol. Rev. 2000, 177, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, K.L.; Broekman, M.J.; Chernoff, A.; Lesznik, G.R.; Drillings, M. Platelet alpha-granule proteins: Studies on release and subcellular localization. Blood 1979, 53, 604–618. [Google Scholar]
- Gear, A.R.L.; Camerini, D. Platelet chemokines and chemokine receptors: Linking hemostasis, inflammation, and host defense. Microcirculation 2003, 10, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Wolf, A.; Lasitschka, F.; Linden, F.; Domschke, G.; Akhavanpoor, M.; Doesch, A.O.; Katus, H.A.; Gleissner, C.A. Prevalence of m4 macrophages within human coronary atherosclerotic plaques is associated with features of plaque instability. Int. J. Cardiol. 2015, 186, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Korosoglou, G.; Ler, P.; Akhavanpoor, M.; Domschke, G.; Linden, F.; Doesch, A.O.; Buss, S.J.; Giannitsis, E.; Katus, H.A.; et al. Cxcl4 plasma levels are not associated with the extent of coronary artery disease or with coronary plaque morphology. PLoS ONE 2015, 10, e0141693. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Shaked, I.; Little, K.M.; Ley, K. Cxc chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J. Immunol. 2010, 184, 4810. [Google Scholar] [CrossRef]
- Falk, E.; Nakano, M.; Bentzon, J.F.; Finn, A.V.; Virmani, R. Update on acute coronary syndromes: The pathologists’ view. Eur. Heart J. 2012, 34, 719–728. [Google Scholar] [CrossRef]
- Virmani, R.; Burke, A.P.; Farb, A.; Kolodgie, F.D. Pathology of the vulnerable plaque. J. Am. Coll. Cardiol. 2006, 47, C13–C18. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Watkins, J.; Abdul-Aziz, A.; Shafat, M.; Calvert, P.A.; Bowles, K.M.; Flather, M.D.; Rushworth, S.A.; Ryding, A.D. Inflammatory differences in plaque erosion and rupture in patients with st-segment elevation myocardial infarction. J. Am. Heart Assoc. 2017, 6, e005868. [Google Scholar] [CrossRef] [PubMed]
- Lafont, A. Basic aspects of plaque vulnerability. Heart 2003, 89, 1262–1267. [Google Scholar] [CrossRef]
- Quillard, T.; Franck, G.; Mawson, T.; Folco, E.; Libby, P. Mechanisms of erosion of atherosclerotic plaques. Curr. Opin. Lipidol. 2017, 28, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Yamamoto, E.; Bryniarski, K.; Xing, L.; Lee, H.; Isobe, M.; Libby, P.; Jang, I.-K. Nonculprit plaque characteristics in patients with acute coronary syndrome caused by plaque erosion vs plaque rupture: A 3-vessel optical coherence tomography studyerosion vs rupture nonculprit plaque morphology in acute coronary syndromeerosion vs rupture nonculprit plaque morphology in acute coronary syndrome. Jama Cardiol. 2018, 3, 207–214. [Google Scholar]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Varga, T.; Mounier, R.; Horvath, A.; Cuvellier, S.; Dumont, F.; Poliska, S.; Ardjoune, H.; Juban, G.; Nagy, L.; Chazaud, B. Highly dynamic transcriptional signature of distinct macrophage subsets during sterile inflammation, resolution, and tissue repair. J. Immunol. 2016, 196, 4771. [Google Scholar] [CrossRef]
- Randolph, G.J. The fate of monocytes in atherosclerosis. J. Thromb. Haemost. 2009, 7, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Uysal, H.; Bockermann, R.; Nandakumar, K.S.; Sehnert, B.; Bajtner, E.; Engström, Å.; Serre, G.; Burkhardt, H.; Thunnissen, M.M.G.M.; Holmdahl, R. Structure and pathogenicity of antibodies specific for citrullinated collagen type ii in experimental arthritis. J. Exp. Med. 2009, 206, 449. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized m2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harbor Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. Il-23 drives a pathogenic t cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector th17 and regulatory t cells. Nature 2006, 441, 235. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Müller, E.; Christopoulos, P.F.; Halder, S.; Lunde, A.; Beraki, K.; Speth, M.; Øynebråten, I.; Corthay, A. Toll-like receptor ligands and interferon-γ synergize for induction of antitumor m1 macrophages. Front. Immunol. 2017, 8, 1383. [Google Scholar] [CrossRef]
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.-T.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE 2010, 5, e8852. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Oh, S.-Y.; Jeon, S.G.; Park, H.-W.; Lee, S.-Y.; Chun, E.-Y.; Bang, B.; Lee, H.-S.; Oh, M.-H.; Kim, Y.-S.; et al. Airway exposure levels of lipopolysaccharide determine type 1 versus type 2 experimental asthma. J. Immunol. 2007, 178, 5375. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The nlrp3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Pesce, J.T.; Ramalingam, T.R.; Mentink-Kane, M.M.; Wilson, M.S.; El Kasmi, K.C.; Smith, A.M.; Thompson, R.W.; Cheever, A.W.; Murray, P.J.; Wynn, T.A. Arginase-1-expressing macrophages suppress th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009, 5, e1000371. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Burgess, D.R.; Vitti, G.F.; Hamilton, J.A. Interleukin-4 stimulates human monocytes to produce tissue-type plasminogen activator. Blood 1989, 74, 1222. [Google Scholar]
- Gratchev, A.; Guillot, P.; Hakiy, N.; Politz, O.; Orfanos, C.E.; Schledzewski, K.; Goerdt, S. Alternatively activated macrophages differentially express fibronectin and its splice variants and the extracellular matrix protein βig-h3. Scand. J. Immunol. 2001, 53, 386–392. [Google Scholar] [CrossRef]
- Borthwick, L.A.; Wynn, T.A.; Fisher, A.J. Cytokine mediated tissue fibrosis. Biochim. Et Biophys. Acta (Bba) Mol. Basis Dis. 2013, 1832, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Van Dyken, S.J.; Locksley, R.M. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu. Rev. Immunol. 2013, 31, 317–343. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac–derived macrophages. J. Exp. Med. 2012, 209, 1167. [Google Scholar] [CrossRef]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via gm-csf. J. Exp. Med. 2013, 210, 1977. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986. [Google Scholar] [CrossRef]
- Stahl, E.C.; Haschak, M.J.; Popovic, B.; Brown, B.N. Macrophages in the aging liver and age-related liver disease. Front. Immunol. 2018, 9, 2795. [Google Scholar] [CrossRef]
- Pinto, A.R.; Godwin, J.W.; Chandran, A.; Hersey, L.; Ilinykh, A.; Debuque, R.; Wang, L.; Rosenthal, N.A. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging 2014, 6, 399–413. [Google Scholar] [CrossRef]
- Sager, H.B.; Hulsmans, M.; Lavine, K.J.; Moreira, M.B.; Heidt, T.; Courties, G.; Sun, Y.; Iwamoto, Y.; Tricot, B.; Khan, O.F.; et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res. 2016, 119, 853–864. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Leid, J.; Carrelha, J.; Boukarabila, H.; Epelman, S.; Jacobsen, S.E.W.; Lavine, K.J. Primitive embryonic macrophages are required for coronary development and maturation. Circ. Res. 2016, 118, 1498–1511. [Google Scholar] [CrossRef] [PubMed]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor ccr2. Nat. Immunol. 2006, 7, 311. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Porrello, E.R.; Tan, W.; Mahmoud, A.I.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; Olson, E.N. Macrophages are required for neonatal heart regeneration. J. Clin. Investig. 2014, 124, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue resident ccr2- and ccr2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.S.; Chudnovskiy, A.; Rauch, P.J.; Figueiredo, J.-L.; Iwamoto, Y.; Gorbatov, R.; Etzrodt, M.; Weber, G.F.; Ueno, T.; van Rooijen, N.; et al. Extramedullary hematopoiesis generates ly-6c(high) monocytes that infiltrate atherosclerotic lesions. Circulation 2012, 125, 364–374. [Google Scholar] [CrossRef]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117, 195–205. [Google Scholar] [CrossRef]
- Tacke, F.; Alvarez, D.; Kaplan, T.J.; Jakubzick, C.; Spanbroek, R.; Llodra, J.; Garin, A.; Liu, J.; Mack, M.; van Rooijen, N.; et al. Monocyte subsets differentially employ ccr2, ccr5, and cx3cr1 to accumulate within atherosclerotic plaques. J. Clin. Investig. 2007, 117, 185–194. [Google Scholar] [CrossRef]
- Combadière, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined inhibition of Ccl2, Cx3cr1 and Ccr5 abrogates Ly6chi and Ly6clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef]
- Pinto, A.R.; Paolicelli, R.; Salimova, E.; Gospocic, J.; Slonimsky, E.; Bilbao-Cortes, D.; Godwin, J.W.; Rosenthal, N.A. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 2012, 7, e36814. [Google Scholar] [CrossRef] [PubMed]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Silva, N.D.; Panizzi, P.; Laan, A.M.v.d.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic sirna silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- DeBerge, M.; Yeap, X.Y.; Dehn, S.; Zhang, S.; Grigoryeva, L.; Misener, S.; Procissi, D.; Zhou, X.; Lee, D.C.; Muller, W.A.; et al. Mertk cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ. Res. 2017, 121, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Ma, Y.; Chiao, Y.A.; Lopez, E.F.; Voorhees, A.P.; Toba, H.; Hall, M.E.; Han, H.-C.; Lindsey, M.L.; Jin, Y.-F. Cardiac aging is initiated by matrix metalloproteinase-9-mediated endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1398–H1407. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Goshorn, D.K.; Squires, C.E.; Escobar, G.P.; Hendrick, J.W.; Mingoia, J.T.; Sweterlitsch, S.E.; Spinale, F.G. Age-dependent changes in myocardial matrix metalloproteinase/tissue inhibitor of metalloproteinase profiles and fibroblast function. Cardiovasc. Res. 2005, 66, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lopez, E.F.; Jin, Y.; Van Remmen, H.; Bauch, T.; Han, H.-C.; Lindsey, M.L. Age-related cardiac muscle sarcopenia: Combining experimental and mathematical modeling to identify mechanisms. Exp. Gerontol. 2008, 43, 296–306. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Ramirez, T.A.; Zamilpa, R.; Okoronkwo, S.M.; Dai, Q.; Zhang, J.; Jin, Y.-F.; Lindsey, M.L. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc. Res. 2012, 96, 444–455. [Google Scholar] [CrossRef]
- DeWood, M.A.; Spores, J.; Notske, R.; Mouser, L.T.; Burroughs, R.; Golden, M.S.; Lang, H.T. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. New Engl. J. Med. 1980, 303, 897–902. [Google Scholar] [CrossRef]
- Bentzon Jacob, F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Jennings Robert, B. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ. Res. 2013, 113, 428–438. [Google Scholar] [CrossRef]
- Sayen, J.J.; Sheldon, W.F.; Peirce, G.; Kuo, P.T. Polarographic oxygen, the epicardial electrocardiogram and muscle contraction in experimental acute regional ischemia of the left ventricle. Circ. Res. 1958, 6, 779–798. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Fujiwara, H.; Matsuda, M.; Onodera, T.; Ishida, M.; Kawamura, A.; Okamoto, Y.; Ban, T.; Kawai, C. Reperfusion injury in dog hearts with permanent occlusion of a coronary artery, probably due to reperfusion via collateral vessels. Int. J. Cardiol. 1991, 30, 275–284. [Google Scholar] [CrossRef]
- Jennings, R.B.; Sommers, H.M.; Smyth, G.A.; Flack, H.A.; Linn, H. Myocardial necrosis induced by temporary occlusion of a coronary artery in the dog. Arch. Pathol. 1960, 70, 68–78. [Google Scholar] [PubMed]
- Jennings, R.; Murry, C.; Steenbergen, C.; Reimer, K.A. Development of cell injury in sustained acute ischemia. Circulation 1990, 82, II2–II12. [Google Scholar]
- Nahrendorf, M.; Pittet, M.J.; Swirski, F.K. Monocytes: Protagonists of infarct inflammation and repair after myocardial infarction. Circulation 2010, 121, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Mukhametshina, R.T.; Kwek, X.Y.; Cabrera-Fuentes, H.A.; Hausenloy, D.J. Inflammation following acute myocardial infarction: Multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol. Ther. 2018, 186, 73–87. [Google Scholar] [CrossRef]
- Andreadou, I.; Cabrera-Fuentes, H.A.; Devaux, Y.; Frangogiannis, N.G.; Frantz, S.; Guzik, T.; Liehn, E.; Gomes, C.P.; Gomes, C.; Schulz, R.; et al. Immune cells as targets for cardioprotection: New players and novel therapeutic opportunities. Cardiovasc. Res. Available online: https://academic.oup.com/cardiovascres/advance-article-abstract/doi/10.1093/cvr/cvz050/5368489 (accessed on 2 March 2019).
- Hernandez-Resendiz, S.; Chinda, K.; Ong, S.B.; Cabrera-Fuentes, H.; Zazueta, C.; Hausenloy, D.J. The role of redox dysregulation in the inflammatory response to acute myocardial ischaemia-reperfusion injury - adding fuel to the fire. Curr. Med. Chem. 2018, 25, 1275–1293. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. New Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael Lloyd, H.; Rollins Barrett, J.; Entman Mark, L.; Frangogiannis Nikolaos, G. Ccl2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef]
- Kaikita, K.; Hayasaki, T.; Okuma, T.; Kuziel, W.A.; Ogawa, H.; Takeya, M. Targeted deletion of cc chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am. J. Pathol. 2004, 165, 439–447. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef]
- Jung, K.; Kim, P.; Leuschner, F.; Gorbatov, R.; Kim, J.K.; Ueno, T.; Nahrendorf, M.; Yun, S.H. Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circ. Res. 2013, 112, 891–899. [Google Scholar] [CrossRef]
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128. [Google Scholar] [CrossRef]
- Troidl, C.; Möllmann, H.; Nef, H.; Masseli, F.; Voss, S.; Szardien, S.; Willmer, M.; Rolf, A.; Rixe, J.; Troidl, K.; et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J. Cell. Mol. Med. 2009, 13, 3485–3496. [Google Scholar] [CrossRef]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef]
- Frantz, S.; Nahrendorf, M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc. Res. 2014, 102, 240–248. [Google Scholar] [CrossRef]
- Wan, E.; Yeap Xin, Y.; Dehn, S.; Terry, R.; Novak, M.; Zhang, S.; Iwata, S.; Han, X.; Homma, S.; Drosatos, K.; et al. Enhanced efferocytosis of apoptotic cardiomyocytes through myeloid-epithelial-reproductive tyrosine kinase links acute inflammation resolution to cardiac repair after infarction. Circ. Res. 2013, 113, 1004–1012. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Inflammation in cardiac injury, repair and regeneration. Curr. Opin. Cardiol. 2015, 30, 240–245. [Google Scholar] [CrossRef]
- Shiraishi, M.; Shintani, Y.; Shintani, Y.; Ishida, H.; Saba, R.; Yamaguchi, A.; Adachi, H.; Yashiro, K.; Suzuki, K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 2016, 126, 2151–2166. [Google Scholar] [CrossRef] [PubMed]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Ludman, A.; Venugopal, V.; Yellon, D.M.; Hausenloy, D.J. Statins and cardioprotection—More than just lipid lowering? Pharmacol. Ther. 2009, 122, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat. Med. 2000, 6, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Ghittoni, R.; Napolitani, G.; Benati, D.; Ulivieri, C.; Patrussi, L.; Laghi Pasini, F.; Lanzavecchia, A.; Baldari, C.T. Simvastatin inhibits the mhc class ii pathway of antigen presentation by impairing ras superfamily gtpases. Eur. J. Immunol. 2006, 36, 2885–2893. [Google Scholar] [CrossRef] [PubMed]
- Di Raimondo, D.; Tuttolomondo, A.; Butta, C.; Miceli, S.; Licata, G.; Pinto, A. Effects of ace-inhibitors and angiotensin receptor blockers on inflammation. Curr. Pharm. Des. 2012, 18, 4385–4413. [Google Scholar] [CrossRef] [PubMed]
- Schreckenberg, R.; Weber, P.; Cabrera-Fuentes, H.A.; Steinert, I.; Preissner, K.T.; Bencsik, P.; Sarkozy, M.; Csonka, C.; Ferdinandy, P.; Schulz, R.; et al. Mechanism and consequences of the shift in cardiac arginine metabolism following ischaemia and reperfusion in rats. Thromb. Haemost. 2015, 113, 482–493. [Google Scholar]
- Bernstein, K.E.; Ong, F.S.; Blackwell, W.L.; Shah, K.H.; Giani, J.F.; Gonzalez-Villalobos, R.A.; Shen, X.Z.; Fuchs, S.; Touyz, R.M. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharm. Rev. 2013, 65, 1–46. [Google Scholar] [CrossRef]
- Leuschner, F.; Panizzi, P.; Chico-Calero, I.; Lee, W.W.; Ueno, T.; Cortez-Retamozo, V.; Waterman, P.; Gorbatov, R.; Marinelli, B.; Iwamoto, Y.; et al. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ. Res. 2010, 107, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Soejima, H.; Ogawa, H.; Yasue, H.; Kaikita, K.; Takazoe, K.; Nishiyama, K.; Misumi, K.; Miyamoto, S.; Yoshimura, M.; Kugiyama, K.; et al. Angiotensin-converting enzyme inhibition reduces monocyte chemoattractant protein-1 and tissue factor levels in patients with myocardial infarction. J. Am. Coll. Cardiol. 1999, 34, 983–988. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science (New York N.Y.) 2011, 331, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Hasan, A.S.; Luo, L.; Yan, C.; Zhang, T.-X.; Urata, Y.; Goto, S.; Mangoura, S.A.; Abdel-Raheem, M.H.; Zhang, S.; Li, T.-S. Cardiosphere-derived cells facilitate heart repair by modulating m1/m2 macrophage polarization and neutrophil recruitment. PLoS ONE 2016, 11, e0165255. [Google Scholar] [CrossRef] [PubMed]
- de Couto, G.; Liu, W.; Tseliou, E.; Sun, B.; Makkar, N.; Kanazawa, H.; Arditi, M.; Marbán, E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J. Clin. Investig. 2015, 125, 3147–3162. [Google Scholar] [CrossRef]
- Smith, R.R.; Barile, L.; Cho, H.C.; Leppo, M.K.; Hare, J.M.; Messina, E.; Giacomello, A.; Abraham, M.R.; Marbán, E. Regenerative potential of cardiosphere-derived cells expanded from percutaneous endomyocardial biopsy specimens. Circulation 2007, 115, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Van Laake, L.W.; Botker, H.E.; Davidson, S.M.; De Caterina, R.; Engel, F.B.; Eschenhagen, T.; Fernandez-Aviles, F.; Hausenloy, D.J.; Hulot, J.S.; et al. Esc working group on cellular biology of the heart: Position paper for cardiovascular research: Tissue engineering strategies combined with cell therapies for cardiac repair in ischaemic heart disease and heart failure. Cardiovasc. Res. 2019, 115, 488–500. [Google Scholar] [CrossRef]
- Kanazawa, H.; Tseliou, E.; Malliaras, K.; Yee, K.; Dawkins, J.F.; De Couto, G.; Smith, R.R.; Kreke, M.; Seinfeld, J.; Middleton, R.C.; et al. Cellular postconditioning. Circ. Heart Fail. 2015, 8, 322–332. [Google Scholar] [CrossRef]
- Shen, D.; Cheng, K.; Marbán, E. Dose-dependent functional benefit of human cardiosphere transplantation in mice with acute myocardial infarction. J. Cell. Mol. Med. 2012, 16, 2112–2116. [Google Scholar] [CrossRef]
- Malliaras, K.; Li, T.-S.; Luthringer, D.; Terrovitis, J.; Cheng, K.; Chakravarty, T.; Galang, G.; Zhang, Y.; Schoenhoff, F.; Van Eyk, J.; et al. Safety and efficacy of allogeneic cell therapy in infarcted rats transplanted with mismatched cardiosphere-derived cells. Circulation 2012, 125, 100–112. [Google Scholar] [CrossRef]
- Li, T.-S.; Cheng, K.; Malliaras, K.; Smith, R.R.; Zhang, Y.; Sun, B.; Matsushita, N.; Blusztajn, A.; Terrovitis, J.; Kusuoka, H.; et al. Direct comparison of different stem cell types and subpopulations reveals superior paracrine potency and myocardial repair efficacy with cardiosphere-derived cells. J. Am. Coll. Cardiol. 2012, 59, 942–953. [Google Scholar] [CrossRef]
- Cheng, K.; Malliaras, K.; Smith, R.R.; Shen, D.; Sun, B.; Blusztajn, A.; Xie, Y.; Ibrahim, A.; Aminzadeh, M.A.; Liu, W.; et al. Human cardiosphere-derived cells from advanced heart failure patients exhibit augmented functional potency in myocardial repair. Jacc. Heart Fail. 2014, 2, 49–61. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yap, J.; Cabrera-Fuentes, H.A.; Irei, J.; Hausenloy, D.J.; Boisvert, W.A. Role of Macrophages in Cardioprotection. Int. J. Mol. Sci. 2019, 20, 2474. https://doi.org/10.3390/ijms20102474
Yap J, Cabrera-Fuentes HA, Irei J, Hausenloy DJ, Boisvert WA. Role of Macrophages in Cardioprotection. International Journal of Molecular Sciences. 2019; 20(10):2474. https://doi.org/10.3390/ijms20102474
Chicago/Turabian StyleYap, Jonathan, Hector A. Cabrera-Fuentes, Jason Irei, Derek J. Hausenloy, and William A. Boisvert. 2019. "Role of Macrophages in Cardioprotection" International Journal of Molecular Sciences 20, no. 10: 2474. https://doi.org/10.3390/ijms20102474
APA StyleYap, J., Cabrera-Fuentes, H. A., Irei, J., Hausenloy, D. J., & Boisvert, W. A. (2019). Role of Macrophages in Cardioprotection. International Journal of Molecular Sciences, 20(10), 2474. https://doi.org/10.3390/ijms20102474