The Enterochromaffin-like [ECL] Cell—Central in Gastric Physiology and Pathology



{kind=link}
Abstract
1. The Regulation of Gastric Acid Secretion and the Cell in Oxyntic Mucosa Producing and Releasing the Histamine Taking Part in This Regulation: A Historical Overview.
2. Embryology of the ECL Cell.
3. Functional and Trophic Regulation of the ECL Cell
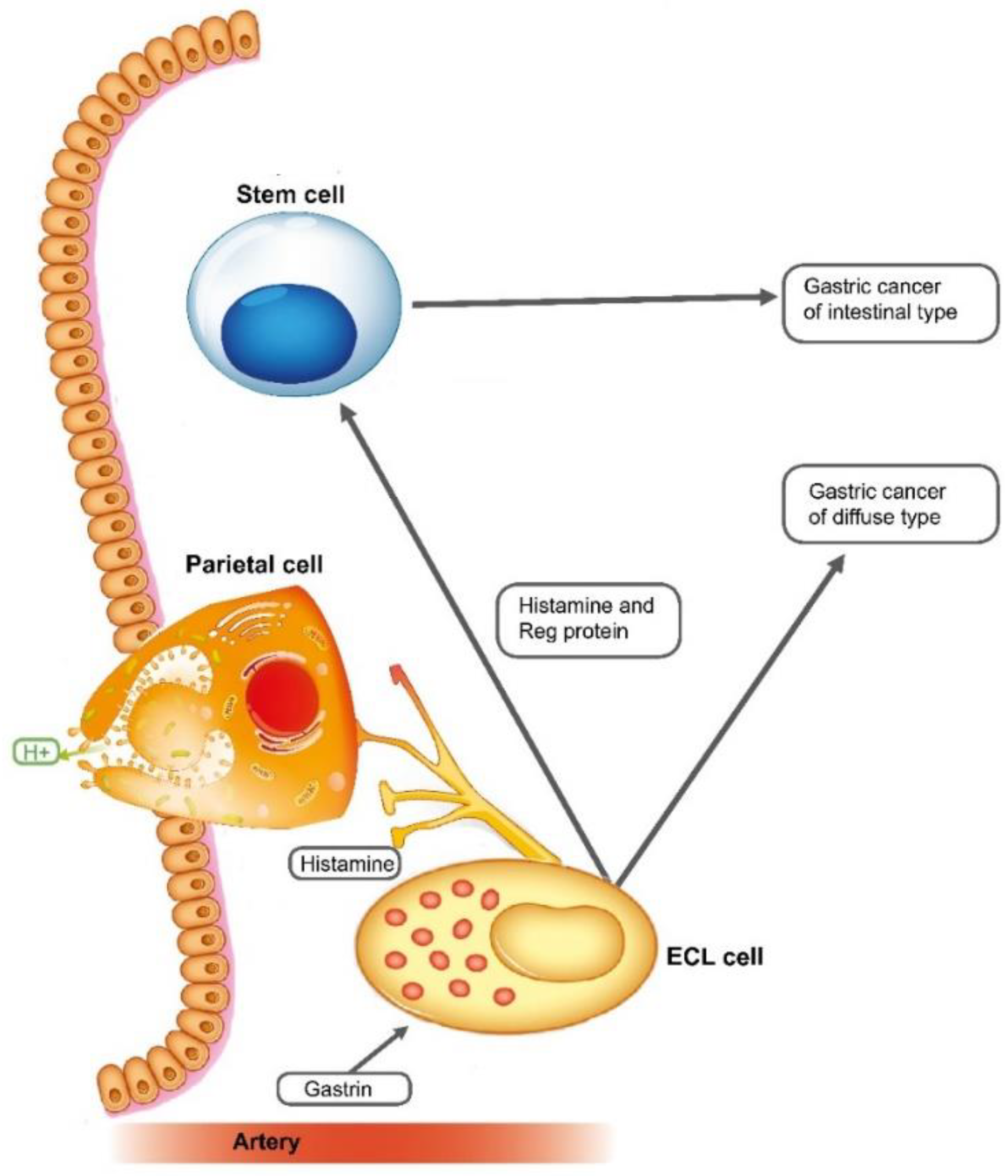
4. Role of the ECL Cell in Disease
4.1. Acid-Related Disorders
4.1.1. Rebound Acid Hypersecretion.
4.1.2. Peptic Ulcer Disease
4.1.3. Gastroesophageal Reflux Disease (GERD)
4.2. Gastric Neoplasia
4.2.1. Neuroendocrine Neoplasms (Neuroendocrine Tumours and Neuroendocrine Carcinomas)
4.2.2. Gastric Adenocarcinomas
5. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Beaumont, W. Experiments and Observation on the Gastric Juice and the Physiology of Digestion; A.P. Allen: Plattsburgh, NY, USA, 1833. [Google Scholar]
- Pavlov, I.P. The Work of the Digestive Glands; Trans. by, W.H. Thompson; Griffin: London, UK, 1902. [Google Scholar]
- Edkins, J. The chemical mechanism of gastric secretion. J. Physiol. 1906, 34, 133–144. [Google Scholar] [CrossRef]
- Popielsky, L. β-Imidazylthylamin und die Organanextrakte. Plüger Archive 1920, 178, 214–236. [Google Scholar] [CrossRef]
- Kahlson, G.; Rosengren, E.; Svahn, D.; Thunberg, R. Mobilization and formation of histamine in the gastric mucosa as related to acid secretion. J. Physiol. 1964, 174, 400–416. [Google Scholar] [CrossRef]
- Hakanson, R.; Owman, C. Argyrophilic reaction of histamine-containing epithelial cells in murine gastric mucosa. Experientia 1969, 25, 625–626. [Google Scholar] [CrossRef] [PubMed]
- Hakanson, R.; Bottcher, G.; Ekblad, E.; Panula, P.; Simonsson, M.; Dohlsten, M.; Hallberg, T.; Sundler, F. Histamine in endocrine cells in the stomach. A survey of several species using a panel of histamine antibodies. Histochemistry 1986, 86, 5–17. [Google Scholar]
- Havu, N. Enterochromaffin-like cell carcinoids of gastric mucosa in rats after life-long inhibition of gastric secretion. Digestion 1986, 35, 42–55. [Google Scholar] [CrossRef]
- Poynter, D.; Selway, S.A.; Papworth, S.A.; Riches, S.R. Changes in the gastric mucosa of the mouse associated with long lasting unsurmountable histamine H2 blockade. Gut 1986, 27, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Hakanson, R.; Sundler, F. Proposed mechanism of induction of gastric carcinoids, the gastrin hypothesis. Eur. J. Clin. Invest. 1990, 20, 65–71. [Google Scholar] [CrossRef]
- Brenna, E.; Waldum, H.L. Studies of isolated parietal and enterochromaffin-like cells from the rat. Scand. J. Gastroenterol. 1991, 26, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Prinz, C.; Kajimura, M.; Scott, D.R.; Mercier, F.; Helander, H.F.; Sachs, G. secretion from rat enterochromaffinlike cells. Gastroenterology 1993, 105, 449–461. [Google Scholar] [CrossRef]
- Waldum, H.L.; Lehy, T.; Brenna, E.; Sandvik, A.K.; Petersen, H.; Søgnen, B.S.; Bonfils, S.; Lewin, M.J. Effect of the histamine-1 antagonist astemizole alone or with omeprazole on rat gastric mucosa. Scand. J. Gastroenterol. 1991, 26, 23–35. [Google Scholar] [CrossRef]
- Kopin, A.S.; Lee, Y.M.; McBride, E.W.; Miller, L.J.; Lu, M.; Lin, H.Y.; Kolakowski LFJr Beinborn, M. Expression, cloning and characterization of the canine parietal cell gastrin receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 3605–3609. [Google Scholar] [CrossRef]
- Bakke, I.; Qvigstad, G.; Sandvik, A.K.; Waldum, H.L. The CCK-2 receptor is located on the ECL cell, but not on the parietal cell. Scand J Gastroenterol 2001, 36, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Athmann, C.; Zeng, N.; Scott, D.R.; Sachs, G. Regulation of parietal cell calcium signaling in gastric glands. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G1048–G1058. [Google Scholar] [CrossRef]
- Friis-Hansen, L.; Sundler, F.; Li, Y.; Gillespie, P.J.; Saunders, T.L.; Greenson, J.K.; Owyang, C.; Rehfeld, J.F.; Samuelson, L.C. Impaired gastric acid secretion in gastrin-deficient mice. Am. J. Physiol. 1998, 274, G561–G568. [Google Scholar] [CrossRef] [PubMed]
- Langhans, N.; Rindi, G.; Chiu, M.; Rehfeld, J.F.; Ardman, B.; Beinborn, M.; Kopin, A.S. Abnormal gastric histology and decreased acid production in cholecystokin-B/gastrin receptor-deficient mice. Gastroenterology 1997, 112, 280–286. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Ishihara, S.; Kadowaki, Y.; Fukui, H.; Chiba, T. Reg protein is a unique growth factor of gastric mucosal cells. J. Gastroenterol. 2004, 39, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, B.I.; Bakke, I.; Hauso, Ø.; Brenna, E.; Waldum, H.L. The enterochromaffinlike cell in rat stomach has elongations making direct contact with parietal cells [abstract]. Third Annu. ENETS Conf. Neuroendocrinol. 2006, 83, 27. [Google Scholar]
- Furness, J.B.; Padbury, R.T.; Baimbridge, K.G.; Skinner, J.M.; Lawson, D.E. Calbindin immunoreactivity is a characteristic of endocrine cells [ECL cells] of the human stomach. Histochemistry 1989, 92, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Bordi CPilato, F.P.; Bertelé, A.; D’Adda, T.; Missale, G. Expression of glycoprotein hormone alpha-subunit by endocrine cells of the oxyntic mucosa is associated with hypergastrinemias. Hum. Pathol. 1988, 19, 550–558. [Google Scholar]
- Pearse, A.G.; Polak, J.M. Neural crest origin of the endocrine polypeptide [APUD] cells of the gastrointestinal tract and pancreas. Gut 1971, 12, 783–788. [Google Scholar] [CrossRef]
- Le Douarin, N.M.; Teillet, M.A. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J. Embryol. Exp. Morphol. 1973, 30, 31–48. [Google Scholar]
- Tielemans, Y.; Willems, G.; Sundler, F.; Håkanson, R. Self-replication of enterochromaffin-like cells in the mouse stomach. Digestion 1990, 45, 138–146. [Google Scholar] [CrossRef]
- Weil, R.J.; Huang, S.; Pack, S.; Vortmeyer, A.O.; Tsokos, M.; Lubensky, I.A.; Oldfield, E.H.; Zhuang, Z. Pluripotent tumor cells in benign pituitary adenomas associated with multiple endocrine neoplasia type 1. Cancer Res. 1998, 58, 4715–4720. [Google Scholar]
- Sei, Y.; Feng, J.; Samsel, L.; White, A.; Zhao, X.; Yun, S.; Citrin, D.; McCoy, J.P.; Sundaresan, S.; Hayes, M.M.; Merchant, J.L.; Leiter, A.; Wank, S.A. Mature enteroendocrine cells contribute to basal and pathological stem cell dynamics in the small intestine. Am. J. Physiol. Gastroentest. Liver Physiol. 2018, 315, G495–G510. [Google Scholar] [CrossRef] [PubMed]
- Jenny, M.; Uhl, C.; Roche, C.; Duluc, I.; Guillermin, V.; Guillemot, F.; Jensen, J.; Kedinger, M.; Gradwohl, G. Neurogenin3 is differentially required for endocrine cell fate specification in the intestinal and gastric epithelium. Embo. J. 2002, 21, 6338–6347. [Google Scholar] [CrossRef]
- Sundler, F. Ontogeny of ECL cells in the rat. Yale J. Biol. Med. 1998, 71, 155–161. [Google Scholar]
- Tømmerås, K.; Hammer, P.; Sundler, F.; Borch, K.; Mårdh, S.; Cabero, J.L. Immunolocalization of cholecystokinin-2 receptors in rat gastric mucosa. Scand. J. Gastroenterol. 2002, 37, 1017–1024. [Google Scholar] [CrossRef]
- Sandvik, A.K.; Waldum, H.L. Rat gastric histamine release, a sensitive gastrin bioassay. Life Sci. 1990, 46, 453–459. [Google Scholar] [CrossRef]
- Brenna, E.; Waldum, H.L. Trophic effect of gastrin on the enterochromaffin like cells of the rat stomach, establishment of a dose response relationship. Gut 1992, 33, 1303–1306. [Google Scholar] [CrossRef]
- Blair, E.L.; Grund, E.R.; Lund, P.K.; Reed, J.D.; Sanders, D.J. Comparison of gastrin bioactivity and immunoreactivity of antral extracts from man, pig and cat. J. Physiol. 1982, 325, 419–421. [Google Scholar] [CrossRef]
- Sjöblom, S.M.; Sipponen, P.; Karonen, S.L.; Järvinen, H.J. Mucosal argyrophil endocrine cells in pernicious anaemia and upper gastrointestinal carcinoid tumours. J. Clin. Pathol. 1989, 42, 371–377. [Google Scholar] [CrossRef]
- Lamberts, R.; Creutzfeldt, W.; Stöckmann, F.; Jacubaschke, U.; Maas, S.; Brunner, G. Long-term omeprazole treatment in man, effects on gastric endocrine cell populations. Digestion 1988, 39, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Waldum, H.L.; Arnestad, J.S.; Brenna, E.; Eide, I.; Syversen, U.; Sandvik, A.K. Marked increase in gastric acid secretory capacity after omeprazole treatment. Gut 1996, 39, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Sandvik, A.K.; Kleveland, P.M.; Waldum, H.L. Muscarinic M2 stimulation releases histamine in the totally isolated, vascularly perfused rat stomach. Scand. J. Gastroenterol. 1988, 23, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Sandvik, A.K.; Cui, G.; Bakke, I.; Munkvold, B.; Waldum, H.L. PACAP stimulates gastric acid secretion in the rat by inducing histamine release. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G997–G1003. [Google Scholar] [CrossRef] [PubMed]
- Håkanson, R.; Vallgren, S.; Ekelund, M.; Rehfeld, J.F. Sundler FThe vagus exerts trophic control of the stomach in the rat. Gastroenterology 1984, 86, 28–32. [Google Scholar] [CrossRef]
- Sandvik, A.K.; Waldum, H.L. The effect of somatostatin on baseline and stimulated acid secretion and vascular histamine release from the totally isolated vascularly perfused rat stomach. Regul. Pept. 1988, 20, 233–239. [Google Scholar] [CrossRef]
- Zamcheck, N.; Grable, E.; Ley, A.; Norman, L. Occurrence of gastric cancer among patients with pernicious anemia at Boston City Hospital. N. Engl. J. Med. 1955, 252, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Vannella, L.; Lahner, E.; Osborn, J.; Annibale, B. Systematic review, gastric cancer incidence in pernicious anaemia. Aliment. Pharmacol. Ther. 2013, 37, 375–382. [Google Scholar] [CrossRef]
- Murphy, G.; Abnet, C.C.; Choo-Wosoba, H.; Vogtmann, E.; Weinstein, S.J.; Taylor, P.R.; Männistö, S.; Albanes, D.; Dawsey, S.M.; Rehfeld, J.F.; Freedman, N.D. Serum gastrin and cholecystokinin are associated with subsequent development of gastric cancer in a prospective cohort of Finnish smokers. Int. J. Epidemiol. 2017, 45, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.S.; Lieu, S.N.; Yamaguchi, D.J.; Tachiki, K.; Lambrecht, N.; Ohning, G.V.; Sachs, G.; Germano, P.M.; Pisegna, J.R. PACAP regulation of secretion and proliferation of pure populations of gastric ECL cell. J. Mol. Neuosci. 2005, 26, 85–97. [Google Scholar] [CrossRef]
- Fykse, V.; Sandvik, A.K.; Qvigstad, G.; Falkmer, S.E.; Syversen, U.; Waldum, H.L. Treatment of ECL cell carcinoids with octreotide LAR. Scand. J. Gastroenterol. 2004, 39, 821–828. [Google Scholar] [CrossRef]
- Bakke, I.; Sandvik, A.K.; Waldum, H.L. Octreotide inhibits the enterochromaffin-like cell but not peroxisome proliferator-induced hypergastrinemia. J. Mol. Endocrinol. 2000, 25, 109–119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reimer, C.; Søndergaard, B.; Hilsted, L.; Bytzer, P. Proton-pump inhibitor therapy induces acid related symptoms in healthy volunteers after withdrawal of therapy. Gastroenterology 2009, 137, 80–87. [Google Scholar] [CrossRef]
- Björnsson, E.; Abrahamsson, H.; Simrén, M.; Mattsson, N.; Jensen, C.; Agerforz, P.; Kilander, A. Discontinuation of proton pump inhibitors in patients on long-term therapy, a double-blind placebo-controlled trial. Aliment. Pharmacol. Ther. 2006, 24, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Qvigstad, G.; Arnestad, J.S.; Brenna, E.; Waldum, H.L. Treatment with proton pump inhibitors induces tolerance to histamine-2 receptor antagonists in Helicobacter pylori negative patients. Scand. J. Gastroenterol. 1998, 33, 1244–1248. [Google Scholar]
- Marshall, B.I.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Levi, S.; Beardshall, K.; Haddad, G.; Playford, R.; Ghosh, P.; Calam, J. Campylobacter pylori and duodenal ulcers, the gastrin link. Lancet 1989, 1, 1157–1158. [Google Scholar]
- Smith, J.T.; Pounder, R.E.; Nwokolo, C.U.; Lanzon-Miller, S.; Evans, D.G.; Graham, D.Y.; Evans, D.J., Jr. Inappropriate hypergastrinemia in asymptomatic healthy subjects infected with Helicobacter pylori. Gut 1990, 31, 522–525. [Google Scholar] [CrossRef]
- Waldum, H.L.; Kleveland, P.M.; Sørdal, Ø.G.F. Helicobacter pylori and gastric acid, an intimate and reciprocal relationship. Ther. Adv. Gastroenterol. 2016, 9, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.E.; Pandol, S.J.; Castell, D.O.; McCarthy, D.M. Gastroesophageal reflux disease in the Zollinger-Ellison syndrome. Ann. Intern. Med. 1981, 95, 37–43. [Google Scholar] [CrossRef]
- Fossmark, R.; Brenna, E.; Waldum, H.L. pH 4.0. Scand. J. Gastroenterol. 2007, 42, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Borch, K.; Renvall, H.; Liedberg, G. Gastric endocrine cell hyperplasia and carcinoid tumors in pernicious anemia. Gastroenterology 1985, 88, 638–648. [Google Scholar] [CrossRef]
- Solcia, E.; Capella, C.; Fiocca, R.; Rindi, G.; Rosai, J. Gastric argyrophil carcinoids in patients with Zollinger-Ellison syndrome due to type 1 multiple endocrine neoplasia. A newly recognized association. Am. J. Surg. Pathol. 1990, 14, 503–513. [Google Scholar] [CrossRef]
- Bordi, C. Carcinoid [ECL cell] tumor of the oxyntic mucosa of the stomach, a hormone dependent neoplasm? Prog. Surg. Pathol. 1988, 8, 177–195. [Google Scholar]
- Tsolakis, A.V.; Portela-Gomes, G.M.; Stridsberg, M.; Grimelius, L.; Sundin, A.; Eriksson, B.K.; Oberg, K.E.; Janson, E.T. Malignant gastric ghrelinoma with hyperghrelinemia. J. Clin. Endocrinol. Metab. 2004, 89, 3739–3744. [Google Scholar] [CrossRef]
- Waldum, H.L.; Haugen, O.A.; Brenna, E. Do neuroendocrine cells, particularly the D cell, play a role in the development of gastric stump cancer? Cancer Detect Prev. 1994, 18, 431–436. [Google Scholar] [PubMed]
- Yang, Z.; Wang, W.; Lu, J.; Pan, G.; Pan, Z.; Chen, Q.; Liu, W.; Zhao, Y. Gastric neuroendocrine tumors [G-Nets], Incidence, prognosis and recent trend toward improved survival. Cell Physiol. Biochem. 2018, 45, 389–396. [Google Scholar] [CrossRef]
- Procter & Gamble Company Astra Zeneca L.P. Omeprazole magnesium tablets. NDA 2002, 21, 229. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2003/21229ltr_Prilosec.pdf (accessed on 16 May 2019).
- Eckhauser, F.E.; Lloyd, R.V.; Thompson, N.W.; Raper, S.E.; Vinik, A.I. Antrectomy for multicentric, argyrophil gastric carcinoids, a preliminary report. Surgery 1988, 104, 1046–1053. [Google Scholar]
- Sato, Y.; Iwafuchi, M.; Ueki, J.; Yoshimura, A.; Mochizuki, T.; Motoyama, H.; Sugimura, K.; Honma, T.; Narisawa, R.; Ichida, T.; Asakura, H.; Van Thiel, D.H. Gastric carcinoid tumors without autoimmune gastritis in Japan, a relationship with Helicobacter pylori. Dig. Dis. Sci. 2002, 47, 579–585. [Google Scholar] [CrossRef]
- Antonodimitrakis, P.; Tsolakis, A.; Welin, S.; Kozlovacki, G.; Oberg, K.; Granberg, D. Gastric carcinoid in a patient infected with Helicobacter pylori, a new entity. World J. Gastroenterol. 2011, 17, 3066–3068. [Google Scholar] [CrossRef]
- Fossmark, R.; Sørdal, Ø.; Jianu, C.S.; Qvigstad, G.; Nordrum, I.S.; Boyce, M.; Waldum, H.L. Treatment of gastric carcinoids type 1 with the gastrin receptor antagonist netazepide [YF476] results in regression of tumours and normalization of serum chromogranin A. Aliment. Pharmacol. Ther. 2012, 36, 1067–1075. [Google Scholar] [CrossRef]
- Jianu, C.S.; Fossmark, R.; Syversen, U.; Hauso, Ø.; Fykse, V.; Waldum, H.L. Five-year follow-up of patients treated for 1 year with octreotide long-acting release for enterochromaffin-like cell carcinoids. Scand. J. Gastroenterol. 2011, 46, 456–463. [Google Scholar] [CrossRef]
- Waldum, H.L.; Sagatun, L.; Mjønes, P. Gastrin and gastric cancer. Front Endocrinol [Lausanne] 2017, 8, 1. [Google Scholar] [CrossRef]
- Rindi, G.; Luinetti, O.; Cornaggia, M.; Capella, C.; Solcia, E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma, a clinicopathologic study. Gastroenterology 1993, 104, 994–1006. [Google Scholar] [CrossRef]
- Qvigstad, G.; Falkmer, S.; Westre, B.; Waldum, H.L. Clinical and histopathological tumour progression in ECL cell carcinoids [“ECLomas”]. APMIS 1999, 107, 1085–1092. [Google Scholar] [CrossRef]
- Calvete, O.; Reyes, J.; Zuñiga, S.; Paumard-Hernández, B.; Fernández, V.; Bujanda, L.; Rodriguez-Pinilla, M.S.; Palacios, J.; Heine-Suñer, D.; Banka, S.; Newman, W.G.; Cañamero, M.; Pritchard, D.M.; Benítez, J. Exome sequencing identifies ATP4A gene as responsible of an atypical familial type I gastric neuroendocrine tumor. Hum. Mol. Genet. 2015, 24, 2914–2922. [Google Scholar] [CrossRef]
- Fossmark, R.; Calvete, O.; Mjønes, P.; Benitez, J.; Waldum, H.L. ECL-cell carcinoids and carcinoma in patients homozygous for an inactivating mutation in the gastric H [+] K[+]ATPase alpha subunit. APMIS 2016, 124, 561–566. [Google Scholar] [CrossRef]
- Capuano, F.; Grami, O.; Pugliese, L.; Paulli, M.; Pietrabissa, A.; Solcia, E.; Vanoli, A. Grade 3 neuroendocrine tumor [G3 NET] in a background of multiple serotonin cell neoplasms of the ileum associated with carcinoid syndrome and aggressive behavior. Endocrin. Pathol. 2018, 29, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Bordi, C.; Rappel, S.; La Rosa, S.; Stolte, M.; Solcia, E. Gastric carcinoids and neuroendocrine carcinomas, pathogenesis, pathology, and behavior. World J. Surg. 1996, 20, 168–172. [Google Scholar] [CrossRef]
- Laurén, P. The two histologicalmain types of gastric carcinoma, diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classifycation. Acta. Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Bosman, F.T.; Carneiro, F.; Hruban, R.H.; Theise, N.D. WHO classification of tumours of the digestive system [No. Ed. 4]. World Health Organization, 2010. Available online: http://apps.who.int/bookorders/anglais/detart1.jsp?codlan=1&codcol=70&codcch=4003 (accessed on 16 May 2019).
- Soga, J.; Tazawa, M.D.; Ito, H. Ultrastructural demonstration of specific secretory granules of Mastomys gastric carcinoids. Acta. Med. Biol. [Niigata] 1969, 17, 119–124. [Google Scholar]
- Poynter, D. Long-term effects of reduced gastric acidity in laboratory animals. Digestion 1985, 31, 174. [Google Scholar]
- Azzopardi, J.G.; Pollock, D.J. Argentaffin and argyrophil cells in gastric carcinoma. J. Pathol. Bacteriol. 1963, 86, 443–451. [Google Scholar] [CrossRef]
- Wilander, E.; El-Salhy, M.; Pitkänen, P. Histopathology of gastric carcinoids, a survey of 42 cases. Histopathology 1984, 8, 183–193. [Google Scholar] [CrossRef]
- Bakkelund, K.; Fossmark, R.; Nordrum, I.; Waldum, H. Signet ring cells in gastric carcinomas are derived from neuroendocrine cells. J. Histochem. Cytochem. 2006, 54, 615–621. [Google Scholar] [CrossRef]
- Sørdal, Ø.; Qvigstad, G.; Nordrum, I.S.; Sandvik, A.K.; Gustafsson, B.I.; Waldum, H. The PAS positive material in gastric cancer cells of signet ring type is not mucin. Exp. Mol. Pathol. 2014, 96, 274–278. [Google Scholar] [CrossRef]
- Qvigstad, G.; Sandvik, A.K.; Brenna, E.; Aase, S.; Waldum, H.L. Detection of chromogranin A in human gastric adenocarcinomas using a sensitive immunohistochemical technique. Histochem. J. 2000, 32, 551–556. [Google Scholar] [CrossRef]
- Fujiyoshi, Y.; Eimoto, T. Chromogranin A expression correlates with tumor cell type and prognosis in signet ring cell carcinoma of the stomach. Histopathology 2008, 52, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Bartley, A.N.; Rashid, A.; Fournier, K.F.; Abraham, S.C. Neuroendocrine and mucinous differentiation in signet ring cell carcinoma of the stomach, evidence for a common cell of origin in composite tumors. Hum. Pathol. 2011, 42, 1420–1429. [Google Scholar] [CrossRef]
- Morson, B.C. Intestinal metaplasia of the gastric mucosa. Br. J. Cancer 1955, 9, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef]
- Pero, R.; Peluso, S.; Angrisano, T.; Tuccillo, C.; Sacchetti, S.; Keller, S.; Tomaiuolo, R.; Bruni, C.B.; Lembo, F.; Chiariotti, L. Chromatin and DNA methylation dynamics of Helicobacter pylori induced COX-2 activation. Int. J. Med. Microbiol. 2011, 301, 140–149. [Google Scholar] [CrossRef]
- Angrisano, T.; Lembo, F.; Peluso, S.; Keller, S.; Chiariotti, L.; Pero, R. Helicobacter pylori regulates INOS promoter by histone modifications in human gastric epithelial cells. Med. Microbiol. Immunol. 2012, 201, 249–257. [Google Scholar] [CrossRef]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Waldum, H.L.; Hauso, Ø.; Sørdal, Ø.F.; Fossmark, R. Gastrin may mediate the carcinogenic effect of Helicobacter pylori infection of the stomach. Dig. Dis. Sci. 2015, 60, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Waldum, H.L.; Sørdal, Ø.F. Classification of epithelial malignant tumors- the differentiation between adenocarcinomas and neuroendocrine carcinomas, Why rely on nonspecific histochemistry and dismiss specific methods like immunohistochemistry and in situ hybridization? Appl. Immunohistochem. Mol. Morphol. 2016, 24, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Mjønes, P.; Nordrum, I.S.; Sørdal, Ø.; Sagatun, L.; Fossmark, R.; Sandvik, A.; Waldum, H.L. Expression of the cholecystokinin-B receptor in neoplastic gastric cells. Horm. Cancer 2018, 9, 40–54. [Google Scholar] [CrossRef]
- Pottegård, A.; Broe, A.; Hallas, J.; de Muckadell, O.B.; Lassen, A.T.; Lødrup, A.B. Use of proton-pump inhibitors among adults, a Danish nationwide drug utilization study. Therap. Adv. Gastroenterol. 2016, 9, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Waldum, H.L.; Hauso, Ø.; Brenna, E.; Qvigstad, G.; Fossmark, R. Does long-term profound acid inhibition of gastric acid secretion increase the risk of ECL-cell derived tumors in man. Scand. J. Gastroenterol. 2016, 51, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.S.; Chan, E.W.; Wong, A.Y.S.; Chen, L.; Wong, I.C.K.; Leung, W.K. Long-term proton pump inhibitors and risk of gastric cancer development after treatment for Helicobacter pylori, a populationbased study. Gut 2018, 67, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Brusselaers, N.; Wahlin, K.; Engstrand, L.; Lagergren, J. Maintenance therapy with proton pump inhibitors and risk of gastric cancer, a nationwide population-based cohort study in Sweden. BMJ Open 2017, 7, e017739. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waldum, H.L.; Sørdal, Ø.F.; Mjønes, P.G. The Enterochromaffin-like [ECL] Cell—Central in Gastric Physiology and Pathology. Int. J. Mol. Sci. 2019, 20, 2444. https://doi.org/10.3390/ijms20102444
Waldum HL, Sørdal ØF, Mjønes PG. The Enterochromaffin-like [ECL] Cell—Central in Gastric Physiology and Pathology. International Journal of Molecular Sciences. 2019; 20(10):2444. https://doi.org/10.3390/ijms20102444
Chicago/Turabian StyleWaldum, Helge L., Øystein F. Sørdal, and Patricia G. Mjønes. 2019. "The Enterochromaffin-like [ECL] Cell—Central in Gastric Physiology and Pathology" International Journal of Molecular Sciences 20, no. 10: 2444. https://doi.org/10.3390/ijms20102444
APA StyleWaldum, H. L., Sørdal, Ø. F., & Mjønes, P. G. (2019). The Enterochromaffin-like [ECL] Cell—Central in Gastric Physiology and Pathology. International Journal of Molecular Sciences, 20(10), 2444. https://doi.org/10.3390/ijms20102444