LT-IIc, A Bacterial Type II Heat-Labile Enterotoxin, Induces Specific Lethality in Triple Negative Breast Cancer Cells by Modulation of Autophagy and Induction of Apoptosis and Necroptosis


Abstract
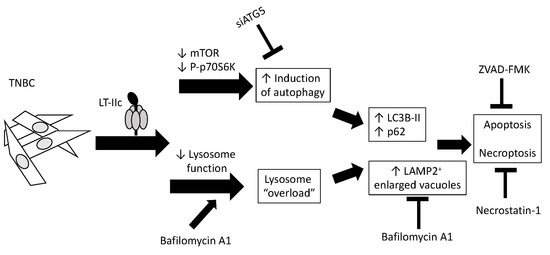
{kind=link}
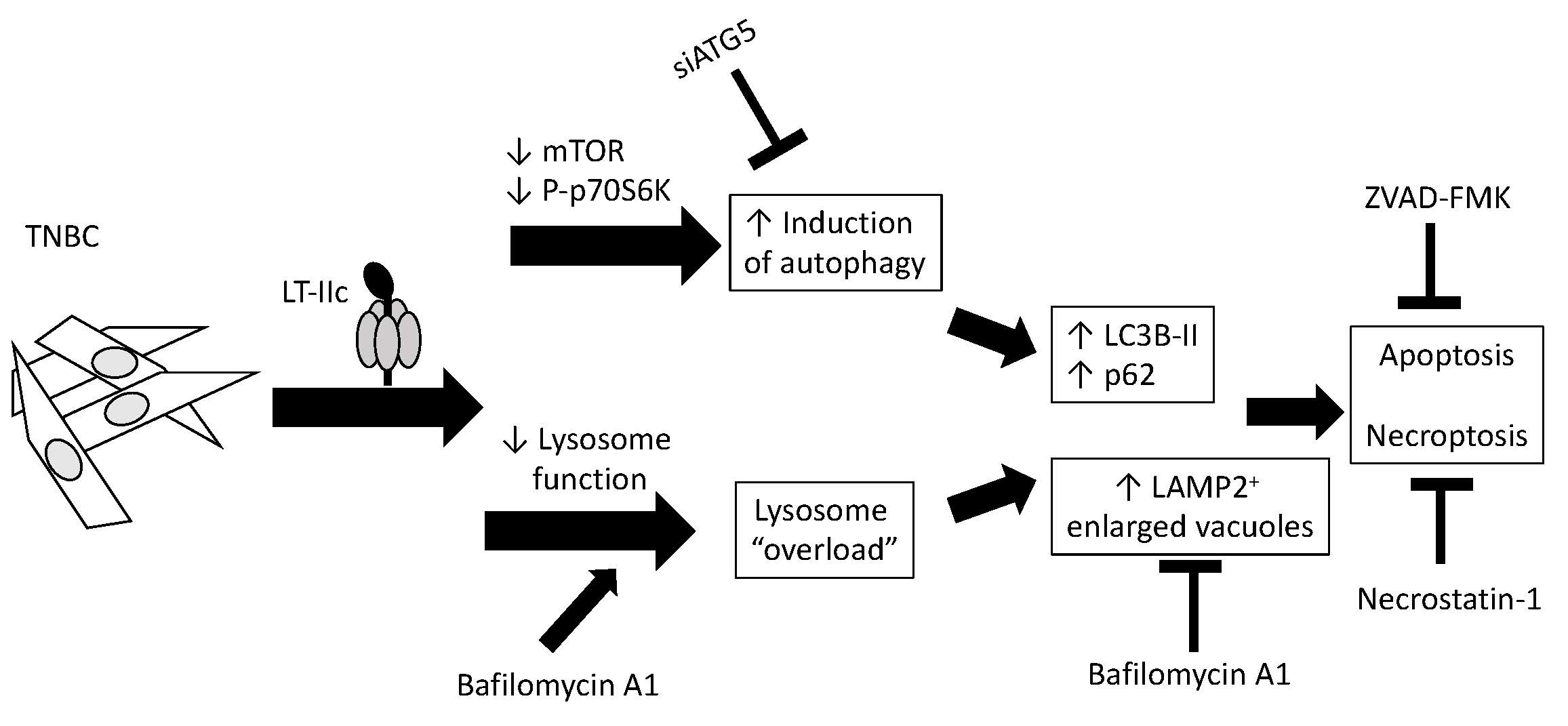
{kind=link}
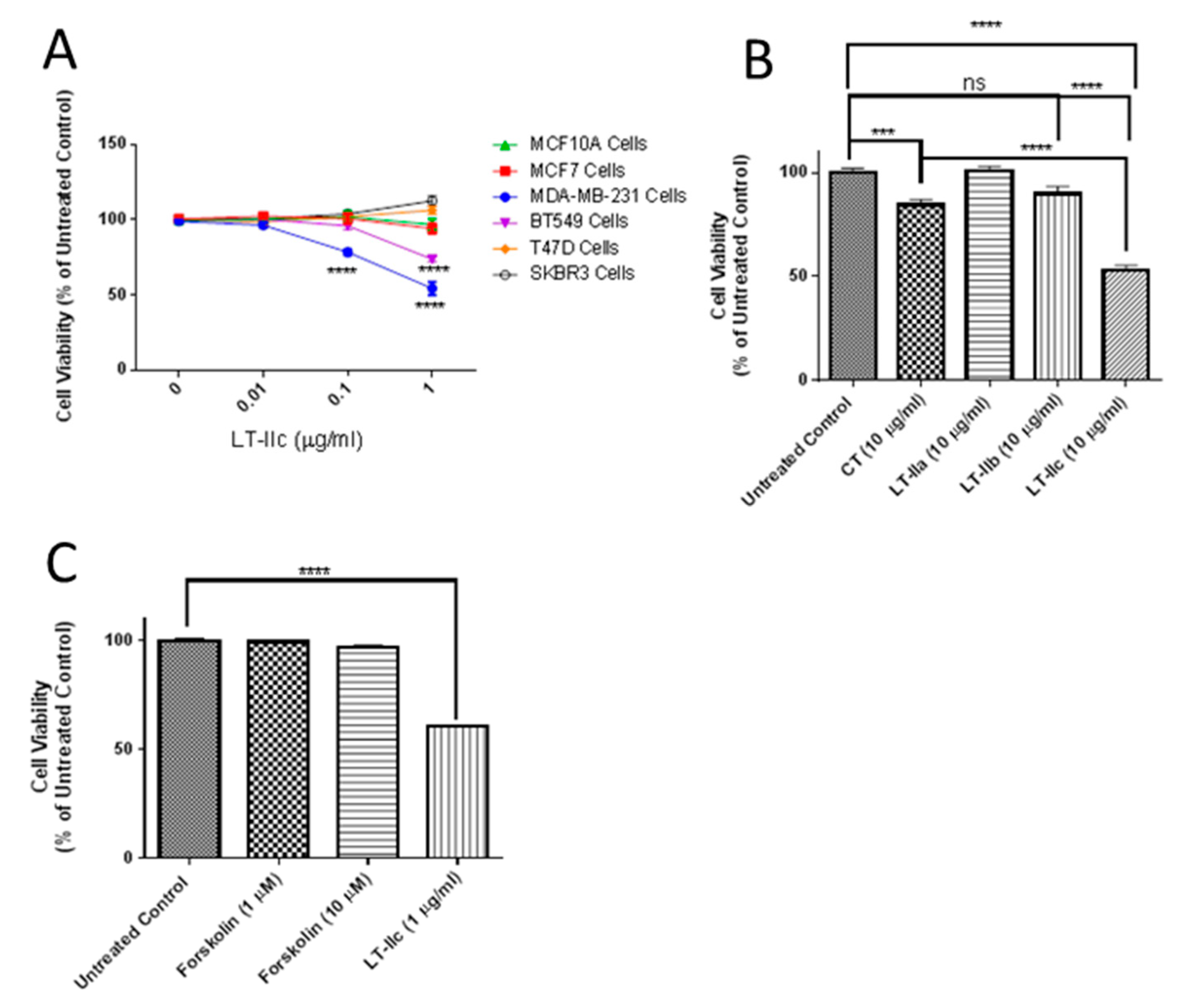
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. LT-IIc Selectively and Irreversibly Induces Cell Death of TNBC Cells
2.2. Requirement for Adenylate Cyclase Activity for Cytotoxic Effects in MDA-MB-231 Cells
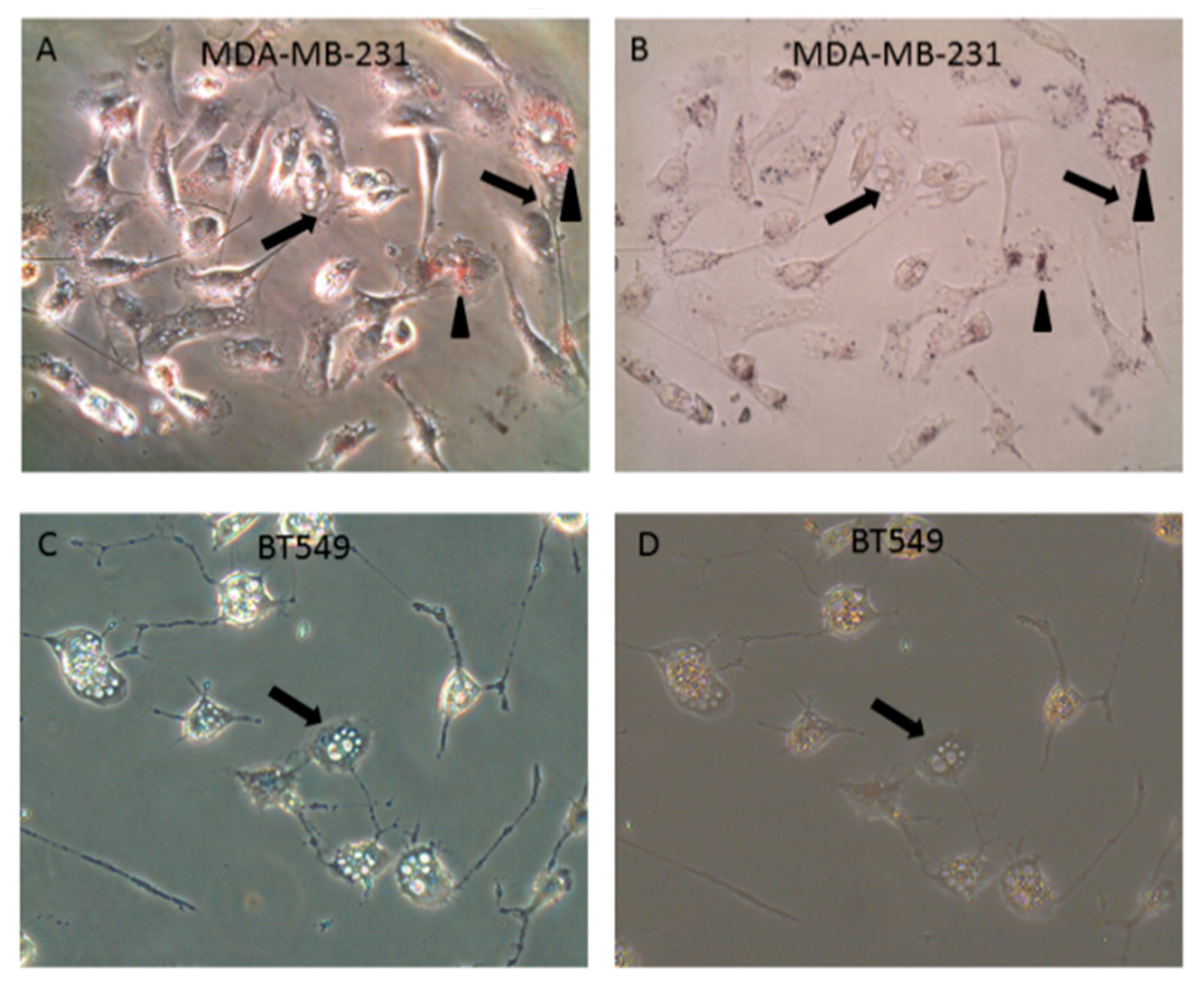
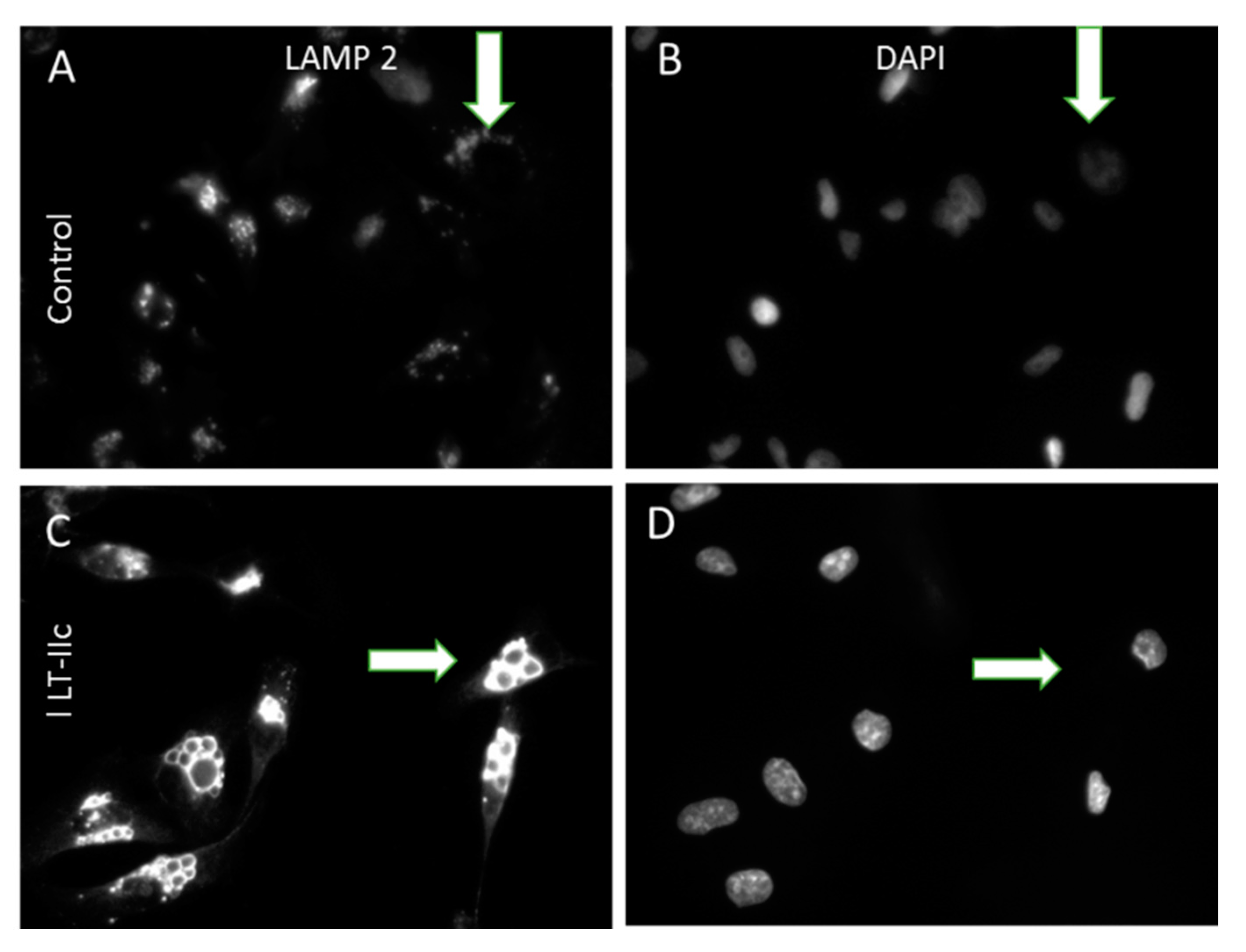
2.3. LT-IIc Induces Accumulation of Autolysosomes Specifically in TNBC
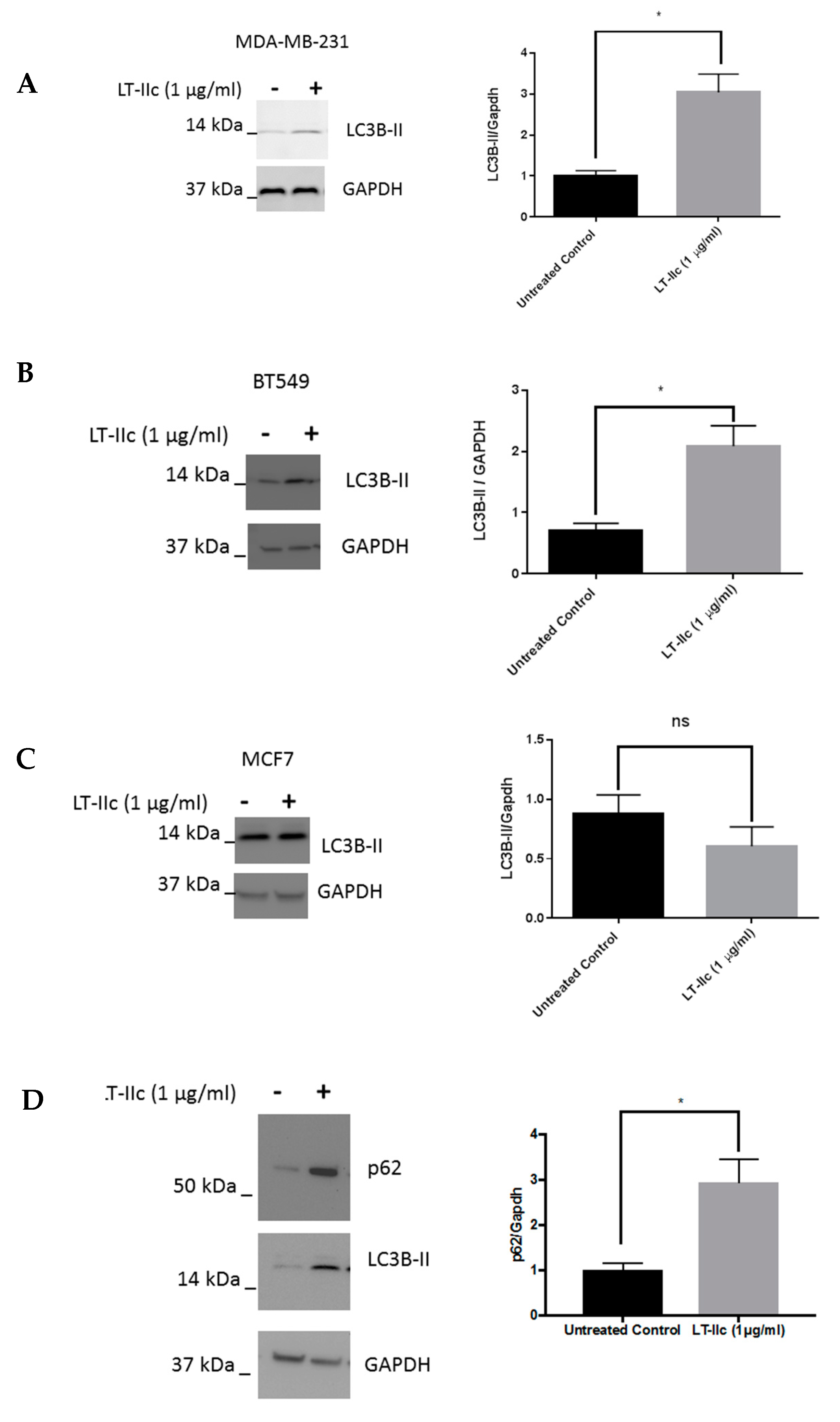
2.4. LT-IIc Alters Markers of Autophagic Flux in TNBC Cell Lines
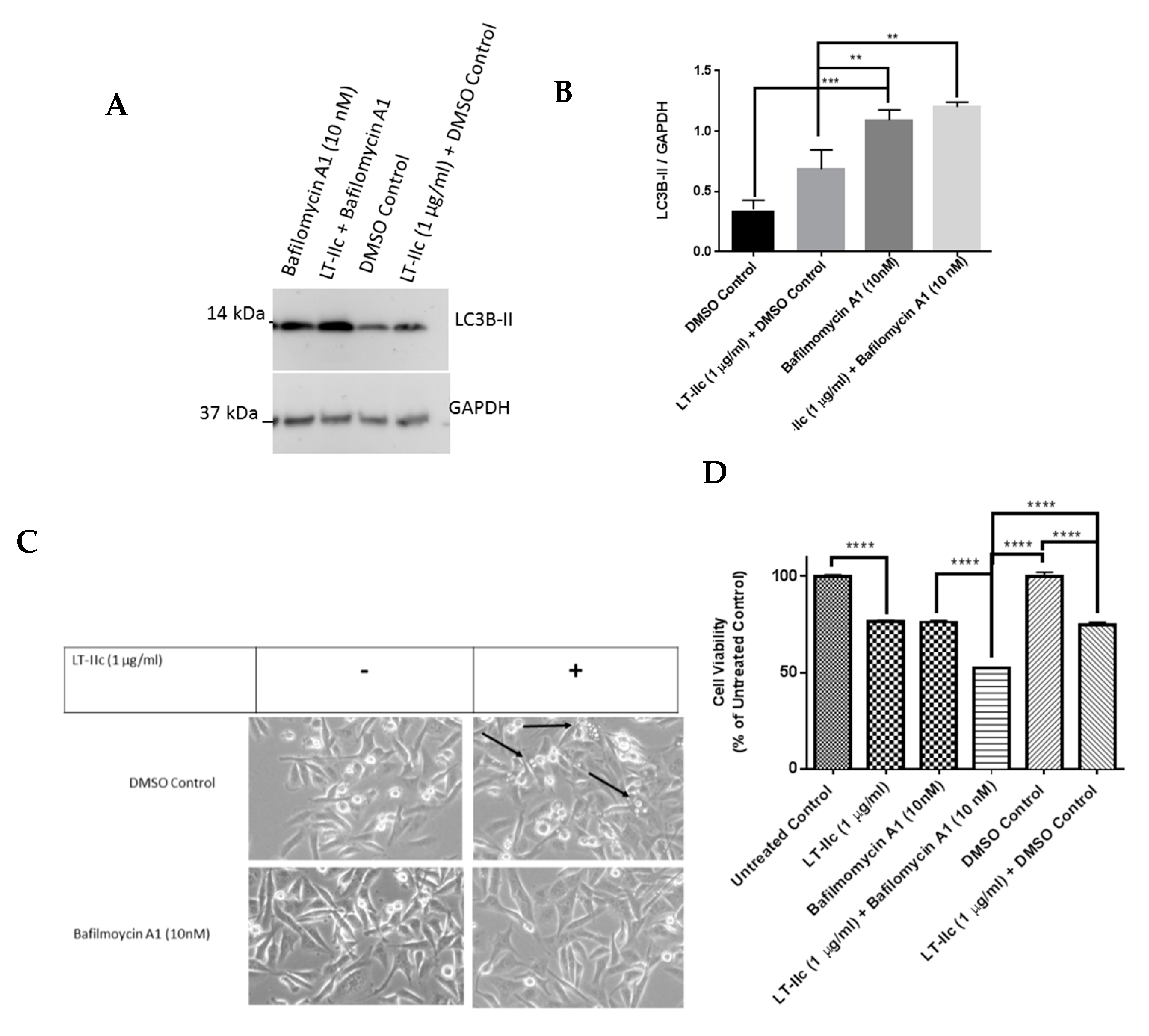
2.5. The Autophagy Inhibitor Bafilomycin A1 Blocks LT-IIc-Mediated Autolysosome Accumulation
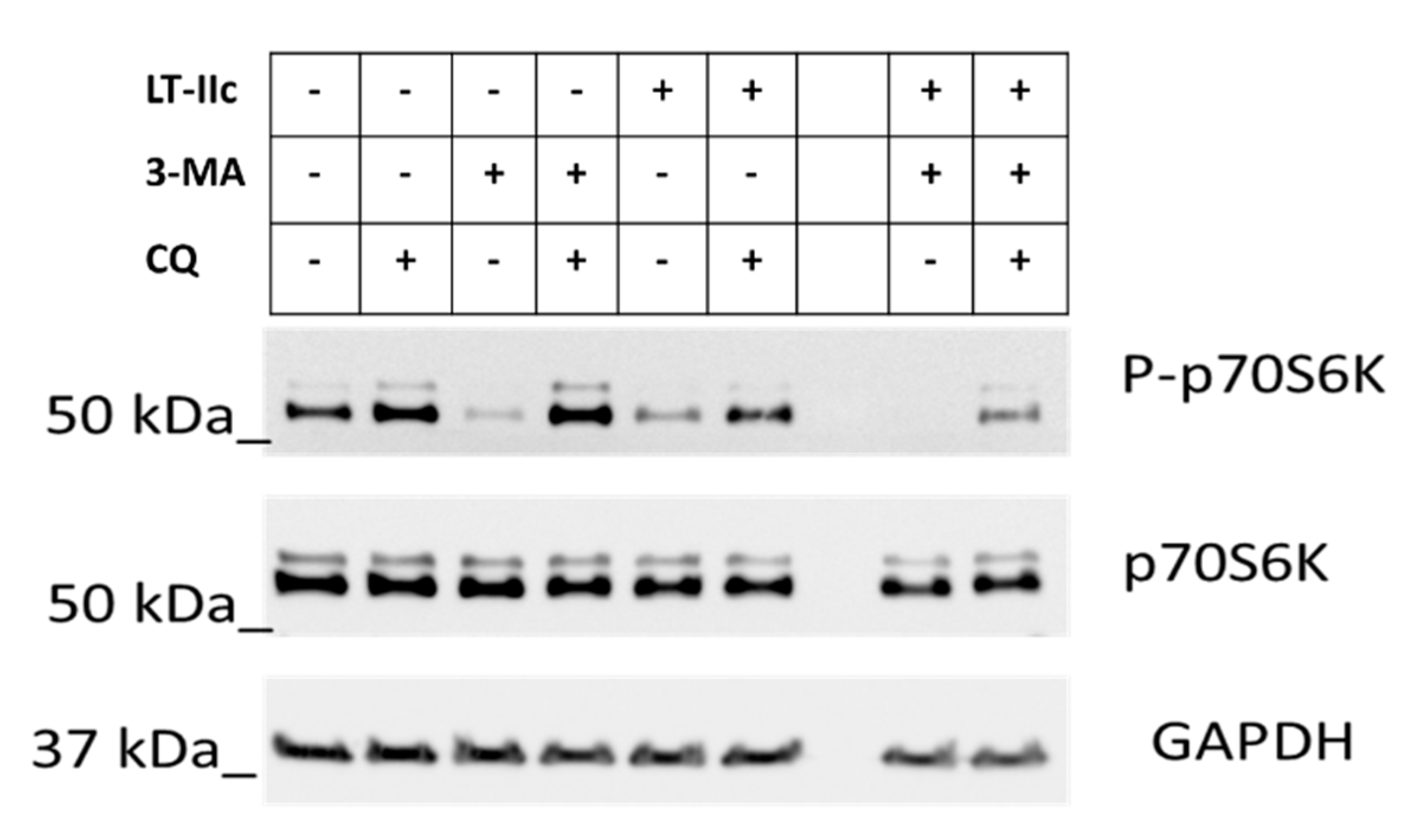
2.6. LT-IIc Induces Autophagy through Inhibition of the mTOR Pathway
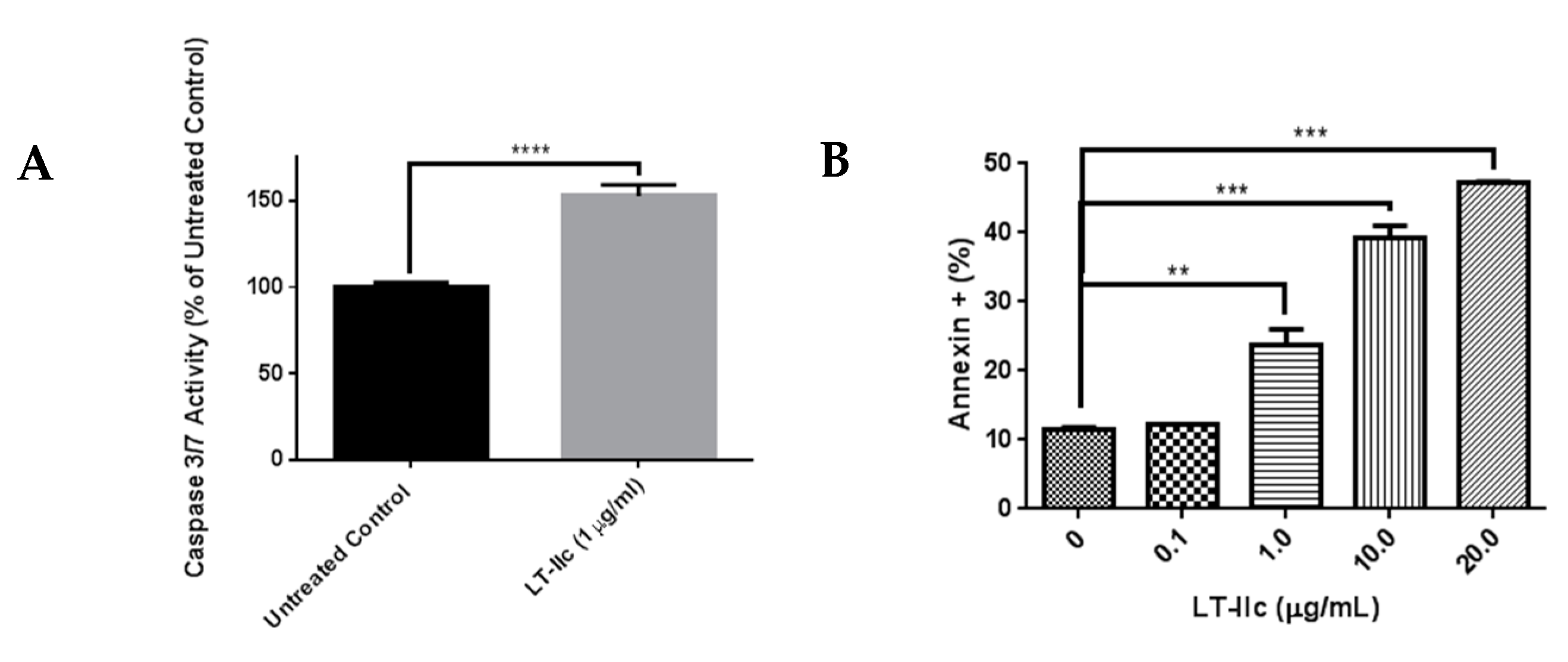
2.7. LT-IIc Induces Apoptotosis in MDA-MB-231 Cells
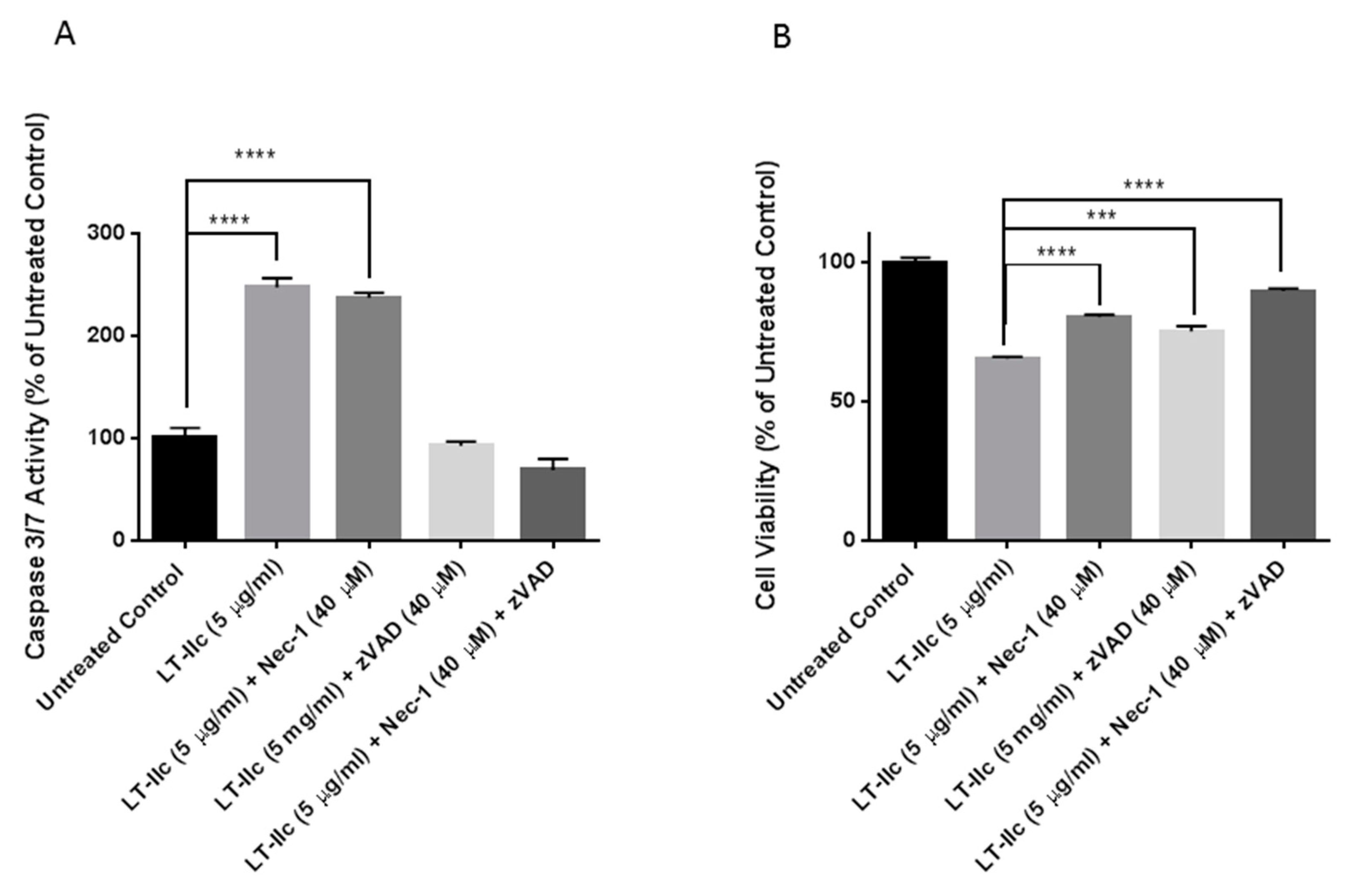
2.8. LT-IIc-Induced Cell Death is Mediated by a Combined Induction of Apoptosis and Necroptosis
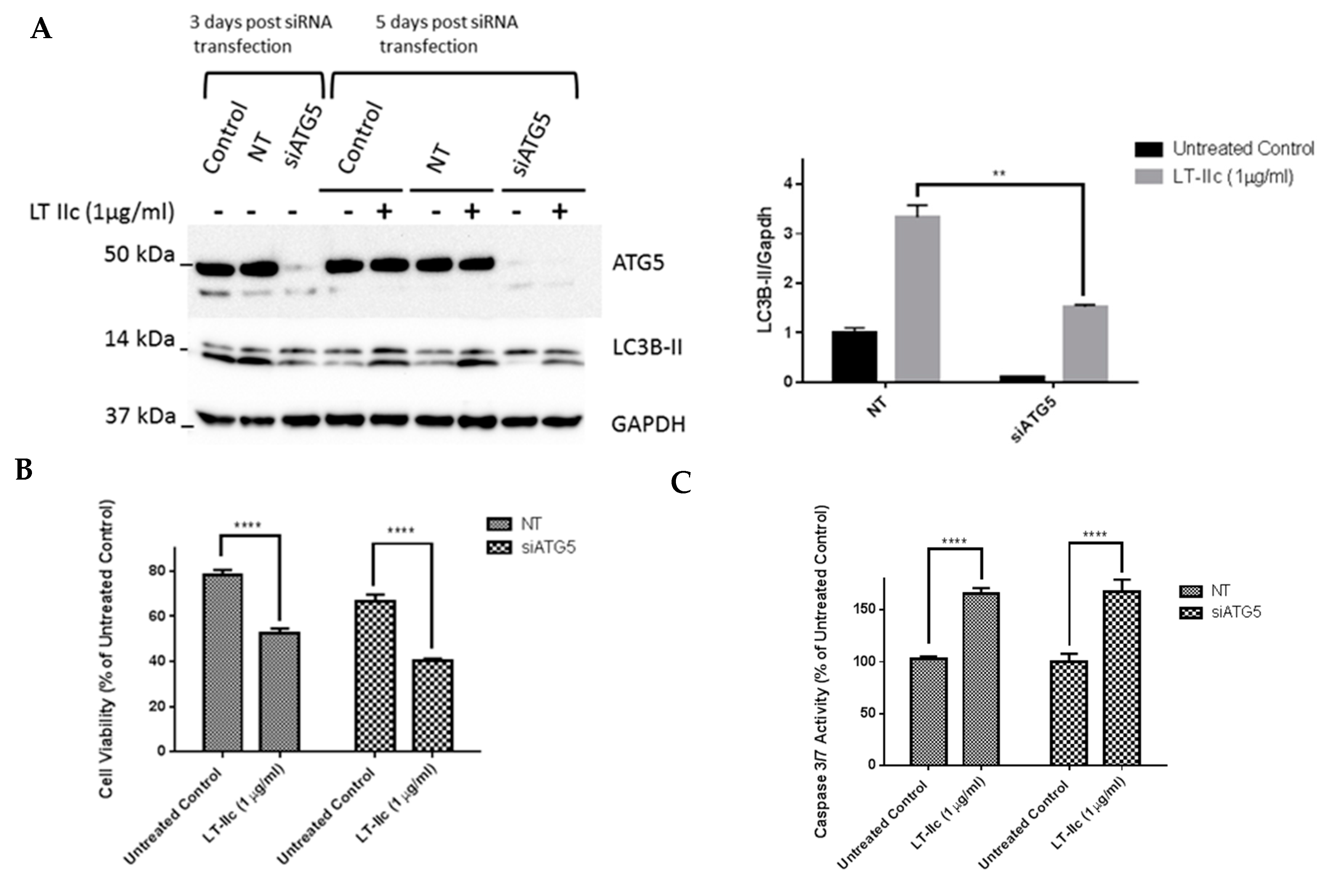
2.9. ATG5 Knockdown Does Not Block LT-IIc-Mediated Cytotoxicity or Levels of LC3B-II
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cell Viability and Caspase 3/7 Activity Assays
4.3. Treatment with ZVAD-FMK and Necrostatin-1
4.4. Annexin V-FITC Apoptosis Assay
4.5. Western Blotting
4.6. Oil Red O Staining
4.7. LAMP-2 Staining
4.8. Cell Imaging
4.9. Cell Transfection with siRNA
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Kalimutho, M.; Parsons, K.; Mittal, D.; Lopez, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef]
- Felgner, S.; Kocijancic, D.; Frahm, M.; Weiss, S. Bacteria in cancer therapy: Renaissance of an old concept. Int. J. Microbiol. 2016, 2016, 8451728. [Google Scholar] [CrossRef]
- Havas, H.F.; Donnelly, A.J. Mixed bacterial toxins in the treatment of tumors. IV. Response of methylcholanthrene-induced, spontaneous, and transplanted tumors in mice. Cancer Res. 1961, 21, 17–25. [Google Scholar]
- Wilde, L.; Tanson, K.; Curry, J.; Martinez-Outschoorn, U. Autophagy in cancer: A complex relationship. Biochem. J. 2018, 475, 1939–1954. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Simon, A.K.; Baehrecke, E.H. Current questions and possible controversies in autophagy. Cell Death Discov. 2015, 1, 15036. [Google Scholar] [CrossRef]
- Bousquet, G.; El Bouchtaoui, M.; Sophie, T.; Leboeuf, C.; de Bazelaire, C.; Ratajczak, P.; Giacchetti, S.; de Roquancourt, A.; Bertheau, P.; Verneuil, L.; et al. Targeting autophagic cancer stem-cells to reverse chemoresistance in human triple negative breast cancer. Oncotarget 2017, 8, 35205–35221. [Google Scholar] [CrossRef]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Mulcahy Levy, J.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-mediated autophagy dependence identifies subtypes of breast cancer where autophagy inhibition can be efficacious. Cancer Res. 2014, 74, 2579–2590. [Google Scholar] [CrossRef]
- Han, Y.; Fan, S.; Qin, T.; Yang, J.; Sun, Y.; Lu, Y.; Mao, J.; Li, L. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int. J. Oncol. 2018, 52, 1057–1070. [Google Scholar] [CrossRef]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The autophagy inhibitor chloroquine targets cancer stem cells in triple negative breast cancer by inducing mitochondrial damage and impairing DNA break repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef]
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.L. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfbeta/Smad Signaling. Cancer Res. 2016, 76, 3397–3410. [Google Scholar] [CrossRef]
- Yu, L.; Lenardo, M.J.; Baehrecke, E.H. Autophagy and caspases: A new cell death program. Cell Cycle 2004, 3, 1124–1126. [Google Scholar] [CrossRef]
- Nawar, H.F.; King-Lyons, N.D.; Hu, J.C.; Pasek, R.C.; Connell, T.D. LT-IIc, a new member of the type II heat-labile enterotoxin family encoded by an Escherichia coli strain obtained from a nonmammalian host. Infect. Immun. 2010, 78, 4705–4713. [Google Scholar] [CrossRef]
- Nawar, H.F.; Greene, C.J.; Lee, C.H.; Mandell, L.M.; Hajishengallis, G.; Connell, T.D. LT-IIc, a new member of the type II heat-labile enterotoxin family, exhibits potent immunomodulatory properties that are different from those induced by LT-IIa or LT-IIb. Vaccine 2011, 29, 721–727. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol. Asp. Med. 2006, 27, 495–502. [Google Scholar] [CrossRef]
- Huynh, K.K.; Eskelinen, E.L.; Scott, C.C.; Malevanets, A.; Saftig, P.; Grinstein, S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007, 26, 313–324. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Chen, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Antoni, F.A. Molecular diversity of cyclic AMP signalling. Front. Neuroendocrinol. 2000, 21, 103–132. [Google Scholar] [CrossRef]
- Totsuka, Y.; Ferdows, M.S.; Nielsen, T.B.; Field, J.B. Effects of forskolin on adenylate cyclase, cyclic AMP, protein kinase and intermediary metabolism of the thyroid gland. Biochim. Biophys. Acta 1983, 756, 319–327. [Google Scholar] [CrossRef]
- Jin, M.; Klionsky, D.J. Regulation of autophagy: Modulation of the size and number of autophagosomes. FEBS Lett. 2014, 588, 2457–2463. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.; Blanz, J.; Von Figura, K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Elazar, Z.; Seglen, P.O.; Rubinsztein, D.C. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 2008, 4, 849–850. [Google Scholar] [CrossRef]
- Chittaranjan, S.; Bortnik, S.; Dragowska, W.H.; Xu, J.; Abeysundara, N.; Leung, A.; Go, N.E.; DeVorkin, L.; Weppler, S.A.; Gelmon, K.; et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin. Cancer Res. 2014, 20, 3159–3173. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Li, M.; Khambu, B.; Zhang, H.; Kang, J.H.; Chen, X.; Chen, D.; Vollmer, L.; Liu, P.Q.; Vogt, A.; Yin, X.M. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J. Biol. Chem. 2013, 288, 35769–35780. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122 Pt 20, 3589–3594. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef]
- Ekert, P.G.; Silke, J.; Vaux, D.L. Caspase inhibitors. Cell Death Differ. 1999, 6, 1081–1086. [Google Scholar] [CrossRef]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Sarotra, P.; Medhi, B. Use of Bacteria in Cancer Therapy. Recent Results Cancer Res. 2016, 209, 111–121. [Google Scholar]
- Patyar, S.; Joshi, R.; Byrav, D.S.; Prakash, A.; Medhi, B.; Das, B.K. Bacteria in cancer therapy: A novel experimental strategy. J. Biomed. Sci. 2010, 17, 21. [Google Scholar] [CrossRef]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Yaghoobi, H.; Bandehpour, M.; Kazemi, B. Apoptotic Effects of the B Subunit of Bacterial Cytolethal Distending Toxin on the A549 Lung Cancer Cell Line. Asian Pac. J. Cancer Prev. 2016, 17, 299–304. [Google Scholar] [CrossRef]
- Fukuhara, H.; Homma, Y.; Todo, T. Oncolytic virus therapy for prostate cancer. Int. J. Urol. 2010, 17, 20–30. [Google Scholar] [CrossRef]
- Goldufsky, J.; Sivendran, S.; Harcharik, S.; Pan, M.; Bernardo, S.; Stern, R.H.; Friedlander, P.; Ruby, C.E.; Saenger, Y.; Kaufman, H.L. Oncolytic virus therapy for cancer. Oncolytic Virother. 2013, 2, 31–46. [Google Scholar] [PubMed]
- Meng, S.; Xu, J.; Wu, Y.; Ding, C. Targeting autophagy to enhance oncolytic virus-based cancer therapy. Expert Opin. Biol. Ther. 2013, 13, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.; Rettedal, E.; Lehouritis, P.; Devoy, C.; Tangney, M. Intratumoural production of TNFalpha by bacteria mediates cancer therapy. PLoS ONE 2017, 12, e0180034. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Saka, H.A.; Chinen, I.; Zoppino, F.C.; Yoshimori, T.; Bocco, J.L.; Colombo, M.I. Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc. Natl. Acad. Sci. USA 2007, 104, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J. Interactions between Autophagy and Bacterial Toxins: Targets for Therapy? Toxins 2015, 7, 2918–2958. [Google Scholar] [CrossRef] [PubMed]
- Buffen, K.; Oosting, M.; Quintin, J.; Ng, A.; Kleinnijenhuis, J.; Kumar, V.; van de Vosse, E.; Wijmenga, C.; van Crevel, R.; Oosterwijk, E.; et al. Autophagy controls BCG-induced trained immunity and the response to intravesical BCG therapy for bladder cancer. PLoS Pathog. 2014, 10, e1004485. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Connell, T.D. Type II heat-labile enterotoxins: Structure, function, and immunomodulatory properties. Vet. Immunol. Immunopathol. 2013, 152, 68–77. [Google Scholar] [CrossRef]
- Pietrocola, F.; Izzo, V.; Niso-Santano, M.; Vacchelli, E.; Galluzzi, L.; Maiuri, M.C.; Kroemer, G. Regulation of autophagy by stress-responsive transcription factors. Semin. Cancer Biol. 2013, 23, 310–322. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Balliet, R.M.; Rivadeneira, D.B.; Chiavarina, B.; Pavlides, S.; Wang, C.; Whitaker-Menezes, D.; Daumer, K.M.; Lin, Z.; Witkiewicz, A.K.; et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010, 9, 3256–3276. [Google Scholar] [CrossRef]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Chiavarina, B.; Pavlides, S.; Whitaker-Menezes, D.; Tsirigos, A.; Witkiewicz, A.; Lin, Z.; Balliet, R.; Howell, A.; et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: Emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol. Ther. 2010, 10, 537–542. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Pavlides, S.; Chiavarina, B.; Bonuccelli, G.; Casey, T.; Tsirigos, A.; Migneco, G.; Witkiewicz, A.; Balliet, R.; et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle 2010, 9, 4297–4306. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Hu, C.A.; Wu, G.; Zhaorigetu, S.; Chand, H.; Sun, K.; Ji, Y.; Wang, B.; Dai, Z.; Walton, B.; et al. Intimacy and a deadly feud: The interplay of autophagy and apoptosis mediated by amino acids. Amino Acids 2015, 47, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Overbye, A.; Brech, A.; Torgersen, M.L.; Jakobsen, I.S.; Sandvig, K.; Llorente, A. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell. Mol. Life Sci. 2016, 73, 4717–4737. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Gupta, D.; Shankar, S.; Srivastava, R.K. Biomolecular characterization of exosomes released from cancer stem cells: Possible implications for biomarker and treatment of cancer. Oncotarget 2015, 6, 3280–3291. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Warshall, C.; Bandyopadhyay, C.; Dutta, D.; Chandran, B. Interactions between exosomes from breast cancer cells and primary mammary epithelial cells leads to generation of reactive oxygen species which induce DNA damage response, stabilization of p53 and autophagy in epithelial cells. PLoS ONE 2014, 9, e97580. [Google Scholar] [CrossRef]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef]
- Wu, W.K.; Coffelt, S.B.; Cho, C.H.; Wang, X.J.; Lee, C.W.; Chan, F.K.; Yu, J.; Sung, J.J. The autophagic paradox in cancer therapy. Oncogene 2012, 31, 939–953. [Google Scholar] [CrossRef]
- Kim, S.; Jung, W.H.; Koo, J.S. Differences in autophagy-related activity by molecular subtype in triple-negative breast cancer. Tumour Biol. 2012, 33, 1681–1694. [Google Scholar] [CrossRef]
- He, Y.; Zhao, X.; Subahan, N.R.; Fan, L.; Gao, J.; Chen, H. The prognostic value of autophagy-related markers beclin-1 and microtubule-associated protein light chain 3B in cancers: A systematic review and meta-analysis. Tumour Biol. 2014, 35, 7317–7326. [Google Scholar] [CrossRef] [PubMed]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Wicinski, J.; Cervera, N.; Finetti, P.; Hur, M.H.; Diebel, M.E.; Monville, F.; Dutcher, J.; et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009, 69, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Iovino, F.; Tarpin, C.; Diebel, M.; Esterni, B.; Houvenaeghel, G.; Extra, J.M.; Bertucci, F.; Jacquemier, J.; et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin. Cancer Res. 2010, 16, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Charafe-Jauffret, E.; Ginestier, C.; Birnbaum, D. Breast cancer stem cells: Tools and models to rely on. BMC Cancer 2009, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.L.; Lin, Y.; Jiang, J.; Tang, Z.; Yang, S.; Lu, L.; Liang, Y.; Liu, X.; Tan, J.; Hu, X.G.; et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J. Pathol. 2017, 243, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Blanco, E.; Kim, Y.S.; Rodriguez, A.A.; Zhao, H.; Huang, T.H.; Chen, C.L.; Jin, G.; Landis, M.D.; Burey, L.A.; et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014, 32, 2309–2323. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Zhu, J.; Mi, M. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS ONE 2014, 9, e102535. [Google Scholar]
- Connell, T.D. Cholera toxin, LT-I, LT-IIa and LT-IIb: The critical role of ganglioside binding in immunomodulation by type I and type II heat-labile enterotoxins. Expert Rev. Vaccines 2007, 6, 821–834. [Google Scholar] [CrossRef]
- Nohara, K.; Wang, F.; Spiegel, S. Glycosphingolipid composition of MDA-MB-231 and MCF-7 human breast cancer cell lines. Breast Cancer Res. Treat. 1998, 48, 149–157. [Google Scholar] [CrossRef]
- Marquina, G.; Waki, H.; Fernandez, L.E.; Kon, K.; Carr, A.; Valiente, O.; Perez, R.; Ando, S. Gangliosides expressed in human breast cancer. Cancer Res. 1996, 56, 5165–5171. [Google Scholar] [PubMed]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masso-Welch, P.; Girald Berlingeri, S.; King-Lyons, N.D.; Mandell, L.; Hu, J.; Greene, C.J.; Federowicz, M.; Cao, P.; Connell, T.D.; Heakal, Y. LT-IIc, A Bacterial Type II Heat-Labile Enterotoxin, Induces Specific Lethality in Triple Negative Breast Cancer Cells by Modulation of Autophagy and Induction of Apoptosis and Necroptosis. Int. J. Mol. Sci. 2019, 20, 85. https://doi.org/10.3390/ijms20010085
Masso-Welch P, Girald Berlingeri S, King-Lyons ND, Mandell L, Hu J, Greene CJ, Federowicz M, Cao P, Connell TD, Heakal Y. LT-IIc, A Bacterial Type II Heat-Labile Enterotoxin, Induces Specific Lethality in Triple Negative Breast Cancer Cells by Modulation of Autophagy and Induction of Apoptosis and Necroptosis. International Journal of Molecular Sciences. 2019; 20(1):85. https://doi.org/10.3390/ijms20010085
Chicago/Turabian StyleMasso-Welch, Patricia, Sofia Girald Berlingeri, Natalie D. King-Lyons, Lorrie Mandell, John Hu, Christopher J. Greene, Matthew Federowicz, Peter Cao, Terry D. Connell, and Yasser Heakal. 2019. "LT-IIc, A Bacterial Type II Heat-Labile Enterotoxin, Induces Specific Lethality in Triple Negative Breast Cancer Cells by Modulation of Autophagy and Induction of Apoptosis and Necroptosis" International Journal of Molecular Sciences 20, no. 1: 85. https://doi.org/10.3390/ijms20010085
APA StyleMasso-Welch, P., Girald Berlingeri, S., King-Lyons, N. D., Mandell, L., Hu, J., Greene, C. J., Federowicz, M., Cao, P., Connell, T. D., & Heakal, Y. (2019). LT-IIc, A Bacterial Type II Heat-Labile Enterotoxin, Induces Specific Lethality in Triple Negative Breast Cancer Cells by Modulation of Autophagy and Induction of Apoptosis and Necroptosis. International Journal of Molecular Sciences, 20(1), 85. https://doi.org/10.3390/ijms20010085