5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
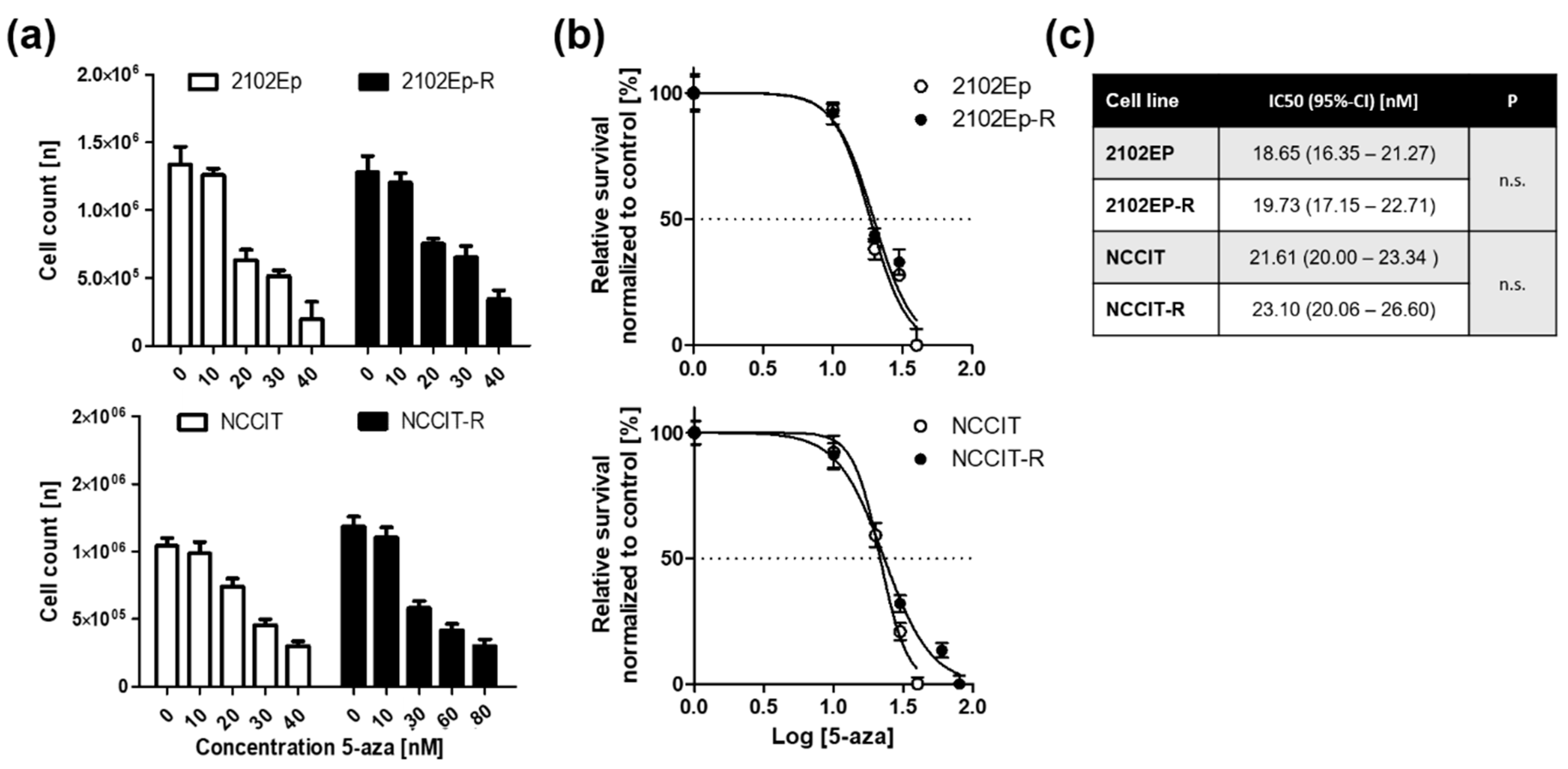
2.1. Embryonal Carcinoma (EC) Cells are Highly Sensitive to 5-Aza at Nanomolar Doses Irrespective of Cisplatin-Sensitivity
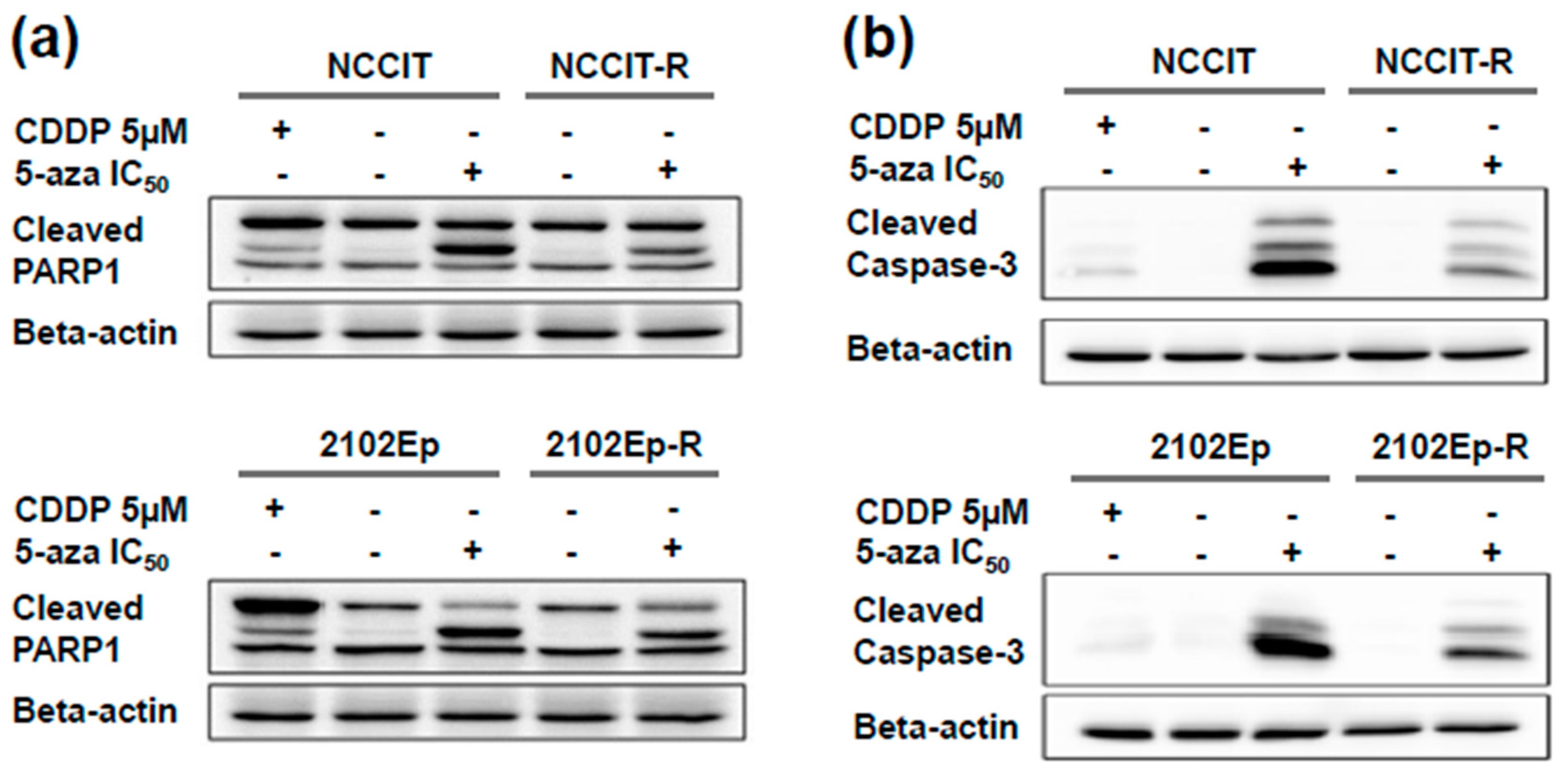
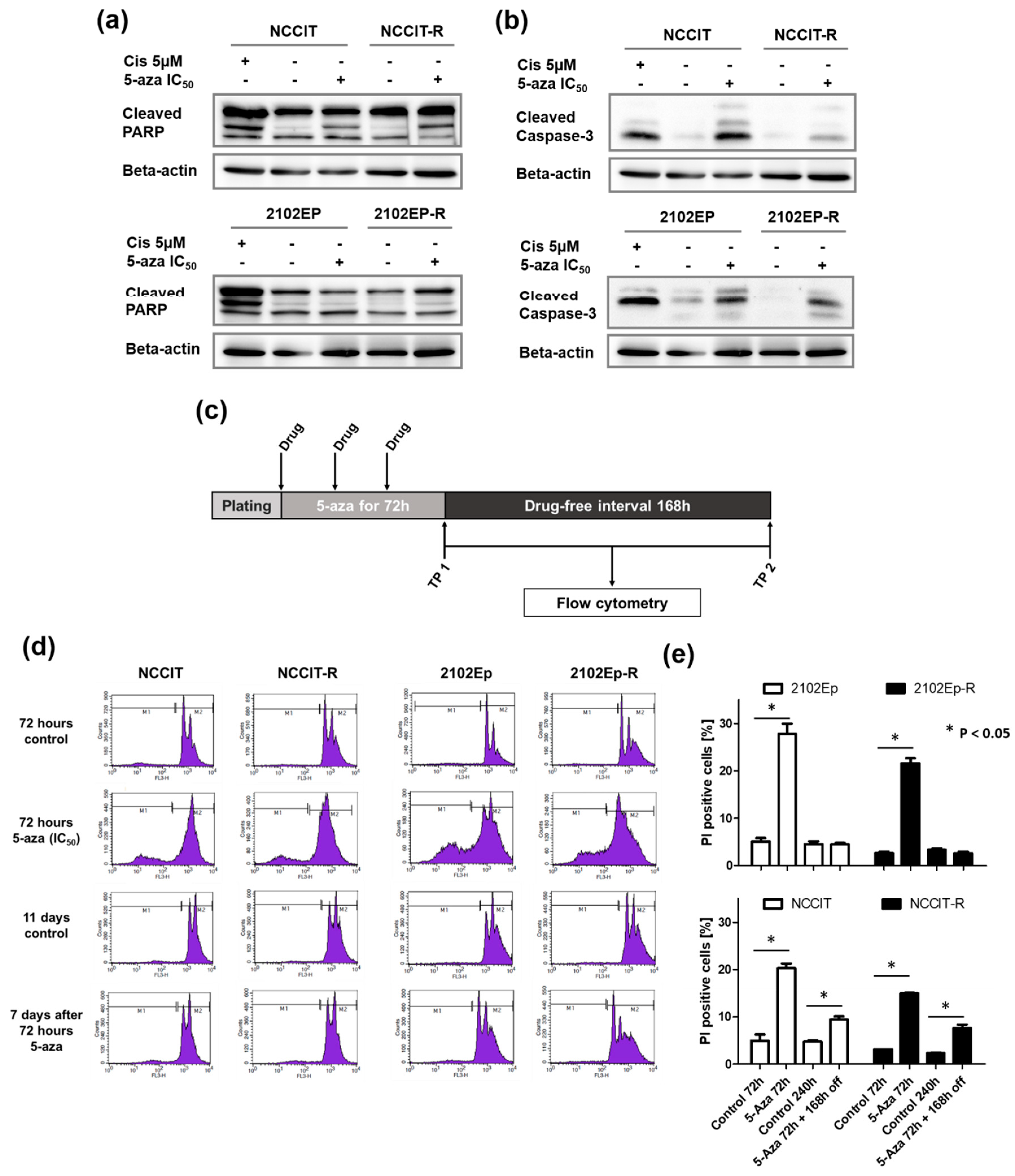
2.2. Exposure to Nanomolar Concentrations of 5-Aza Induces a Strong and Prolonged Apoptotic Response in EC Cells
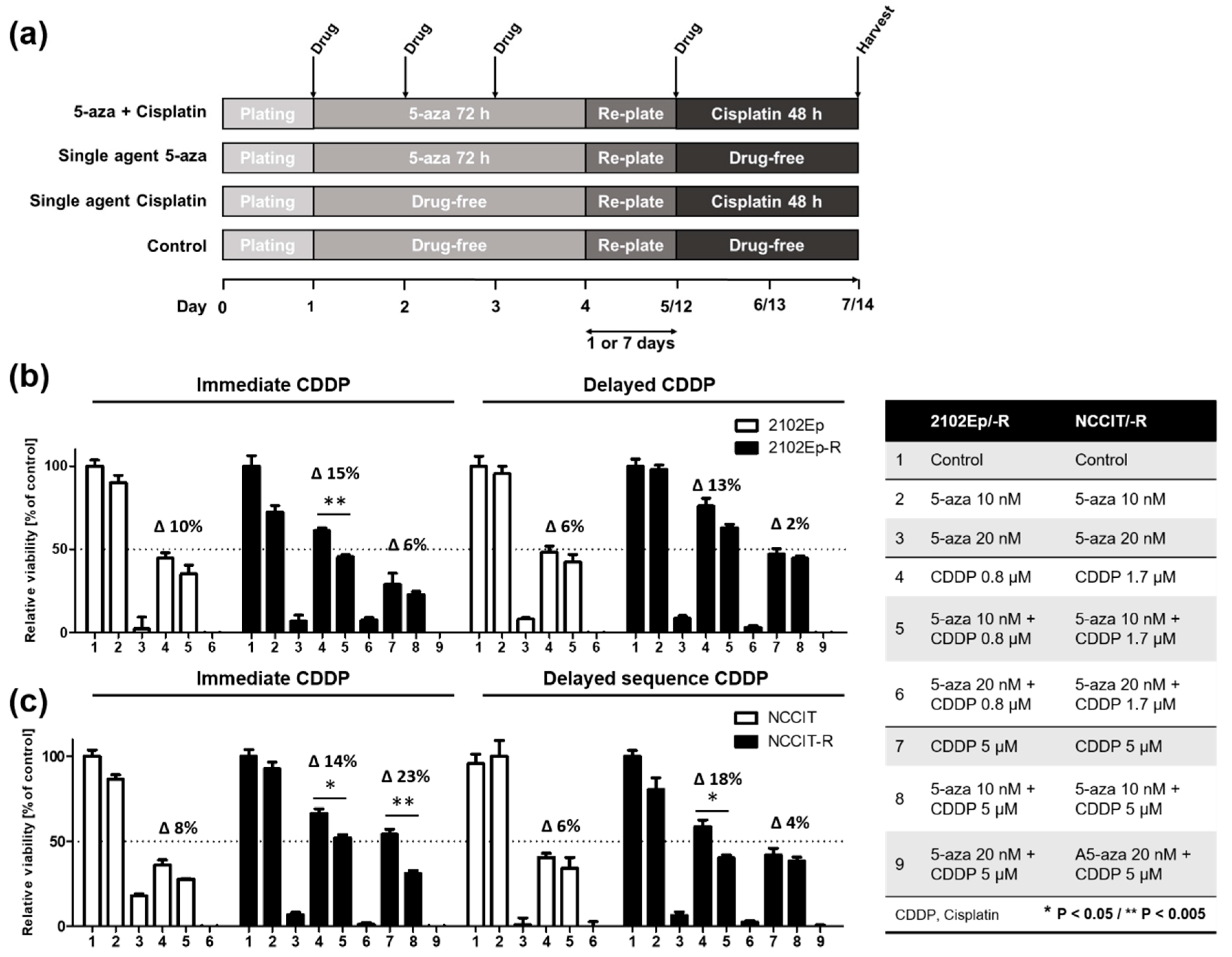
2.3. Nanomolar Concentration of 5-Aza Overcomes Cisplatin-Resistance in EC cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culturing Conditions
4.2. Cytotoxic Treatment
4.3. Cell Viability Assay
4.4. Flow Cytometry
4.5. Western Blotting
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-aza | 5-azacitidine |
| 5-mC | 5’-methyl cytosine |
| CDDP | Cisplatin |
| DAC | 5-aza-2’-deoxycitidine, decitabine |
| DNMT | DNA methyl transferase |
| DNMTI | DNA methyl transferase inhibitor |
| EC | Embryonal carcinoma |
| FDA | Food and Drug Administration |
| GCNIS | Germ cell neoplasia in situ |
| GCT | Germ cell tumor |
| PARP1 | Poly-(ADP-ribose) polymerase 1 |
References
- Einhorn, L.H. Treatment of testicular cancer: A new and improved model. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1990, 8, 1777–1781. [Google Scholar] [CrossRef] [PubMed]
- International Prognostic Factors Study, Group; Lorch, A.; Beyer, J.; Bascoul-Mollevi, C.; Kramar, A.; Einhorn, L.H.; Necchi, A.; Massard, C.; De Giorgi, U.; Flechon, A.; et al. Prognostic factors in patients with metastatic germ cell tumors who experienced treatment failure with cisplatin-based first-line chemotherapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 4906–4911. [Google Scholar]
- Oing, C.; Alsdorf, W.H.; von Amsberg, G.; Oechsle, K.; Bokemeyer, C. Platinum-refractory germ cell tumors: An update on current treatment options and developments. World J. Urol. 2017, 35, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, C.; Honecker, F. Cisplatin resistance in germ cell tumours: Models and mechanisms. Andrology 2015, 3, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; et al. Role of promoter hypermethylation in cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seidel, C.; Florean, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Chromatin-modifying agents in anti-cancer therapy. Biochimie 2012, 94, 2264–2279. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [CrossRef]
- van der Zwan, Y.G.; Rijlaarsdam, M.A.; Rossello, F.J.; Notini, A.J.; de Boer, S.; Watkins, D.N.; Gillis, A.J.; Dorssers, L.C.; White, S.J.; Looijenga, L.H. Seminoma and embryonal carcinoma footprints identified by analysis of integrated genome-wide epigenetic and expression profiles of germ cell cancer cell lines. PLoS ONE 2014, 9, e98330. [Google Scholar] [CrossRef]
- Wermann, H.; Stoop, H.; Gillis, A.J.; Honecker, F.; van Gurp, R.J.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Global DNA methylation in fetal human germ cells and germ cell tumours: Association with differentiation and cisplatin resistance. J. Pathol. 2010, 221, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Beyrouthy, M.J.; Garner, K.M.; Hever, M.P.; Freemantle, S.J.; Eastman, A.; Dmitrovsky, E.; Spinella, M.J. High DNA methyltransferase 3b expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 2009, 69, 9360–9366. [Google Scholar] [CrossRef] [PubMed]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, P.W.; Nguyen, A.N.; Brady, H.; Williams, M.; Ning, Y.; Richard, N.; Krushel, L.; Aukerman, S.L.; Heise, C.; MacBeth, K.J. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE 2010, 5, e9001. [Google Scholar] [CrossRef] [PubMed]
- Oechsle, K.; Honecker, F.; Cheng, T.; Mayer, F.; Czaykowski, P.; Winquist, E.; Wood, L.; Fenner, M.; Glaesener, S.; Hartmann, J.T.; et al. Preclinical and clinical activity of sunitinib in patients with cisplatin-refractory or multiply relapsed germ cell tumors: A canadian urologic oncology group/german testicular cancer study group cooperative study. Ann. Oncol. Off. J. Eur. Soc. Med Oncol./ESMO 2011, 22, 2654–2660. [Google Scholar] [CrossRef] [PubMed]
- Urien, S.; Lokiec, F. Population pharmacokinetics of total and unbound plasma cisplatin in adult patients. Br. J. Clin. Pharmacol. 2004, 57, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Oing, C.; Bokemeyer, C.; Russell, K.; Millis, S.Z.; Bender, R.; Gatalica, Z.; Voss, A. Molecular profiling of cisplatin-resistant testicular germ cell tumors. Oncol. Res. Treat. 2015, 38, 168. [Google Scholar]
- Bagrodia, A.; Lee, B.H.; Lee, W.; Cha, E.K.; Sfakianos, J.P.; Iyer, G.; Pietzak, E.J.; Gao, S.P.; Zabor, E.C.; Ostrovnaya, I.; et al. Genetic determinants of cisplatin resistance in patients with advanced germ cell tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4000–4007. [Google Scholar] [CrossRef]
- Brait, M.; Maldonado, L.; Begum, S.; Loyo, M.; Wehle, D.; Tavora, F.F.; Looijenga, L.H.; Kowalski, J.; Zhang, Z.; Rosenbaum, E.; et al. DNA methylation profiles delineate epigenetic heterogeneity in seminoma and non-seminoma. Br. J. Cancer 2012, 106, 414–423. [Google Scholar] [CrossRef]
- Furukawa, S.; Haruta, M.; Arai, Y.; Honda, S.; Ohshima, J.; Sugawara, W.; Kageyama, Y.; Higashi, Y.; Nishida, K.; Tsunematsu, Y.; et al. Yolk sac tumor but not seminoma or teratoma is associated with abnormal epigenetic reprogramming pathway and shows frequent hypermethylation of various tumor suppressor genes. Cancer Sci. 2009, 100, 698–708. [Google Scholar] [CrossRef]
- Albany, C.; Hever-Jardine, M.P.; von Herrmann, K.M.; Yim, C.Y.; Tam, J.; Warzecha, J.M.; Shin, L.; Bock, S.E.; Curran, B.S.; Chaudhry, A.S.; et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget 2017, 8, 2949–2959. [Google Scholar] [CrossRef] [PubMed]
- Biswal, B.K.; Beyrouthy, M.J.; Hever-Jardine, M.P.; Armstrong, D.; Tomlinson, C.R.; Christensen, B.C.; Marsit, C.J.; Spinella, M.J. Acute hypersensitivity of pluripotent testicular cancer-derived embryonal carcinoma to low-dose 5-aza deoxycytidine is associated with global DNA damage-associated p53 activation, anti-pluripotency and DNA demethylation. PLoS ONE 2012, 7, e53003. [Google Scholar]
- Karpf, A.R.; Moore, B.C.; Ririe, T.O.; Jones, D.A. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2’-deoxycytidine. Mol. Pharmacol. 2001, 59, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase iii study. Lancet. Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lay, F.; Han, H.; Jones, P.A. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol. Sci. 2010, 31, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.J.; Elson, P.; Sledge, G.W., Jr.; Einhorn, L.H.; Trump, D.L. 5-azacytidine (nsc 102816) in refractory germ cell tumors. A phase ii trial of the eastern cooperative oncology group. Investig. New Drugs 1993, 11, 201–202. [Google Scholar] [CrossRef]
- Pleyer, L.; Greil, R. Digging deep into "dirty" drugs—Modulation of the methylation machinery. Drug Metab. Rev. 2015, 47, 252–279. [Google Scholar] [CrossRef]
- Gailhouste, L.; Liew, L.C.; Hatada, I.; Nakagama, H.; Ochiya, T. Epigenetic reprogramming using 5-azacytidine promotes an anti-cancer response in pancreatic adenocarcinoma cells. Cell Death Dis. 2018, 9, 468. [Google Scholar] [CrossRef]
- Gailhouste, L.; Liew, L.C.; Yasukawa, K.; Hatada, I.; Tanaka, Y.; Nakagama, H.; Ochiya, T. Differentiation therapy by epigenetic reconditioning exerts antitumor effects on liver cancer cells. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 1840–1854. [Google Scholar] [CrossRef]
- Khan, G.N.; Kim, E.J.; Shin, T.S.; Lee, S.H. Azacytidine-induced chemosensitivity to doxorubicin in human breast cancer mcf7 cells. Anticancer Res. 2017, 37, 2355–2364. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Fang, F.; Shen, C.; Schilder, J.; Arnold, A.; Zeng, Y.; Berry, W.A.; Huang, T.; Nephew, K.P. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012, 72, 2197–2205. [Google Scholar] [CrossRef] [PubMed]
- Matei, D.; Ghamande, S.; Roman, L.; Alvarez Secord, A.; Nemunaitis, J.; Markham, M.J.; Nephew, K.P.; Jueliger, S.; Oganesian, A.; Naim, S.; et al. A phase i clinical trial of guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer: Clinical, pharmacokinetic, and pharmacodynamic analyses. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Munck, J.; Tang, J.; Taverna, P.; Wang, Y.; Miller, D.F.; Pilrose, J.; Choy, G.; Azab, M.; Pawelczak, K.S.; et al. The novel, small-molecule DNA methylation inhibitor sgi-110 as an ovarian cancer chemosensitizer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6504–6516. [Google Scholar] [CrossRef] [PubMed]
- Fenske, A.E.; Glaesener, S.; Bokemeyer, C.; Thomale, J.; Dahm-Daphi, J.; Honecker, F.; Dartsch, D.C. Cisplatin resistance induced in germ cell tumour cells is due to reduced susceptibility towards cell death but not to altered DNA damage induction or repair. Cancer Lett. 2012, 324, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Pelageev, D.N.; Dyshlovoy, S.A.; Pokhilo, N.D.; Denisenko, V.A.; Borisova, K.L.; Keller-von Amsberg, G.; Bokemeyer, C.; Fedorov, S.N.; Honecker, F.; Anufriev, V.P. Quinone-carbohydrate nonglucoside conjugates as a new type of cytotoxic agents: Synthesis and determination of in vitro activity. Eur. J. Med. Chem. 2014, 77, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Dyshlovoy, S.A.; Naeth, I.; Venz, S.; Preukschas, M.; Sievert, H.; Jacobsen, C.; Shubina, L.K.; Gesell Salazar, M.; Scharf, C.; Walther, R.; et al. Proteomic profiling of germ cell cancer cells treated with aaptamine, a marine alkaloid with antiproliferative activity. J. Proteome Res. 2012, 11, 2316–2330. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oing, C.; Verem, I.; Mansour, W.Y.; Bokemeyer, C.; Dyshlovoy, S.; Honecker, F. 5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells. Int. J. Mol. Sci. 2019, 20, 21. https://doi.org/10.3390/ijms20010021
Oing C, Verem I, Mansour WY, Bokemeyer C, Dyshlovoy S, Honecker F. 5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells. International Journal of Molecular Sciences. 2019; 20(1):21. https://doi.org/10.3390/ijms20010021
Chicago/Turabian StyleOing, Christoph, Izudin Verem, Wael Y. Mansour, Carsten Bokemeyer, Sergey Dyshlovoy, and Friedemann Honecker. 2019. "5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells" International Journal of Molecular Sciences 20, no. 1: 21. https://doi.org/10.3390/ijms20010021
APA StyleOing, C., Verem, I., Mansour, W. Y., Bokemeyer, C., Dyshlovoy, S., & Honecker, F. (2019). 5-Azacitidine Exerts Prolonged Pro-Apoptotic Effects and Overcomes Cisplatin-Resistance in Non-Seminomatous Germ Cell Tumor Cells. International Journal of Molecular Sciences, 20(1), 21. https://doi.org/10.3390/ijms20010021