Detection of Low-Frequency Mutations and Identification of Heat-Induced Artifactual Mutations Using Duplex Sequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
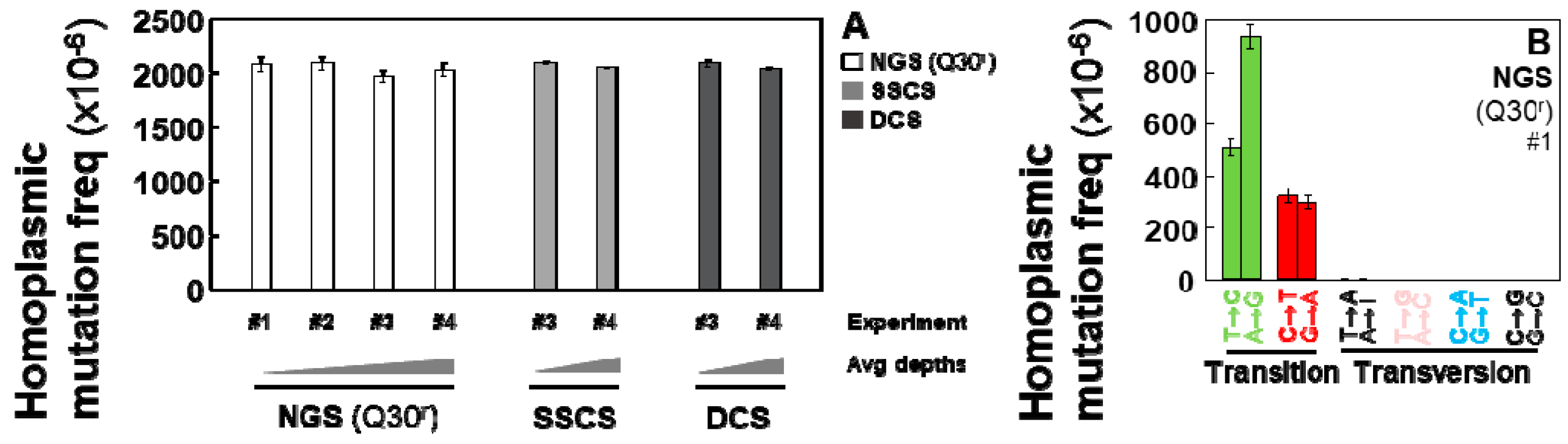
2.1. Homoplasmic Mutations are Detectable by All Three Methods (Conventional NGS, Tag-Based Single Strand Sequencing, and Duplex Sequencing) with the Similar Accuracy and Reproducibility
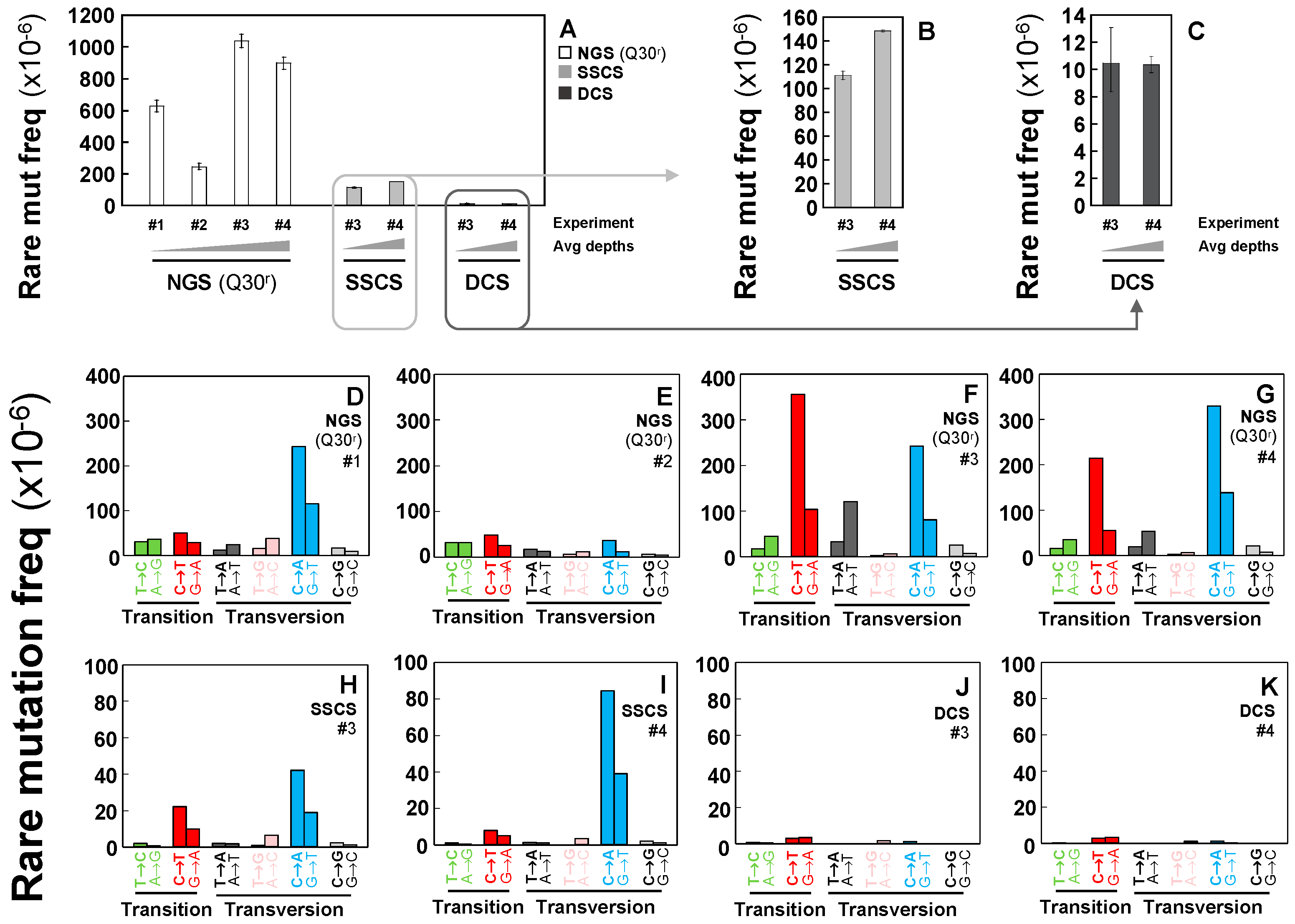
2.2. Rarely Occurring Mutations are Neither Accurately Detectable by Conventional NGS Methods nor Tag-Based Single Strand DNA Sequencing, but are Accurately Detectable by Duplex Sequencing
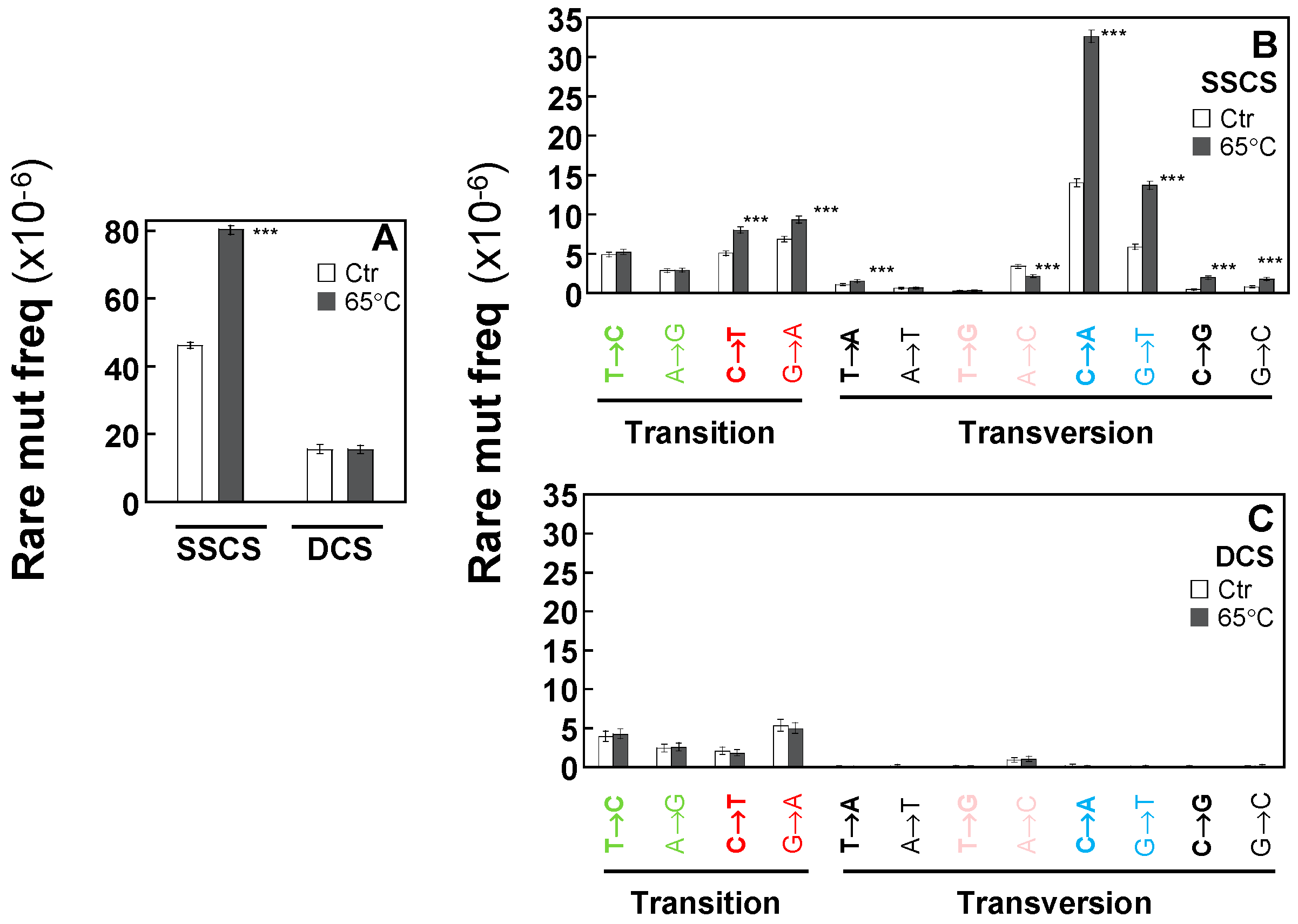
2.3. Duplex Sequencing Identifies and Corrects the Heat-Induced Artifactual Variants Introduced During DNA Sample Preparation
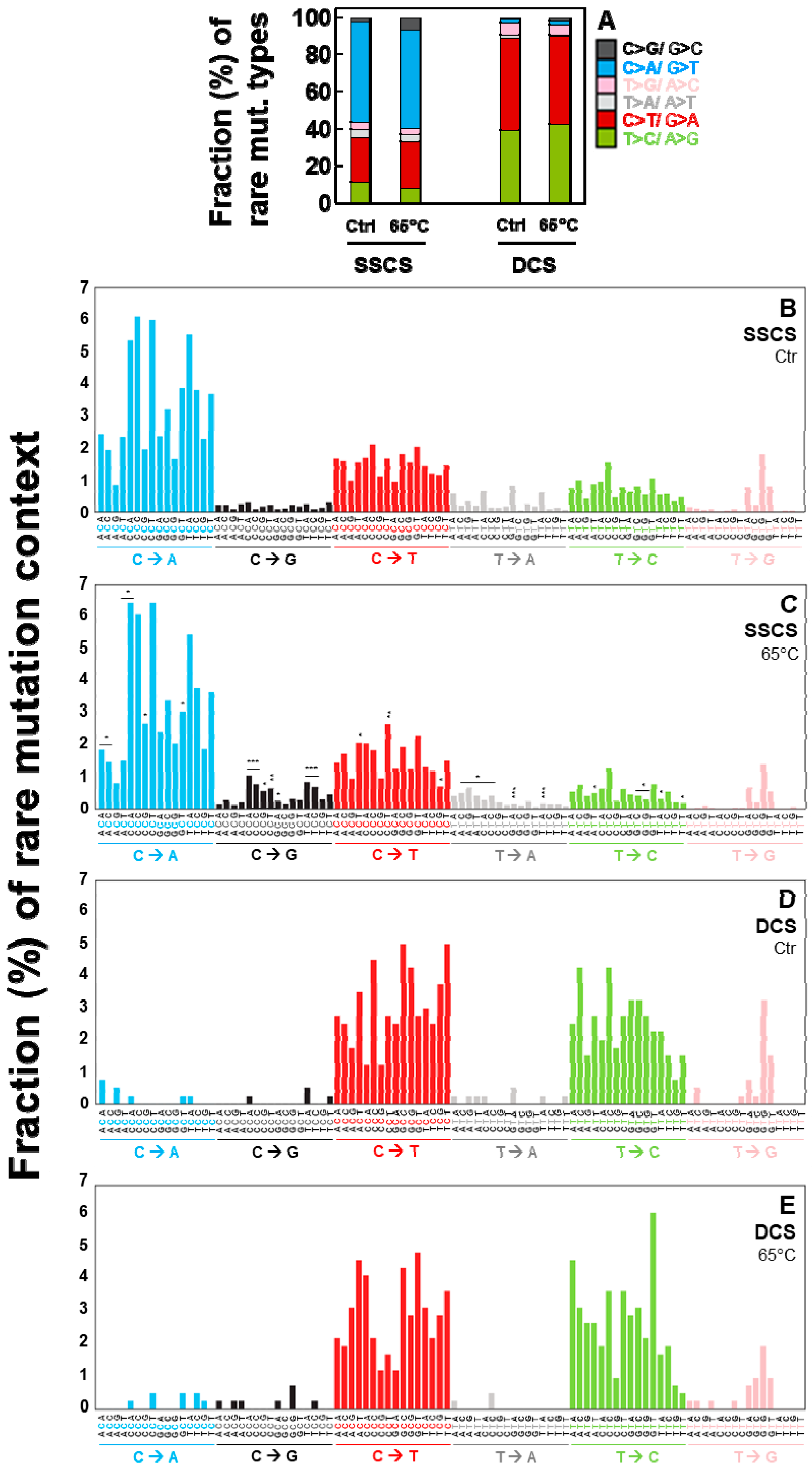
2.4. Duplex Sequencing Identifies the Specific Mutation Spectra of Heat-Induced Artifacts
2.5. Independent Experiments of Duplex Sequencing Reproducibly Identify the Heat-Induced Artifactual Mutaiton Profiles
2.6. All Identified Heat-Induced Artifactual Variants are Stochastically Occurring Variants Throughout the Whole Mitochondrial Genome
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. DNA Extraction, Adapter Synthesis, and Library Preparation
4.3. Heat-Induced DNA Damage
4.4. Data Processing
4.4.1. Conventional NGS Datasets and conventional NGS, SSCS and DCS Data Processing
4.4.2. Base Quality and PCR Duplicates
4.4.3. Comparison of Variant Positions
4.4.4. Counting Mutations
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DCS | Duplex Consensus Sequence |
| DS | Duplex Sequencing |
| Mt | Mitochondrial |
| NGS | Next-generation Sequencing |
| SSCS | Single Strand Consensus Sequence |
References
- Wetterstrand, K.A. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP); National Human Genome Research Institute: Bethesda, MD, USA, 2013. [Google Scholar]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Lou, D.I.; Hussmann, J.A.; McBee, R.M.; Acevedo, A.; Andino, R.; Press, W.H.; Sawyer, S.L. High-throughput DNA sequencing errors are reduced by orders of magnitude using circle sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 19872–19877. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.J.; Reid-Bayliss, K.S.; Emond, M.J.; Loeb, L.A. Accuracy of Next Generation Sequencing Platforms. Next Gener. Seq. Appl. 2014, 1. [Google Scholar] [CrossRef]
- Loeb, L.A. Human Cancers Express a Mutator Phenotype: Hypothesis, Origin, and Consequences. Cancer Res. 2016, 76, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.J.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.-C.; Risques, R.-A.; et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 2014, 9, 2586–2606. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Fox, E.J.; Prindle, M.J.; Reid-Bayliss, K.S.; True, L.D.; Radich, J.P.; Loeb, L.A. Sequencing small genomic targets with high efficiency and extreme accuracy. Nat. Methods 2015, 12, 423–425. [Google Scholar] [CrossRef]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Arbeithuber, B.; Makova, K.D.; Tiemann-Boege, I. Artifactual mutations resulting from DNA lesions limit detection levels in ultrasensitive sequencing applications. DNA Res. 2016, 23, 547–559. [Google Scholar] [CrossRef]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Shinde, J.; Alexandrov, L.B.; Assié, G.; André, T.; Hélias-Rodzewicz, Z.; Ducoudray, R.; Le Corre, D.; Zucman-Rossi, J.; Emile, J.-F.; et al. Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas: Mutational signature associated with MUTYH deficiency in cancers. J. Pathol. 2017, 242, 10–15. [Google Scholar] [CrossRef] [PubMed]
- MITOMAP A Human Mitochondrial Genome Database. Available online: http://www.mitomap.org/ (accessed on 3 May 2018).
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G-T and A-C substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [PubMed]
- Bruskov, V.I.; Malakhova, L.V.; Masalimov, Z.K.; Chernikov, A.V. Heat-induced formation of reactive oxygen species and 8-oxoguanine, a biomarker of damage to DNA. Nucl. Acids Res. 2002, 30, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Slimen, I.B.; Najar, T.; Ghram, A.; Dabbebi, H.; Ben Mrad, M.; Abdrabbah, M. Reactive oxygen species, heat stress and oxidative-induced mitochondrial damage. A review. Int. J. Hyperth. 2014, 30, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Parkin, B.; Giraldez, M.D.; Tewari, M. Mutant DNA quantification by digital PCR can be confounded by heating during DNA fragmentation. BioTechniques 2016, 60, 175–176. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef]
- Costello, M.; Pugh, T.J.; Fennell, T.J.; Stewart, C.; Lichtenstein, L.; Meldrim, J.C.; Fostel, J.L.; Friedrich, D.C.; Perrin, D.; Dionne, D.; et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucl. Acids Res. 2013, 41, e67. [Google Scholar] [CrossRef]
- Chen, L.; Liu, P.; Evans, T.C.; Ettwiller, L.M. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science 2017, 355, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Nomata, K.; Oakley, C.S.; Welsch, C.W.; Chang, C.C. Two types of normal human breast epithelial cells derived from reduction mammoplasty: Phenotypic characterization and response to SV40 transfection. Carcinogenesis 1995, 16, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.S.; Morita, I.; Cruz, A.; Jeon, Y.J.; Trosko, J.E.; Chang, C.C. Expression of estrogen receptors in a normal human breast epithelial cell type with luminal and stem cell characteristics and its neoplastically transformed cell lines. Carcinogenesis 1997, 18, 251–257. [Google Scholar] [CrossRef]
- Chang, C.C.; Sun, W.; Cruz, A.; Saitoh, M.; Tai, M.H.; Trosko, J.E. A human breast epithelial cell type with stem cell characteristics as target cells for carcinogenesis. Radiat. Res. 2001, 155, 201–207. [Google Scholar] [CrossRef]
- Park, J.-S.; Noh, D.-Y.; Kim, S.-H.; Kim, S.-H.; Kong, G.; Chang, C.-C.; Lee, Y.-S.; Trosko, J.E.; Kang, K.-S. Gene expression analysis in SV40-immortalized human breast luminal epithelial cells with stem cell characteristics using a cDNA microarray. Int. J. Oncol. 2004, 24, 1545–1558. [Google Scholar]
- Tai, M.-H.; Chang, C.-C.; Kiupel, M.; Webster, J.D.; Olson, L.K.; Trosko, J.E. Oct4 expression in adult human stem cells: Evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis 2005, 26, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Chang, C.-C.; Schroeder, J.J. Evaluation of sphinganine and sphingosine as human breast cancer chemotherapeutic and chemopreventive agents. Exp. Biol. Med. 2006, 231, 1664–1672. [Google Scholar] [CrossRef]
- Ahn, E.H.; Lee, S.H.; Kim, J.Y.; Chang, C.-C.; Loeb, L.A. Decreased Mitochondrial Mutagenesis during Transformation of Human Breast Stem Cells into Tumorigenic Cells. Cancer Res. 2016, 76, 4569–4578. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Hirohata, K.; Kohrn, B.F.; Fox, E.J.; Chang, C.-C.; Loeb, L.A. Detection of Ultra-Rare Mitochondrial Mutations in Breast Stem Cells by Duplex Sequencing. PLoS ONE 2015, 10, e0136216. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Shahin, H.; Elkan-Miller, T.; Lee, M.K.; Thornton, A.M.; Roeb, W.; Abu Rayyan, A.; Loulus, S.; Avraham, K.B.; King, M.-C.; et al. Whole Exome Sequencing and Homozygosity Mapping Identify Mutation in the Cell Polarity Protein GPSM2 as the Cause of Nonsyndromic Hearing Loss DFNB82. Am. J. Hum. Genet. 2010, 87, 90–94. [Google Scholar] [CrossRef]
- Gulsuner, S.; Walsh, T.; Watts, A.C.; Lee, M.K.; Thornton, A.M.; Casadei, S.; Rippey, C.; Shahin, H.; Consortium on the Genetics of Schizophrenia (COGS); PAARTNERS Study Group; et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 2013, 154, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Griffin, H.R.; Pyle, A.; Blakely, E.L.; Alston, C.L.; Duff, J.; Hudson, G.; Horvath, R.; Wilson, I.J.; Santibanez-Koref, M.; Taylor, R.W.; et al. Accurate mitochondrial DNA sequencing using off-target reads provides a single test to identify pathogenic point mutations. Genet. Med. 2014, 16, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Cliften, P. Chapter 7: Base Calling, Read Mapping, and Coverage Analysis. In Clinical Genomics, 1st ed.; Kulkarni, S., Pfeifer, J., Eds.; Elsevier Inc.: New York, NY, USA, 2015; Volume 1, pp. 91–107. ISBN 9780124041488. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, E.H.; Lee, S.H. Detection of Low-Frequency Mutations and Identification of Heat-Induced Artifactual Mutations Using Duplex Sequencing. Int. J. Mol. Sci. 2019, 20, 199. https://doi.org/10.3390/ijms20010199
Ahn EH, Lee SH. Detection of Low-Frequency Mutations and Identification of Heat-Induced Artifactual Mutations Using Duplex Sequencing. International Journal of Molecular Sciences. 2019; 20(1):199. https://doi.org/10.3390/ijms20010199
Chicago/Turabian StyleAhn, Eun Hyun, and Seung Hyuk Lee. 2019. "Detection of Low-Frequency Mutations and Identification of Heat-Induced Artifactual Mutations Using Duplex Sequencing" International Journal of Molecular Sciences 20, no. 1: 199. https://doi.org/10.3390/ijms20010199
APA StyleAhn, E. H., & Lee, S. H. (2019). Detection of Low-Frequency Mutations and Identification of Heat-Induced Artifactual Mutations Using Duplex Sequencing. International Journal of Molecular Sciences, 20(1), 199. https://doi.org/10.3390/ijms20010199