Structural Determinants of the Prion Protein N-Terminus and Its Adducts with Copper Ions

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
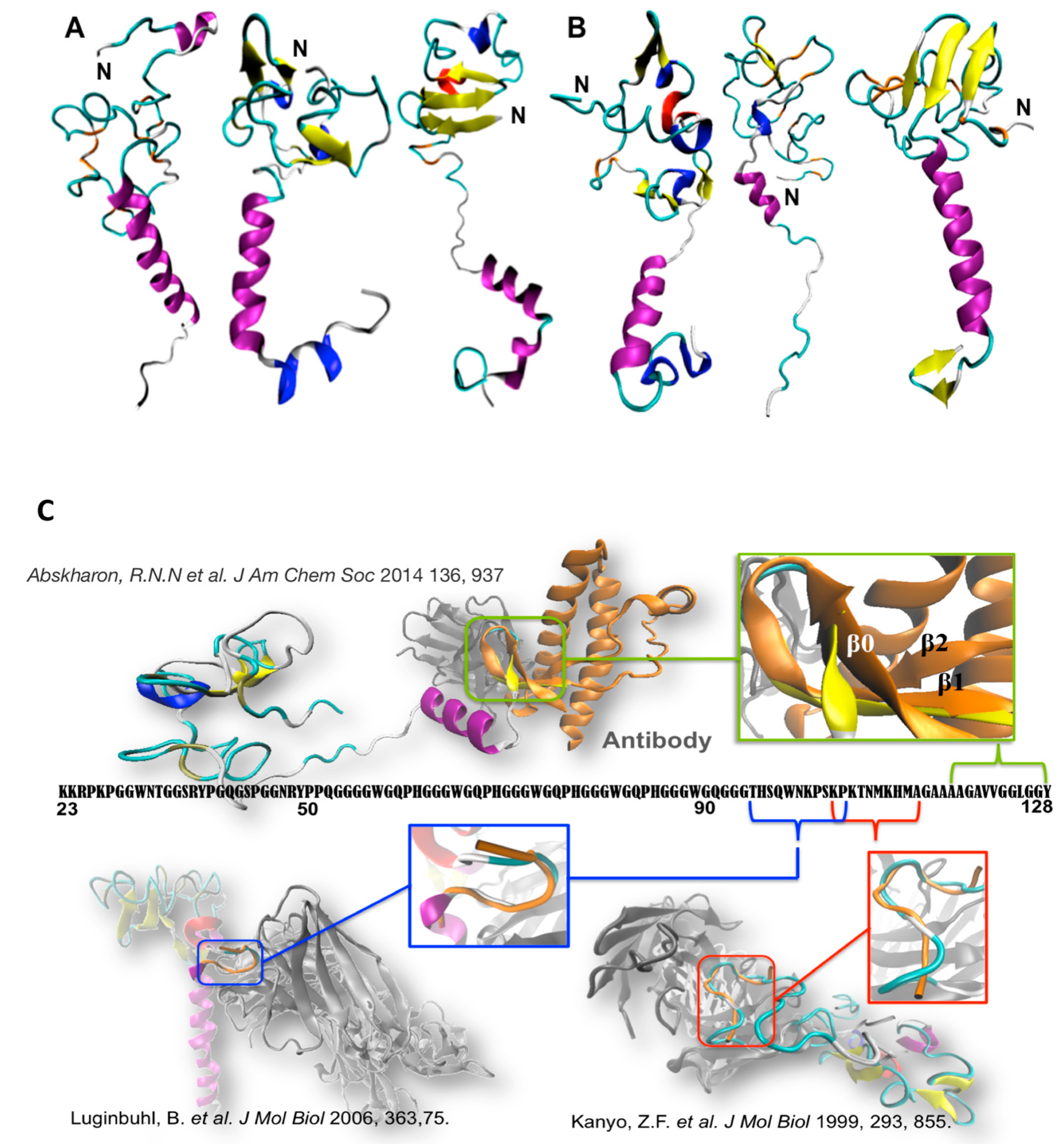
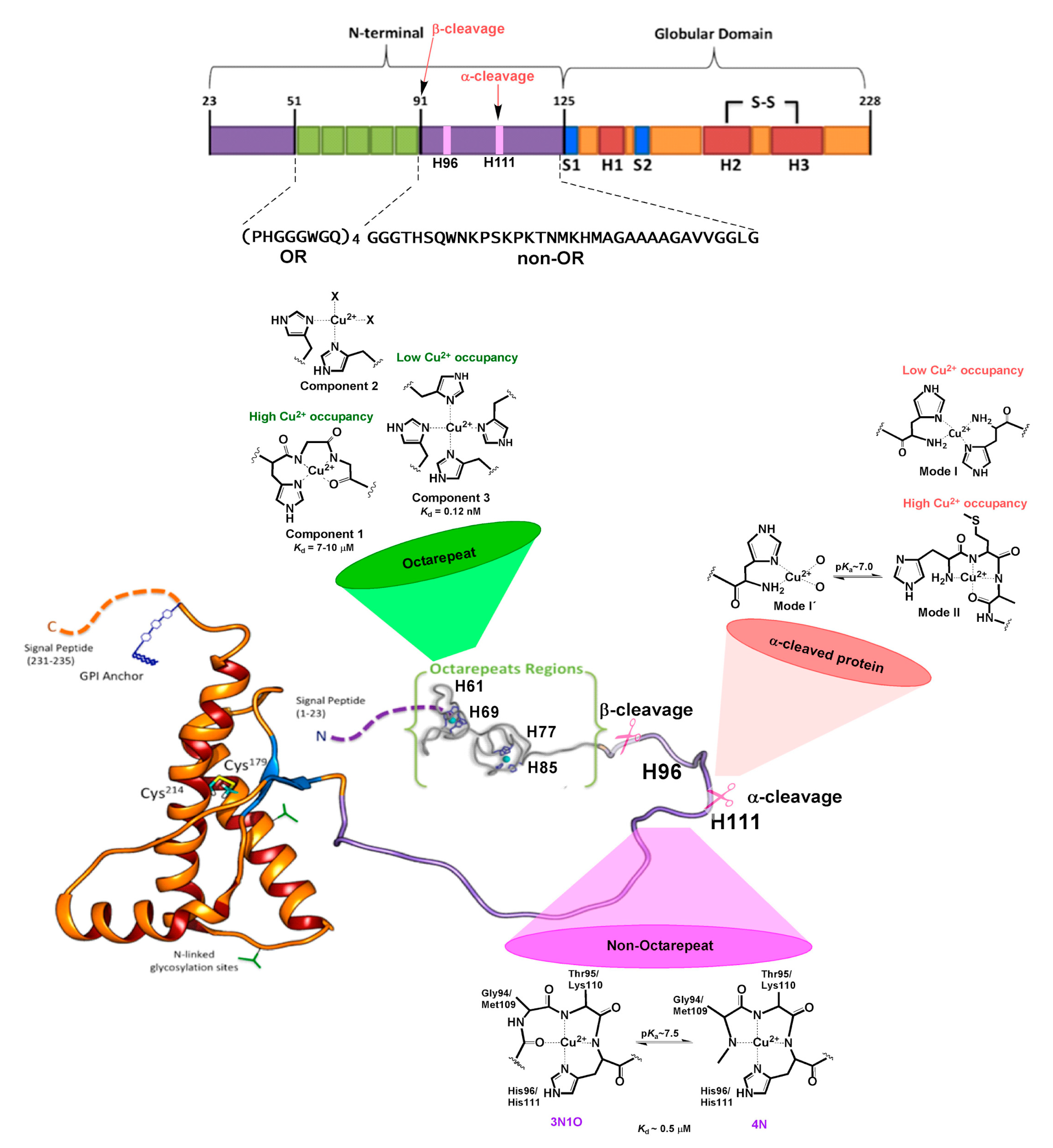
2. The N-Term: Function and Structural Determinants
3. Copper Binding
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Molecular biology and pathogenesis of prion diseases. Trends Biochem. Sci. 1996, 21, 482–487. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Gendoo, D.M.A.; Harrison, P.M. Discordant and chameleon sequences: Their distribution and implications for amyloidogenicity. Protein Sci. 2011, 20, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, I.B.; Rackovsky, S. Comparative computational analysis of prion proteins reveals two fragments with unusual structural properties and a pattern of increase in hydrophobicity associated with disease-promoting mutations. Protein Sci. 2004, 13, 3230–3244. [Google Scholar] [CrossRef] [PubMed]
- Dima, R.; Thirumalai, D. Exploring the propensities of helices in PrPc to form beta sheet using NMR structures and sequence alignments. Biophys. J. 2002, 83, 1268–1280. [Google Scholar] [CrossRef]
- Dima, R.I.; Thirumalai, D. Probing the instabilities in the dynamics of helical fragments from mouse PrPC. Proc. Natl. Acad. Sci. USA 2004, 101, 15335–15340. [Google Scholar] [CrossRef] [PubMed]
- Adrover, M.; Pauwels, K.; Prigent, S.; de Chiara, C.; Xu, Z.; Chapuis, C.; Pastore, A.; Rezaei, H. Prion Fibrillization Is Mediated by a Native Structural Element That Comprises Helices H2 and H3. J. Biol. Chem. 2010, 285, 21004–21012. [Google Scholar] [CrossRef]
- Priola, S.A.; Meade-White, K.; Lawson, V.A.; Chesebro, B. Flexible N-terminal region of prion protein influences conformation of protease-resistant prion protein isoforms associated with cross-species scrapie infection in vivo and in vitro. J. Biol. Chem. 2004, 279, 13689–13695. [Google Scholar] [CrossRef]
- Rudd, P.M.; Merry, A.H.; Wormald, M.R.; Dwek, R.A. Glycosylation and prion protein. Curr. Opin. Struct. Biol. 2002, 12, 578–586. [Google Scholar] [CrossRef]
- Abid, K.; Soto, C. The intriguing prion disorders. Cell. Mol. Life Sci. 2006, 63, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Swietnicki, W.; Petersen, R.; Gambetti, P.; Surewicz, W.K. pH-dependent stability and conformation of the recombinant human prion protein PrP(90–231). J. Biol. Chem. 1997, 272, 27517–27520. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.S.; Hosszu, L.L.; Power, A.; Hill, A.F.; Kenney, J.; Saibil, H.; Craven, C.J.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science 1999, 283, 1935–1937. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.J.; Kanthasamy, A.; Anantharam, V.; Kanthasamy, A.G. Interaction of metals with prion protein: Possible role of divalent cations in the pathogenesis of prion diseases. Neurotoxicology 2006, 27, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S. Metal ions and prion diseases. Curr. Opin. Chem. Biol. 2002, 6, 187–192. [Google Scholar] [CrossRef]
- Jin, T.; Gu, Y.; Zanusso, G.; Sy, M.; Kumar, A.; Cohen, M.; Gambetti, P.; Singh, N. The chaperone protein BiP binds to a mutant prion protein and mediates its degradation by the proteasome. J. Biol. Chem. 2000, 275, 38699–38704. [Google Scholar] [CrossRef]
- Hachiya, N.S.; Imagawa, M.; Kaneko, K. The possible role of protein X, a putative auxiliary factor in pathological prion replication, in regulating a physiological endoproteolytic cleavage of cellular prion protein. Med. Hypotheses 2007, 68, 670–673. [Google Scholar] [CrossRef]
- Dossena, S.; Imeri, L.; Mangieri, M.; Garofoli, A.; Ferrari, L.; Senatore, A.; Restelli, E.; Baiducci, C.; Fiordaliso, F.; Salio, M.; et al. Mutant Prion Protein Expression Causes Motor and Memory Deficits and Abnormal Sleep Patterns in a Transgenic Mouse Model. Neuron 2008, 60, 598–609. [Google Scholar] [CrossRef]
- Antonyuk, S.V.; Trevitt, C.R.; Strange, R.W.; Jackson, G.S.; Sangar, D.; Batchelor, M.; Cooper, S.; Fraser, C.; Jones, S.; Georgiou, T.; et al. Crystal structure of human prion protein bound to a therapeutic antibody. Proc. Natl. Acad. Sci. USA 2009, 106, 2554–2558. [Google Scholar] [CrossRef]
- Friedman-Levi, Y.; Meiner, Z.; Canello, T.; Frid, K.; Kovacs, G.G.; Budka, H.; Avrahami, D.; Gabizon, R. Fatal Prion Disease in a Mouse Model of Genetic E200K Creutzfeldt-Jakob Disease. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Trabattoni, G.; Hainfellner, J.A.; Ironside, J.W.; Knight, R.S.G.; Budka, H. Mutations of the prion protein gene phenotypic spectrum. J. Neurol. 2002, 249, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Campana, V.; Sarnataro, D.; Zurzolo, C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Linden, R.; Martins, V.R.; Prado, M.A.M.; Cammarota, M.N.; Izquierdo, I.N.; Brentani, R.R. Physiology of the Prion Protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.C.; Strome, R.; Fraser, P.E.; Kruck, T.; Von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Zahn, R.; Liu, A.Z.; Luhrs, T.; Riek, R.; von Schroetter, C.; Garcia, F.L.; Billeter, M.; Calzolai, L.; Wider, G.; Wuthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V.; Legname, G.; Prusiner, S.B. Folding of prion protein to its native α-helical conformation is under kinetic control. J. Biol. Chem. 2001, 276, 19687–19690. [Google Scholar] [CrossRef] [PubMed]
- Surewicz, W.K.; Apostol, M.I. Prion Protein and Its Conformational Conversion: A Structural Perspective. In Prion Proteins; Springer: Berlin/Heidelberg, Germany, 2011; Volume 305, pp. 135–167. [Google Scholar]
- Govaerts, C.; Wille, H.; Prusiner, S.B.; Cohen, F.E. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc. Natl. Acad. Sci. USA 2004, 101, 8342–8347. [Google Scholar] [CrossRef] [PubMed]
- Cobb, N.J.; Sonnichsen, F.D.; McHaourab, H.; Surewicz, W.K. Molecular architecture of human prion protein amyloid: A parallel, in-register beta-structure. Proc. Natl. Acad. Sci. USA 2007, 104, 18946–18951. [Google Scholar] [CrossRef]
- DeMarco, M.L.; Silveira, J.; Caughey, B.; Daggett, V. Structural properties of prion protein protofibrils and fibrils: An experimental assessment of atomic models. Biochemistry 2006, 45, 15573–15582. [Google Scholar] [CrossRef]
- Rossetti, G.; Carloni, P. Structural Modeling of Human Prion Protein’s Point Mutations. Prog. Mol. Biol. Transl. Sci. 2017, 150, 105–122. [Google Scholar] [CrossRef]
- Rossetti, G.; Bongarzone, S.; Carloni, P. Computational studies on the prion protein. Curr. Top. Med. Chem. 2013, 13, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Espinoza, R.; Soto, C. High-resolution structure of infectious prion protein: The final frontier. Nat. Struct. Mol. Biol. 2012, 19, 370–377. [Google Scholar] [CrossRef]
- Aguzzi, A.; Calella, A.M. Prions: Protein aggregation and infectious diseases. Physiol. Rev. 2009, 89, 1105–1152. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Nazabal, A.; Hornemann, S.; Aguzzi, A.; Zenobi, R. Hydrogen/deuterium exchange mass spectrometry identifies two highly protected regions in recombinant full-length prion protein amyloid fibrils. J. Mass Spectrom. 2009, 44, 965–977. [Google Scholar] [CrossRef]
- Sim, V.; Caughey, B. Ultrastructures and strain comparison of under-glycosylated scrapie prion fibrils. Neurobiol. Aging 2009, 30, 2031–2042. [Google Scholar] [CrossRef] [PubMed]
- Stoehr, J.; Weinmann, N.; Wille, H.; Kaimann, T.; Nagel-Steger, L.; Birkmann, E.; Panza, G.; Prusiner, S.B.; Eigen, M.; Riesner, D. Mechanisms of prion protein assembly into amyloid. Proc. Natl. Acad. Sci. USA 2008, 105, 2409–2414. [Google Scholar] [CrossRef]
- Hegde, R.S.; Mastrianni, J.A.; Scott, M.R.; DeFea, K.A.; Tremblay, P.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998, 279, 827–834. [Google Scholar] [CrossRef]
- Li, A.M.; Christensen, H.M.; Stewart, L.R.; Roth, K.A.; Chiesa, R.; Harris, D.A. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007, 26, 548–558. [Google Scholar] [CrossRef]
- Ott, C.M.; Akhavan, A.; Lingappa, V.R. Specific features of the prion protein transmembrane domain regulate nascent chain orientation. J. Biol. Chem. 2007, 282, 11163–11171. [Google Scholar] [CrossRef]
- Chakrabarti, O.; Ashok, A.; Hegde, R.S. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem. Sci. 2009, 34, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Beland, M.; Roucou, X. The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J. Neurochem. 2012, 120, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Vieira, T.C.R.G.; Gomes, M.P.B.; Rangel, L.P.; Scapin, S.M.N.; Cordeiro, Y. Experimental approaches to the interaction of the prion protein with nucleic acids and glycosaminoglycans: Modulators of the pathogenic conversion. Methods 2011, 53, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Hajj, G.N.M.; Lopes, M.H.; Mercadante, A.F.; Veiga, S.S.; da Silveira, R.B.; Santos, T.G.; Ribeiro, K.C.B.; Juliano, M.A.; Jacchieri, S.G.; Zanata, S.M.; et al. Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J. Cell Sci. 2007, 120, 1915–1926. [Google Scholar] [CrossRef]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.M.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.O.; Cabral, A.L.B.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1184. [Google Scholar] [CrossRef]
- Nicoll, A.J.; Panico, S.; Freir, D.B.; Wright, D.; Terry, C.; Risse, E.; Herron, C.E.; O’Malley, T.; Wadsworth, J.D.; Farrow, M.A.; et al. Amyloid-beta nanotubes are associated with prion protein-dependent synaptotoxicity. Nat. Commun. 2013, 4, 2416. [Google Scholar] [CrossRef]
- Chen, S.G.; Yadav, S.P.; Surewicz, W.K. Interaction between Human Prion Protein and Amyloid-beta (A beta) Oligomers Role of N-Terminal Residues. J. Biol. Chem. 2010, 285, 26377–26383. [Google Scholar] [CrossRef]
- Parkyn, C.J.; Vermeulen, E.G.; Mootoosamy, R.C.; Sunyach, C.; Jacobsen, C.; Oxvig, C.; Moestrup, S.; Liu, Q.; Bu, G.; Jen, A.; et al. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J. Cell Sci. 2008, 121, 773–783. [Google Scholar] [CrossRef]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef]
- Cong, X.; Casiraghi, N.; Rossetti, G.; Mohanty, S.; Giachin, G.; Legname, G.; Carloni, P. Role of Prion Disease-Linked Mutations in the Intrinsically Disordered N-Terminal Domain of the Prion Protein. J. Chem. Theory Comput. 2013, 9, 5158–5167. [Google Scholar] [CrossRef] [PubMed]
- Calzolai, L.; Zahn, R. Influence of pH on NMR structure and stability of the human prion protein globular domain. J. Boil. Chem. 2003, 278, 35592–35596. [Google Scholar] [CrossRef] [PubMed]
- Degioia, L.; Selvaggini, C.; Ghibaudi, E.; Diomede, L.; Bugiani, O.; Forloni, G.; Tagliavini, F.; Salmona, M. Conformational Polymorphism of the Amyloidogenic and Neurotoxic Peptide Homologous to Residues-106–126 of the Prion Protein. J. Biol. Chem. 1994, 269, 7859–7862. [Google Scholar]
- Miura, T.; Yoda, M.; Takaku, N.; Hirose, T.; Takeuchi, H. Clustered negative charges on the lipid membrane surface induce beta-sheet formation of prion protein fragment 106–126. Biochemistry 2007, 46, 11589–11597. [Google Scholar] [CrossRef] [PubMed]
- Satheeshkumar, K.S.; Jayakumar, R. Conformational polymorphism of the amyloidogenic peptide homologous to residues 113–127 of the prion protein. Biophys. J. 2003, 85, 473–483. [Google Scholar] [CrossRef]
- Zahn, R. The octapeptide repeats in mammalian prion protein constitute a pH-dependent folding and aggregation site. J. Mol. Biol. 2003, 334, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Van der Kamp, M.W.; Daggett, V. The consequences of pathogenic mutations to the human prion protein. Protein Eng. Des. Sel. 2009, 22, 461–468. [Google Scholar] [CrossRef]
- Rossetti, G.; Cong, X.J.; Caliandro, R.; Legname, G.; Carloni, P. Common Structural Traits across Pathogenic Mutants of the Human Prion Protein and Their Implications for Familial Prion Diseases. J. Mol. Biol. 2011, 411, 700–712. [Google Scholar] [CrossRef]
- Ilc, G.; Giachin, G.; Jaremko, M.; Jaremko, L.; Benetti, F.; Plavec, J.; Zhukov, I.; Legname, G. NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features. PLoS ONE 2010, 5, e11715. [Google Scholar] [CrossRef]
- Van der Kamp, M.W.; Daggett, V. Pathogenic Mutations in the Hydrophobic Core of the Human Prion Protein Can Promote Structural Instability and Misfolding. J. Mol. Biol. 2010, 404, 732–748. [Google Scholar] [CrossRef]
- Apetri, A.C.; Surewicz, K.; Surewicz, W.K. The effect of disease-associated mutations on the folding pathway of human prion protein. J. Biol. Chem. 2004, 279, 18008–18014. [Google Scholar] [CrossRef] [PubMed]
- Liemann, S.; Glockshuber, R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry 1999, 38, 3258–3267. [Google Scholar] [CrossRef] [PubMed]
- Swietnicki, W.; Petersen, R.B.; Gambetti, P.; Surewicz, W.K. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J. Biol. Chem. 1998, 273, 31048–31052. [Google Scholar] [CrossRef] [PubMed]
- Evans, E.G.B.; Millhauser, G.L. Copper- and Zinc-Promoted Interdomain Structure in the Prion Protein: A Mechanism for Autoinhibition of the Neurotoxic N-Terminus. In Prion Protein; Elsevier: Amsterdam, The Netherlands, 2017; Volume 150, pp. 35–56. [Google Scholar]
- Hegde, R.S.; Kang, S.W. The concept of translocational regulation. J. Cell Biol. 2008, 182, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Hornemann, S.; von Schroetter, C.; Damberger, F.F.; Wuthrich, K. Prion Protein-Detergent Micelle Interactions Studied by NMR in Solution. J. Biol. Chem. 2009, 284, 22713–22721. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Kovacs, I.; Hajos, F.; Kalman, M.; Simonyi, M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar] [CrossRef]
- Walter, E.D.; Chattopadhyay, M.; Millhauser, G.L. The affinity of copper binding to the prion protein octarepeat domain: Evidence for negative cooperativity. Biochemistry 2006, 45, 13083–13092. [Google Scholar] [CrossRef]
- Lee, K.S.; Magalhaes, A.C.; Zanata, S.M.; Brentani, R.R.; Martins, V.R.; Prado, M.A.M. Internalization of mammalian fluorescent cellular prion protein and N-terminal deletion mutants in living cells. J. Neurochem. 2001, 79, 79–87. [Google Scholar] [CrossRef]
- Pauly, P.C.; Harris, D.A. Copper Stimulates Endocytosis of the Prion Protein. J. Biol. Chem. 1998, 273, 33107–33110. [Google Scholar] [CrossRef]
- Ren, K.; Gao, C.; Zhang, J.; Wang, K.; Xu, Y.; Wang, S.-B.; Wang, H.; Tian, C.; Shi, Q.; Dong, X.-P. Flotillin-1 Mediates PrPC Endocytosis in the Cultured Cells During Cu2+ Stimulation Through Molecular Interaction. Mol. Neurobiol. 2013, 48, 631–646. [Google Scholar] [CrossRef]
- Sumudhu, W.; Perera, S.; Hooper, N.M. Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr. Biol. 2001, 11, 519–523. [Google Scholar] [CrossRef]
- Huang, S.; Chen, L.; Bladen, C.; Stys, P.K.; Zamponi, G.W. Differential modulation of NMDA and AMPA receptors by cellular prion protein and copper ions. Mol. Brain 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Tsutsui, S.; Hameed, S.; Kannanayakal, T.J.; Chen, L.; Xia, P.; Engbers, J.D.T.; Lipton, S.A.; Stys, P.K.; Zamponi, G.W. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-d-aspartate receptors. Proc. Natl. Acad. Sci. USA 2012, 109, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Stys, P.K.; You, H.; Zamponi, G.W. Copper-dependent regulation of NMDA receptors by cellular prion protein: Implications for neurodegenerative disorders. J. Physiol. 2012, 590, 1357–1368. [Google Scholar] [CrossRef]
- Gasperini, L.; Meneghetti, E.; Pastore, B.; Benetti, F.; Legname, G. Prion Protein and Copper Cooperatively Protect Neurons by Modulating NMDA Receptor Through S-nitrosylation. Antioxid. Redox Signal. 2015, 22, 772–784. [Google Scholar] [CrossRef]
- Aronoff-Spencer, E.; Burns, C.S.; Avdievich, N.I.; Gerfen, G.J.; Peisach, J.; Antholine, W.E.; Ball, H.L.; Cohen, F.E.; Prusiner, S.B.; Millhauser, G.L. Identification of the Cu2+ binding sites in the N-terminal domain of the prion protein by EPR and CD spectroscopy. Biochemistry 2000, 39, 13760–13771. [Google Scholar] [CrossRef]
- Millhauser, G.L. Copper Binding in the Prion Protein. Acc. Chem. Res. 2004, 37, 79–85. [Google Scholar] [CrossRef]
- Millhauser, G.L. Copper and the Prion Protein: Methods, Structures, Function, and Disease. Annu. Rev. Phys. Chem. 2007, 58, 299–320. [Google Scholar] [CrossRef]
- Quintanar, L.; Rivillas-Acevedo, L.; Grande-Aztatzi, R.; Gómez-Castro, C.Z.; Arcos-López, T.; Vela, A. Copper coordination to the prion protein: Insights from theoretical studies. Coord. Chem. Rev. 2013, 257, 429–444. [Google Scholar] [CrossRef]
- Wells, M.A.; Jelinska, C.; Hosszu, L.L.; Craven, C.J.; Clarke, A.R.; Collinge, J.; Waltho, J.P.; Jackson, G.S. Multiple forms of copper(II) co-ordination occur throughout the disordered N-terminal region of the prion protein at pH 7.4. Biochem. J. 2006, 400, 501–510. [Google Scholar] [CrossRef]
- Wopfner, F.; Weidenhöfer, G.; Schneider, R.; von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schätzl, H.M. Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, M.; Walter, E.D.; Newell, D.J.; Jackson, P.J.; Aronoff-Spencer, E.; Peisach, J.; Gerfen, G.J.; Bennett, B.; Antholine, W.E.; Millhauser, G.L. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J. Am. Chem. Soc. 2005, 127, 12647–12656. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.S.; Aronoff-Spencer, E.; Legname, G.; Prusiner, S.B.; Antholine, W.E.; Gerfen, G.J.; Peisach, J.; Millhauser, G.L. Copper coordination in the full-length, recombinant prion protein. Biochemistry 2003, 42, 6794–6803. [Google Scholar] [CrossRef] [PubMed]
- Burns, C.S.; Aronoff-Spencer, E.; Dunham, C.M.; Lario, P.; Avdievich, N.I.; Antholine, W.E.; Olmstead, M.M.; Vrielink, A.; Gerfen, G.J.; Peisach, J.; et al. Molecular features of the copper binding sites in the octarepeat domain of the prion protein. Biochemistry 2002, 41, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Jiang, D.; McDonald, A.; Hao, Y.; Millhauser, G.L.; Zhou, F. Copper redox cycling in the prion protein depends critically on binding mode. J. Am. Chem. Soc. 2011, 133, 12229–12237. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Millhauser, G.L. The rich electrochemistry and redox reaacions of the copper sites in the cellular prion protein. Coord. Chem. Rev. 2012, 256, 2285–2296. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.S.; Murray, I.; Hosszu, L.L.; Gibbs, N.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 8531–8535. [Google Scholar] [CrossRef]
- Walter, E.D.; Stevens, D.J.; Spevacek, A.R.; Visconte, M.P.; Dei Rossi, A.; Millhauser, G.L. Copper binding extrinsic to the octarepeat region in the prion protein. Curr. Protein Pept. Sci. 2009, 10, 529–535. [Google Scholar] [CrossRef]
- Giachin, G.; Mai, P.T.; Tran, H.N.; Salzano, G.; Benetti, F.; Migliorati, V.; Arcovito, A.; Della Longa, S.; Mancini, G.; D’Angelo, P.; et al. The non-octarepeat copper binding site of the prion protein is a key regulator of prion conversion. Sci. Rep. 2015, 5, 15253. [Google Scholar] [CrossRef]
- Klewpatinond, M.; Davies, P.; Bowen, S.; Brown, D.R.; Viles, J.H. Deconvoluting the Cu(2+) binding modes of full-length prion protein. J. Biol. Chem. 2008, 283, 1870–1881. [Google Scholar] [CrossRef]
- Grande-Aztatzi, R.; Rivillas-Acevedo, L.; Quintanar, L.; Vela, A. Structural models for Cu(II) bound to the fragment 92-96 of the human prion protein. J. Phys. Chem. B 2013, 117, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Hureau, C.; Charlet, L.; Dorlet, P.; Gonnet, F.; Spadini, L.; Anxolabéhère-Mallart, E.; Girerd, J.J. A spectroscopic and voltammetric study of the pH-dependent Cu(II) coordination to the peptide GGGTH: Relevance to the fifth Cu(II) site in the prion protein. J. Boil. Inorg. Chem. 2006, 11, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Hureau, C.; Mathé, C.; Faller, P.; Mattioli, T.A.; Dorlet, P. Folding of the prion peptide GGGTHSQW around the copper(II) ion: Identifying the oxygen donor ligand at neutral pH and probing the proximity of the tryptophan residue to the copper ion. J. Biol. Inorg. Chem. 2008, 13, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Rivillas-Acevedo, L.; Grande-Aztatzi, R.; Lomelí, I.; García, J.E.; Barrios, E.; Teloxa, S.; Vela, A.; Quintanar, L. Spectroscopic and electronic structure studies of copper(II) binding to His111 in the human prion protein fragment 106–115: Evaluating the role of protons and methionine residues. Inorg. Chem. 2011, 50, 1956–1972. [Google Scholar] [CrossRef] [PubMed]
- Arcos-López, T.; Qayyum, M.; Rivillas-Acevedo, L.; Miotto, M.C.; Grande-Aztatzi, R.; Fernández, C.O.; Hedman, B.; Hodgson, K.O.; Vela, A.; Solomon, E.I.; et al. Spectroscopic and Theoretical Study of Cu(I) Binding to His111 in the Human Prion Protein Fragment 106–115. Inorg. Chem. 2016, 55, 2909–2922. [Google Scholar] [CrossRef] [PubMed]
- Berti, F.; Gaggelli, E.; Guerrini, R.; Janicka, A.; Kozlowski, H.; Legowska, A.; Miecznikowska, H.; Migliorini, C.; Pogni, R.; Remelli, M.; et al. Structural and dynamic characterization of copper(II) binding of the human prion protein outside the octarepeat region. Chem. Eur. J. 2007, 13, 1991–2001. [Google Scholar] [CrossRef] [PubMed]
- DiNatale, G.; Ösz, K.; Nagy, Z.; Sanna, D.; Micera, G.; Pappalardo, G.; Sóvágó, I.; Rizzarell, E. Interaction of Copper(II) with the Prion Peptide Fragment HuPrP(76–114) Encompassing Four Histidyl Residues within and outside the Octarepeat Domain. Inorg. Chem. 2009, 48, 4239–4250. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.E.; Klewpatinond, M.; Abdelraheim, S.R.; Brown, D.R.; Viles, J.H. Probing copper2+ binding to the prion protein using diamagnetic nickel2+ and 1H NMR: The unstructured N terminus facilitates the coordination of six copper2+ ions at physiological concentrations. J. Mol. Biol. 2005, 346, 1393–1407. [Google Scholar] [CrossRef] [PubMed]
- Nadal, R.C.; Davies, P.; Brown, D.R.; Viles, J.H. Evaluation of copper2+ affinities for the prion protein. Biochemistry 2009, 48, 8929–8931. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, C.; Rivillas-Acevedo, L.; Cruz-Vásquez, O.; Quintanar, L. Methionine 109 plays a key role in Cu(II) binding to His111 in the 92–115 fragment of the human prion protein. Inorg. Chim. Acta 2018, 481, 87–97. [Google Scholar] [CrossRef]
- Evans, E.G.B.; Pushie, M.J.; Markham, K.A.; Lee, H.-W.; Millhauser, G.L. Interaction between Prion Protein’s Copper-Bound Octarepeat Domain and a Charged C-Terminal Pocket Suggests a Mechanism for N-Terminal Regulation. Structure 2016, 24, 1057–1067. [Google Scholar] [CrossRef] [PubMed]
- Spevacek, A.R.; Evans, E.G.B.; Miller, J.L.; Meyer, H.C.; Pelton, J.G.; Millhauser, G.L. Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 2013, 21, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.K.; Srivastava, A.K.; Srinivas, V.; Chary, K.V.; Rao, C.M. Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J. Biol. Chem. 2011, 286, 38533–38545. [Google Scholar] [CrossRef] [PubMed]
- Younan, N.D.; Klewpatinond, M.; Davies, P.; Ruban, A.V.; Brown, D.R.; Viles, J.H. Copper(II)-induced secondary structure changes and reduced folding stability of the prion protein. J. Mol. Biol. 2011, 410, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.S.; Botelho, H.M.; Gomes, C.M. Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev. 2012, 256, 2253–2270. [Google Scholar] [CrossRef]
- D’angelo, P.; Della Longa, S.; Arcovito, A.; Mancini, G.; Zitolo, A.; Chillemi, G.; Giachin, G.; Legname, G.; Benetti, F. Effects of the Pathological Q212P Mutation on Human Prion Protein Non-Octarepeat Copper-Binding Site. Biochemistry 2012, 51, 6068–6079. [Google Scholar] [CrossRef]
- Altmeppen, H.C.; Puig, B.; Dohler, F.; Thurm, D.K.; Falker, C.; Krasemann, S.; Glatzel, M. Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 2012, 1, 15–31. [Google Scholar]
- McMahon, H.E.; Mangé, A.; Nishida, N.; Créminon, C.; Casanova, D.; Lehmann, S. Cleavage of the amino terminus of the prion protein by reactive oxygen species. J. Biol. Chem. 2001, 276, 2286–2291. [Google Scholar] [CrossRef]
- Watt, N.T.; Taylor, D.R.; Gillott, A.; Thomas, D.A.; Perera, W.S.S.; Hooper, N.M. Reactive Oxygen Species-mediated β-Cleavage of the Prion Protein in the Cellular Response to Oxidative Stress. J. Biol. Chem. 2005, 280, 35914–35921. [Google Scholar] [CrossRef]
- McDonald, A.J.; Dibble, J.P.; Evans, E.G.; Millhauser, G.L. A new paradigm for enzymatic control of alpha-cleavage and beta-cleavage of the prion protein. J. Biol. Chem. 2014, 289, 803–813. [Google Scholar] [CrossRef]
- McDonald, A.J.; Millhauser, G.L. PrP overdrive. Does inhibition of alpha-cleavage contribute to PrPC toxicity and prion disease? Prion 2014, 8, 183–191. [Google Scholar] [CrossRef]
- Oliveira-Martins, J.B.; Yusa, S.; Calella, A.M.; Bridel, C.; Baumann, F.; Dametto, P.; Aguzzi, A. Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS ONE 2010, 5, e9107. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; Lewis, V.A.; Vella, L.J.; Masters, C.L.; Hill, A.F.; Lawson, V.A.; Collins, S.J. PrPC-related signal transduction is influenced by copper, membrane integrity and the alpha cleavage site. Cell Res. 2009, 19, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, C.; Fernández, C.O.; Quintanar, L. Neuroprotective alpha-cleavage of the human prion protein significantly impacts Cu(II) coordination at its His111 site. Dalton Trans. 2018, 47, 9274–9282. [Google Scholar] [CrossRef] [PubMed]
- Black, S.A.G.; Stys, P.K.; Zamponi, G.W.; Tsutsui, S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Villar-Piqué, A.; Lopes da Fonseca, T.; Sant’Anna, R.; Szegö, E.M.; Fonseca-Ornelas, L.; Pinho, R.; Carija, A.; Gerhardt, E.; Masaracchia, C.; Abad Gonzalez, E.; et al. Environmental and genetic factors support the dissociation between α-synuclein aggregation and toxicity. Proc. Natl. Acad. Sci. USA 2016, 113, 6506–65115. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-López, C.; Rossetti, G.; Quintanar, L.; Carloni, P. Structural Determinants of the Prion Protein N-Terminus and Its Adducts with Copper Ions. Int. J. Mol. Sci. 2019, 20, 18. https://doi.org/10.3390/ijms20010018
Sánchez-López C, Rossetti G, Quintanar L, Carloni P. Structural Determinants of the Prion Protein N-Terminus and Its Adducts with Copper Ions. International Journal of Molecular Sciences. 2019; 20(1):18. https://doi.org/10.3390/ijms20010018
Chicago/Turabian StyleSánchez-López, Carolina, Giulia Rossetti, Liliana Quintanar, and Paolo Carloni. 2019. "Structural Determinants of the Prion Protein N-Terminus and Its Adducts with Copper Ions" International Journal of Molecular Sciences 20, no. 1: 18. https://doi.org/10.3390/ijms20010018
APA StyleSánchez-López, C., Rossetti, G., Quintanar, L., & Carloni, P. (2019). Structural Determinants of the Prion Protein N-Terminus and Its Adducts with Copper Ions. International Journal of Molecular Sciences, 20(1), 18. https://doi.org/10.3390/ijms20010018