Targeting Multiple Receptors to Increase Checkpoint Blockade Efficacy


Abstract
{kind=link}
{kind=link}
1. Introduction
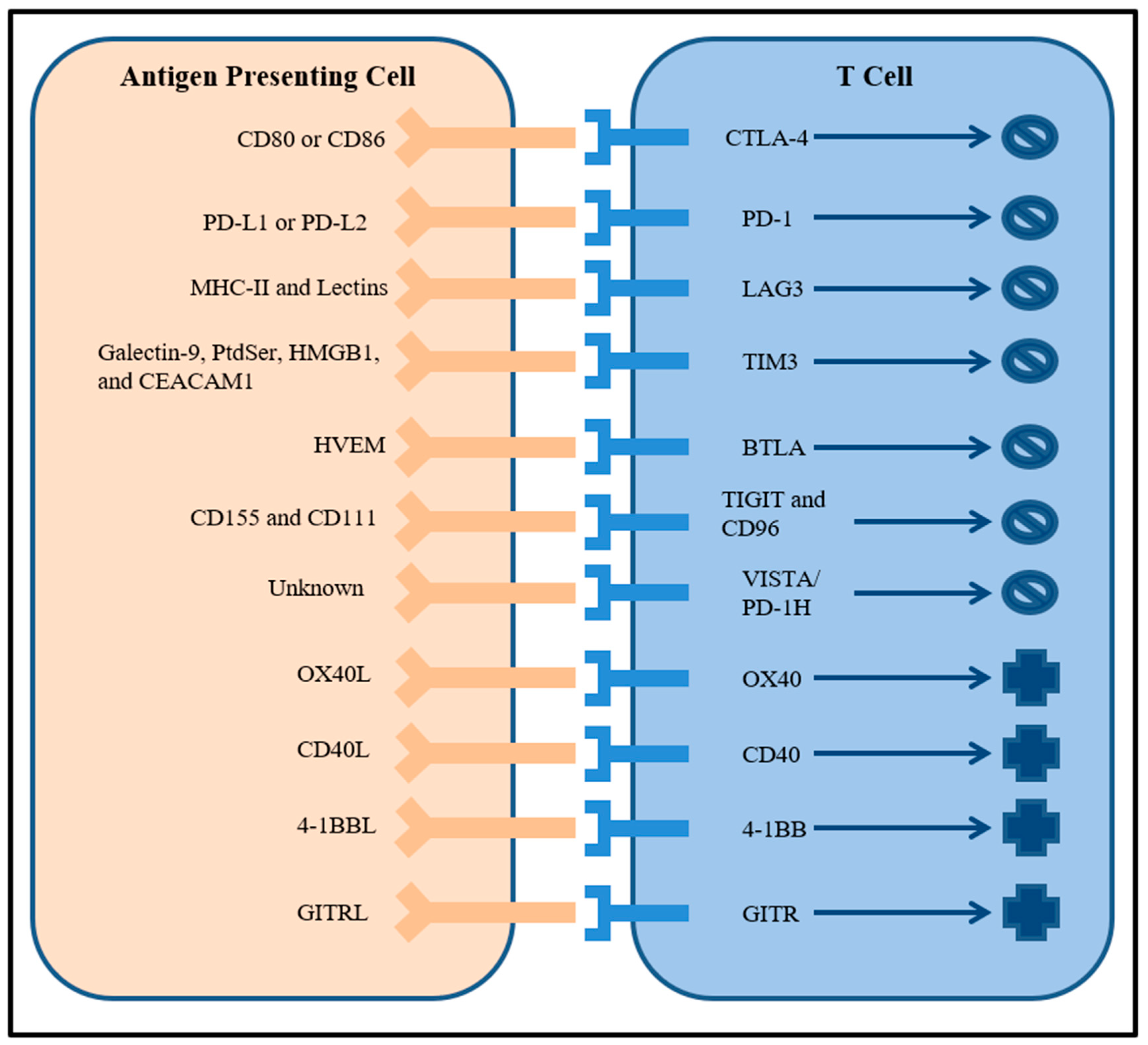
2. Checkpoint Blockade
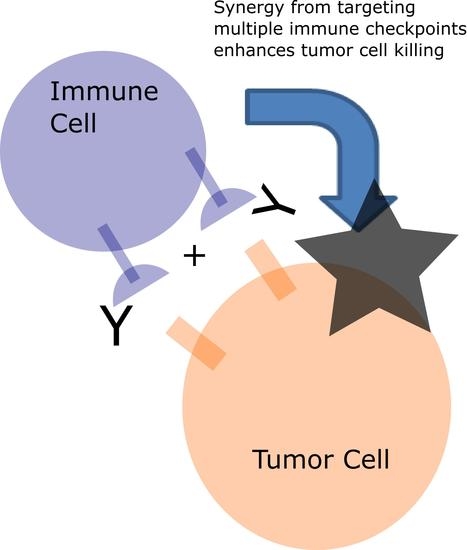
3. Targeting Multiple Immune Checkpoints
4. Combining Checkpoint Blockade with Other Receptor Targets
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hargadon, K.; Johnson, C.; Williams, C. Immune Checkpoint Blockade Therapy for Cancer: An Overview of FDA-Approved Immune Checkpoint Inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.; Hodi, F.; Kaufman, H.; Wigginton, J.; Wolchok, J. Combination Immunotherapy: A Road Map. J. Immunother. Cancer 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Karamouzis, M.; Papavassiliou, A. Combination of Checkpoint Inhibitors with Other Agents as a Strategy to Improve Anti-Cancer Effect—A Glimpse to the Future. Expert Opin. Investig. Drugs 2018, 27, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.; Thompson, C. At the Bench: Preclinical Rationale for CTLA-4 and PD-1 Blockade as Cancer Immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.; Baker, J.; Jeffery, L.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.; Allison, J. CTLA-4 Engagement Inhibits IL-2 Accumulation and Cell Cycle Progression Upon Activation of Resting T Cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Chambers, C.; Sullivan, T.; Truong, T.; Allison, J. Secondary but Not Primary T Cell Responses Are Enhanced in CTLA-4-Deficient CD8+ T Cells. Eur. J. Immunol. 1998, 28, 3137–3143. [Google Scholar] [CrossRef]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 Control Over Foxp3+ Regulatory T Cell Function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.; Krummel, M.; Allison, J. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Hodi, F.; O’Day, S.; McDermott, D.; Weber, R.; Sosman, J.; Haanen, J.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Topalian, S.; Drake, C.; Pardoll, D. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Page, D.; Postow, M.; Callahan, M.; Allison, J.; Wolchok, J. Immune Modulation in Cancer with Antibodies. Annu. Rev. Med. 2014, 65, 185–202. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-Like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Topalian, S.; Hodi, F.; Brahmer, J.; Gettinger, S.; Smith, D.; McDermott, D.; Powderly, J.; Carvajal, R.; Sosman, J.; Atkins, M.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Sznol, M.; Chen, L. Antagonist Antibodies to PD-1 and B7-H1 (PD-L1) in the Treatment of Advanced Human Cancer—Response. Clin. Cancer Res. 2013, 19, 5542. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; D’Angelo, S.; Minor, D.; Hodi, F.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.; Miller, W.; Lao, C.; et al. Nivolumab Versus Chemotherapy in Patients with Advanced Melanoma Who Progressed after Anti-CTLA-4 Treatment (Checkmate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.; Baas, P.; Crinò, L.; Eberhardt, W.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.; Holgado, E.; et al. Nivolumab Versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.; Escudier, B.; McDermott, D.; George, S.; Hammers, H.; Srinivas, S.; Tykodi, S.; Sosman, J.; Procopio, G.; Plimack, E.; et al. Nivolumab Versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.; Schachter, J.; Pavlick, A.; Lewis, K.; et al. Pembrolizumab Versus Investigator-Choice Chemotherapy for Ipilimumab-Refractory Melanoma (KEYNOTE-002): A Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Fuchs, C.; Doi, T.; Jang, R.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.; Shah, M.; Metges, J.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Abdin, S.; Zaher, D.; Arafa, E.; Omar, H. Tackling Cancer Resistance by Immunotherapy: Updated Clinical Impact and Safety of PD-1/PD-L1 Inhibitors. Cancers 2018, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Gaulard, P.; Faure, F.; Hercend, T.; Triebel, F. Cellular Expression and Tissue Distribution of the Human LAG-3-Encoded Protein, an MHC Class II Ligand. Immunogenetics 1994, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. Lsectin Expressed on Melanoma Cells Promotes Tumor Progression by Inhibiting Antitumor T-Cell Responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [PubMed]
- Sierro, S.; Romero, P.; Speiser, D. The CD4-Like Molecule LAG-3, Biology and Therapeutic Applications. Expert Opin. Ther. Targets 2010, 15, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Duffy, C.; Allison, J. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.; Blazar, B.; Kuchroo, V.; Anderson, A. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.; Lopez-Verges, S.; Barbour, J.; Jones, R.; Jha, A.; Long, B.; Schoeffler, E.; Fujita, T.; Nixon, D.; Lanier, L. Tim-3 Marks Human Natural Killer Cell Maturation and Suppresses Cell-Mediated Cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.; Clark, H.; et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat. Immunol. 2008, 10, 48–57. [Google Scholar] [CrossRef]
- Dougall, W.; Kurtulus, S.; Smyth, M.; Anderson, A. TIGIT and CD96: New Checkpoint Receptor Targets for Cancer Immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Blake, S.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.; Mittal, D.; Aguilera, A.; Miles, J.; Lutzky, V.; de Andrade, L.; et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef]
- Zhu, Y.; Paniccia, A.; Schulick, A.; Chen, W.; Koenig, M.; Byers, J.; Yao, S.; Bevers, S.; Edil, B. Identification of CD112R as a Novel Checkpoint for Human T Cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Sunderland, A.; Zhou, Y.; Schulick, R.; Edil, B.; Zhu, Y. Blockade of CD112R and TIGIT Signaling Sensitizes Human Natural Killer Cell Functions. Cancer Immunol. Immunother. 2017, 66, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Nelson, C.; Šedý, J. Balancing Co-Stimulation and Inhibition with BTLA and HVEM. Nat. Rev. Immunol. 2006, 6, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Dong, C. New Checkpoints in Cancer Immunotherapy. Immunol. Rev. 2017, 276, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, M.; Shah, U.; Liu, W.; Zhao, A.; Schoenberg, M.; Zang, X. The Third Group of the B7-CD28 Immune Checkpoint Family: HHLA2, TMIGD2, B7x, and B7-H3. Immunol. Rev. 2017, 276, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Byrne, K.; Vonderheide, R. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep. 2016, 15, 2719–2732. [Google Scholar] [CrossRef]
- Korman, A.; Chen, B.; Wang, C.; Wu, L.; Cardarelli, P.; Selby, M. Activity of anti-PD-1 in murine tumor models: Role of “host” PD-L1 and synergistic effect of anti-PD-1 and anti-CTLA-4. J. Immunol. 2007, 178, 48.1–48.40. [Google Scholar]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.; Cowey, C.; Lao, C.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Postow, M.; Chesney, J.; Pavlick, A.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.; Meyer, N.; Giguere, J.; Agarwala, S.; et al. Nivolumab and Ipilimumab Versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef]
- Nolan, E.; Savas, P.; Policheni, A.; Darcy, P.; Vaillant, F.; Mintoff, C.; Dushyanthen, S.; Mansour, M.; Pang, J.; Fox, S.; et al. Combined Immune Checkpoint Blockade as a Therapeutic Strategy for BRCA1-Mutated Breast Cancer. Sci. Transl. Med. 2017, 9, eaal4922. [Google Scholar] [CrossRef]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety Profiles of Anti-CTLA-4 and Anti-PD-1 Antibodies Alone and in Combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8 + T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yuan, Y.; Chen, W.; Putra, J.; Suriawinata, A.; Schenk, A.; Miller, H.; Guleria, I.; Barth, R.; Huang, Y.; et al. Immune-Checkpoint Proteins VISTA and PD-1 Nonredundantly Regulate Murine T-Cell Responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6682–6687. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.; Kim, M.; Montalvo, W.; Al-Shamkhani, A.; Allison, J. Combination CTLA-4 Blockade and 4-1BB Activation Enhances Tumor Rejection by Increasing T-Cell Infiltration, Proliferation, and Cytokine Production. PLoS ONE 2011, 6, e19499. [Google Scholar] [CrossRef] [PubMed]
- Shuford, W.; Klussman, K.; Tritchler, D.; Loo, D.; Chalupny, J.; Siadak, A.; Brown, T.; Emswiler, J.; Raecho, H.; Larsen, C.; et al. 4-1BB Costimulatory Signals Preferentially Induce CD8+T Cell Proliferation and Lead to the Amplification In Vivo of Cytotoxic T Cell Responses. J. Exp. Med. 1997, 186, 47–55. [Google Scholar] [CrossRef]
- Kohrt, H.; Houot, R.; Goldstein, M.; Weiskopf, K.; Alizadeh, A.; Brody, J.; Muller, A.; Pachynski, R.; Czerwinski, D.; Coutre, S.; et al. CD137 Stimulation Enhances the Antilymphoma Activity of Anti-CD20 Antibodies. Blood 2010, 117, 2423–2432. [Google Scholar] [CrossRef]
- Shimizu, J.; Yamazaki, S.; Takahashi, T.; Ishida, Y.; Sakaguchi, S. Stimulation of CD25+CD4+ Regulatory T Cells Through GITR Breaks Immunological Self-Tolerance. Nat. Immunol. 2002, 3, 135–142. [Google Scholar] [CrossRef]
- Siu, L.; Steeghs, N.; Meniawy, T.; Joerger, M.; Spratlin, J.; Rottey, S.; Nagrial, A.; Cooper, A.; Meier, R.; Guan, X.; et al. Preliminary Results of a Phase I/Iia Study of BMS-986156 (Glucocorticoid-Induced Tumor Necrosis Factor Receptor–Related Gene [GITR] Agonist), Alone and in Combination with Nivolumab in Pts with Advanced Solid Tumors. J. Clin. Oncol. 2017, 35, 104. [Google Scholar] [CrossRef]
- Melero, I.; Berman, D.; Aznar, M.; Korman, A.; Gracia, J.; Haanen, J. Evolving Synergistic Combinations of Targeted Immunotherapies to Combat Cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef]
- Leone, R.; Sun, I.; Oh, M.; Sun, I.; Wen, J.; Englert, J.; Powell, J. Inhibition of the Adenosine A2a Receptor Modulates Expression of T Cell Coinhibitory Receptors and Improves Effector Function for Enhanced Checkpoint Blockade and ACT in Murine Cancer Models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, V.; Hermann-Kleiter, N.; Do-Dinh, P.; Jakic, B.; Offermann, A.; Efremova, M.; Sopper, S.; Rieder, D.; Krogsdam, A.; Gamerith, G.; et al. Nuclear Receptor NR2F6 Inhibition Potentiates Responses to PD-L1/PD-1 Cancer Immune Checkpoint Blockade. Nat. Commun. 2018, 9, 1538. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. Tgfβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.; Schaer, D.; Li, Y.; Castaneda, S.; Murphy, M.; Xu, X.; Inigo, I.; Dobkin, J.; Manro, J.; Iversen, P.; et al. Targeting the Tgfβ Pathway with Galunisertib, a Tgfβri Small Molecule Inhibitor, Promotes Anti-Tumor Immunity Leading to Durable, Complete Responses, as Monotherapy and in Combination with Checkpoint Blockade. J. Immunother. Cancer 2018, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Umemura, N.; Saio, M.; Suwa, T.; Kitoh, Y.; Bai, J.; Nonaka, K.; Ouyang, G.; Okada, M.; Balazs, M.; Adany, R.; et al. Tumor-Infiltrating Myeloid-Derived Suppressor Cells are Pleiotropic-Inflamed Monocytes/Macrophages That Bear M1- and M2-Type Characteristics. J. Leukoc. Biol. 2008, 83, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.; Brachfeld, A.; Gasmi, B.; Jones, D.; Mattar, M.; Doman, T.; Murphy, M.; Schaer, D.; Wolchok, J.; Merghoub, T. Timing of CSF-1/CSF-1R Signaling Blockade Is Critical to Improving Responses to CTLA-4 Based Immunotherapy. OncoImmunology 2016, 5, e1151595. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.; Motz, G.; Busch, J.; Coukos, G. Angiogenesis and the Tumor Vasculature as Antitumor Immune Modulators: The Role of Vascular Endothelial Growth Factor and Endothelin. Curr. Top. Microbiol. Immunol. 2010, 129–148. [Google Scholar] [CrossRef]
- Ohm, J.; Carbone, D. VEGF as a Mediator of Tumor-Associated Immunodeficiency. Immunol. Res. 2001, 23, 263–272. [Google Scholar] [CrossRef]
- Ott, P.; Hodi, F.; Buchbinder, E. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef]
- Läubli, H.; Müller, P.; D’Amico, L.; Buchi, M.; Kashyap, A.; Zippelius, A. The Multi-Receptor Inhibitor Axitinib Reverses Tumor-Induced Immunosuppression and Potentiates Treatment with Immune-Modulatory Antibodies in Preclinical Murine Models. Cancer Immunol. Immunother. 2018, 67, 815–824. [Google Scholar] [CrossRef]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous Blockade of Programmed Death 1 and Vascular Endothelial Growth Factor Receptor 2 (VEGFR2) Induces Synergistic Anti-Tumour Effectin Vivo. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef]
- Balachandran, V.; Cavnar, M.; Zeng, S.; Bamboat, Z.; Ocuin, L.; Obaid, H.; Sorenson, E.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib Potentiates Antitumor T Cell Responses in Gastrointestinal Stromal Tumor Through the Inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Soo, R.; Lim, S.; Syn, N.; Teng, R.; Soong, R.; Mok, T.; Cho, B. Immune Checkpoint Inhibitors in Epidermal Growth Factor Receptor Mutant Non-Small Cell Lung Cancer: Current Controversies and Future Directions. Lung Cancer 2018, 115, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Loi, S.; Divisekera, U.; Ngiow, S.; Duret, H.; Yagita, H.; Teng, M.; Smyth, M. Anti-Erbb-2 Mab Therapy Requires Type I and II Interferons and Synergizes with Anti-PD-1 or Anti-CD137 Mab Therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 7142–7147. [Google Scholar] [CrossRef]
- Smith, J.; Wang, S.; Nadella, S.; Jablonski, S.; Weiner, L. Cholecystokinin Receptor Antagonist Alters Pancreatic Cancer Microenvironment and Increases Efficacy of Immune Checkpoint Antibody Therapy in Mice. Cancer Immunol. Immunother. 2017, 67, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Khajeh Alizadeh Attar, M.; Anwar, M.; Eskian, M.; Keshavarz-Fathi, M.; Choi, S.; Rezaei, N. Basic Understanding and Therapeutic Approaches to Target Toll-Like Receptors in Cancerous Microenvironment and Metastasis. Med. Res. Rev. 2017, 38, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Wang, G.; Yin, B.; Ozao, J.; Ku, T.; Divino, C.; Chen, S. Reversion of Immune Tolerance in Advanced Malignancy: Modulation of Myeloid-Derived Suppressor Cell Development by Blockade of Stem-Cell Factor Function. Blood 2007, 111, 219–228. [Google Scholar] [CrossRef]
- Garton, A.; Seibel, S.; Lopresti-Morrow, L.; Crew, L.; Janson, N.; Mandiyan, S.; Trombetta, E.; Pankratz, S.; LaVallee, T.; Gedrich, R. Anti-KIT Monoclonal Antibody Treatment Enhances the Antitumor Activity of Immune Checkpoint Inhibitors by Reversing Tumor-Induced Immunosuppression. Mol. Cancer Ther. 2017, 16, 671–680. [Google Scholar] [CrossRef]
- Akalu, Y.; Rothlin, C.; Ghosh, S. TAM Receptor Tyrosine Kinases as Emerging Targets of Innate Immune Checkpoint Blockade for Cancer Therapy. Immunol. Rev. 2017, 276, 165–177. [Google Scholar] [CrossRef]
- Matlung, H.; Szilagyi, K.; Barclay, N.; van den Berg, T. The CD47-Sirpα Signaling Axis as an Innate Immune Checkpoint in Cancer. Immunol. Rev. 2017, 276, 145–164. [Google Scholar] [CrossRef]
- Wu, L.; Yu, G.; Deng, W.; Mao, L.; Yang, L.; Ma, S.; Bu, L.; Kulkarni, A.; Zhang, W.; Zhang, L.; et al. Anti-CD47 Treatment Enhances Anti-Tumor T-Cell Immunity and Improves Immunosuppressive Environment in Head and Neck Squamous Cell Carcinoma. OncoImmunology 2018, 7, e1397248. [Google Scholar] [CrossRef] [PubMed]
- Ring, N.; Herndler-Brandstetter, D.; Weiskopf, K.; Shan, L.; Volkmer, J.; George, B.; Lietzenmayer, M.; McKenna, K.; Naik, T.; McCarty, A.; et al. Anti-Sirpα Antibody Immunotherapy Enhances Neutrophil and Macrophage Antitumor Activity. Proc. Natl. Acad. Sci. USA 2017, 114, E10578–E10585. [Google Scholar] [CrossRef] [PubMed]
- Fucà, G.; de Braud, F.; Di Nicola, M. Immunotherapy-Based Combinations. Curr. Opin. Oncol. 2018, 30, 345–351. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahavi, D.J.; Weiner, L.M. Targeting Multiple Receptors to Increase Checkpoint Blockade Efficacy. Int. J. Mol. Sci. 2019, 20, 158. https://doi.org/10.3390/ijms20010158
Zahavi DJ, Weiner LM. Targeting Multiple Receptors to Increase Checkpoint Blockade Efficacy. International Journal of Molecular Sciences. 2019; 20(1):158. https://doi.org/10.3390/ijms20010158
Chicago/Turabian StyleZahavi, David J., and Louis M. Weiner. 2019. "Targeting Multiple Receptors to Increase Checkpoint Blockade Efficacy" International Journal of Molecular Sciences 20, no. 1: 158. https://doi.org/10.3390/ijms20010158
APA StyleZahavi, D. J., & Weiner, L. M. (2019). Targeting Multiple Receptors to Increase Checkpoint Blockade Efficacy. International Journal of Molecular Sciences, 20(1), 158. https://doi.org/10.3390/ijms20010158