Chloramphenicol Induces Autophagy and Inhibits the Hypoxia Inducible Factor-1 Alpha Pathway in Non-Small Cell Lung Cancer Cells

 ,
,
Abstract
1. Introduction
2. Results
2.1. Short Incubation with Chloramphenicol Does Not Affect Cell Viability in Normoxia or Hypoxia Conditions
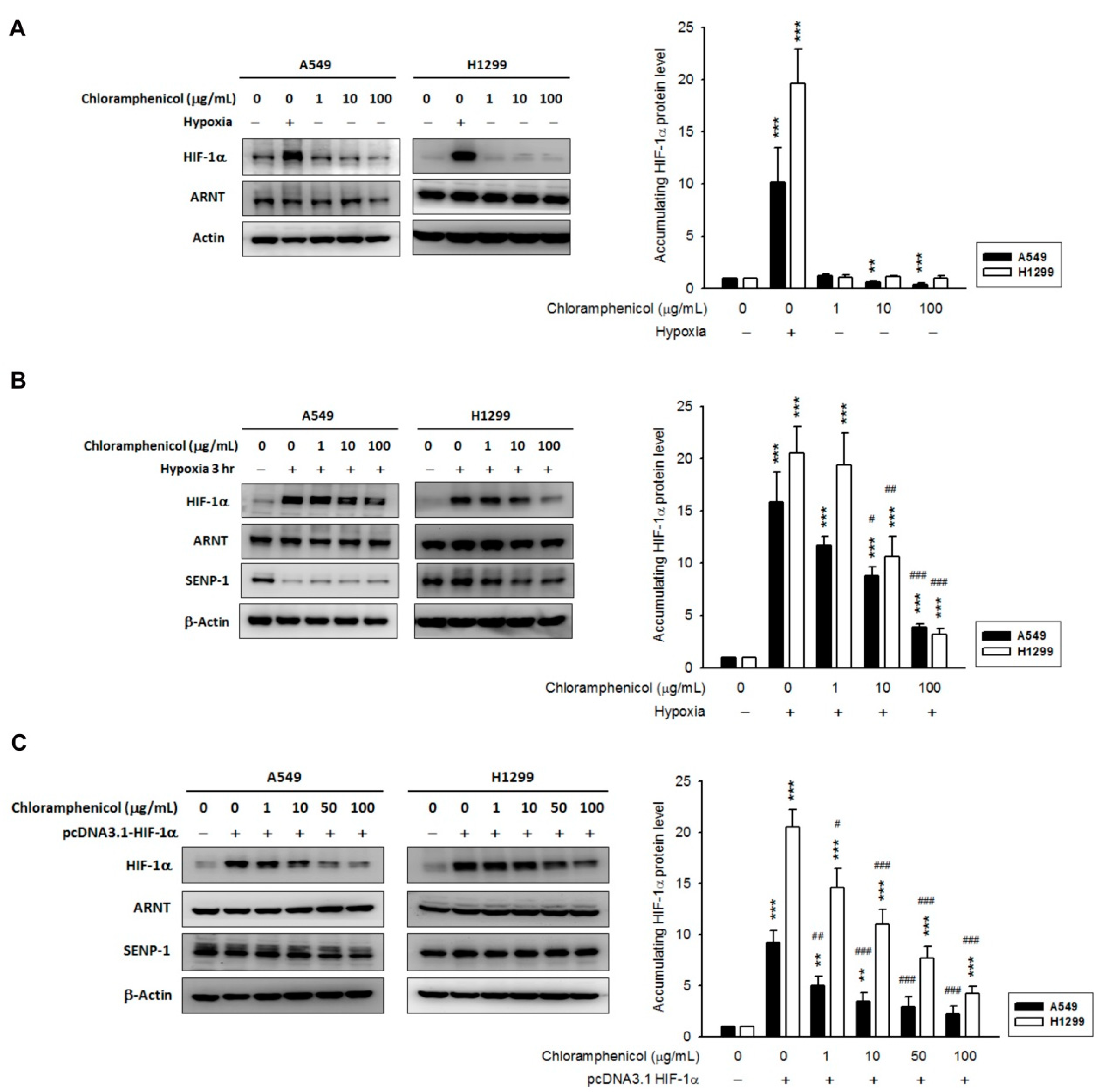
2.2. Chloramphenicol Inhibited HIF-1α Protein Accumulation in Non-Small Cell Lung Cancer (NSCLC) Cells in a Concentration-Dependent Manner
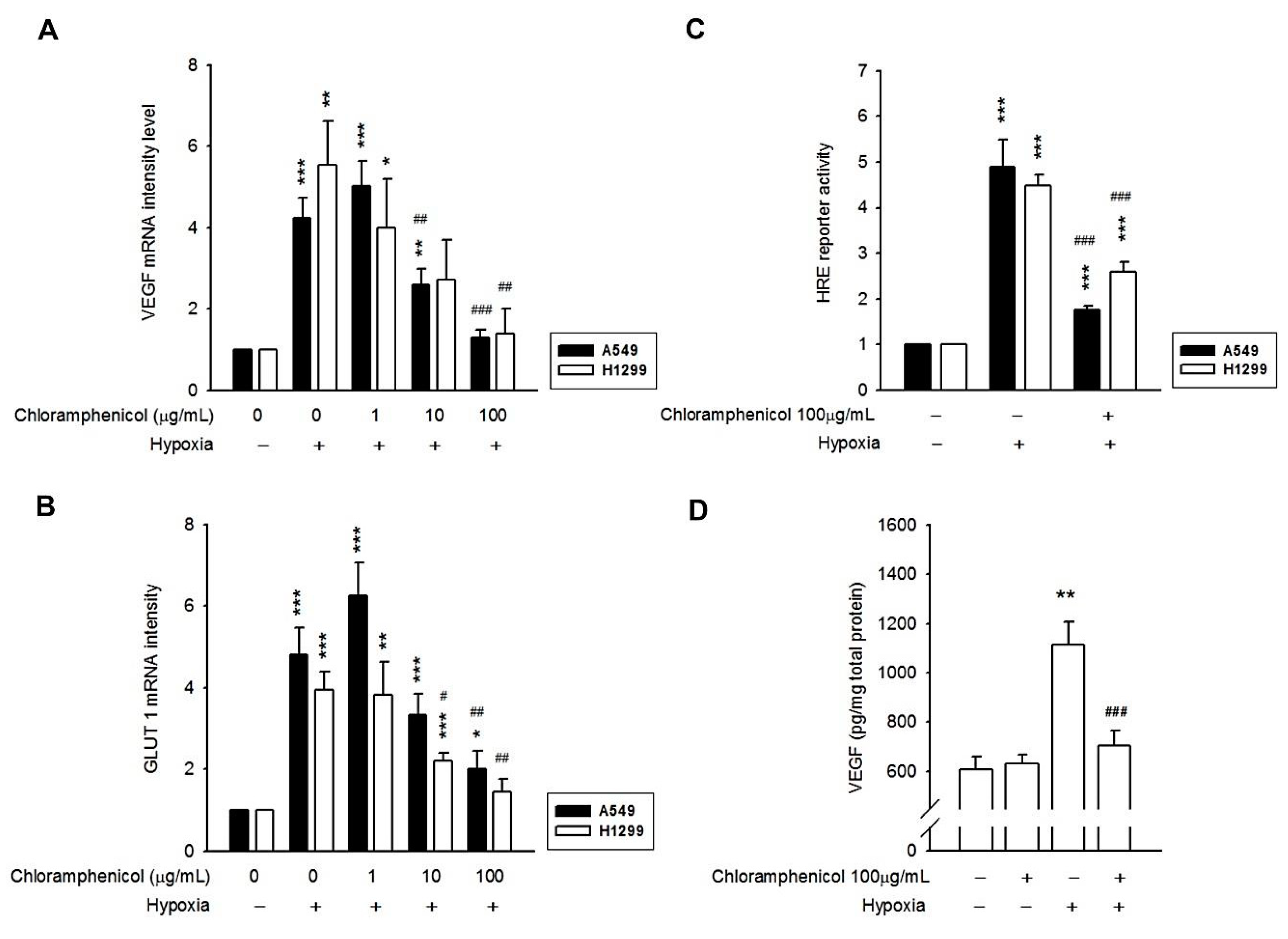
2.3. Hypoxia-Induced mRNA Expression of GLUT-1 and VEGF, and the Secretion of VEGF Protein Were Inhibited by Chloramphenicol Treatment
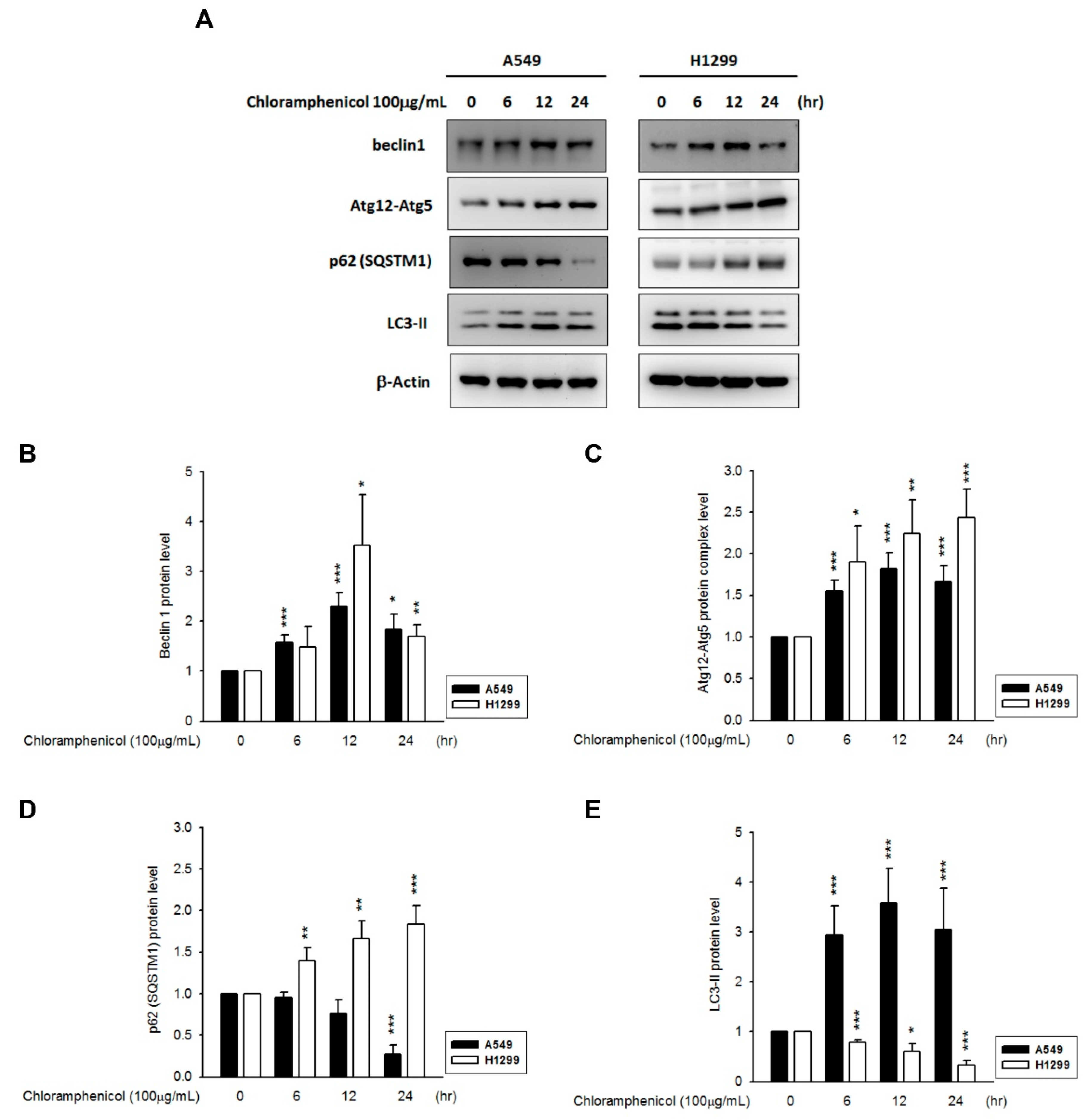
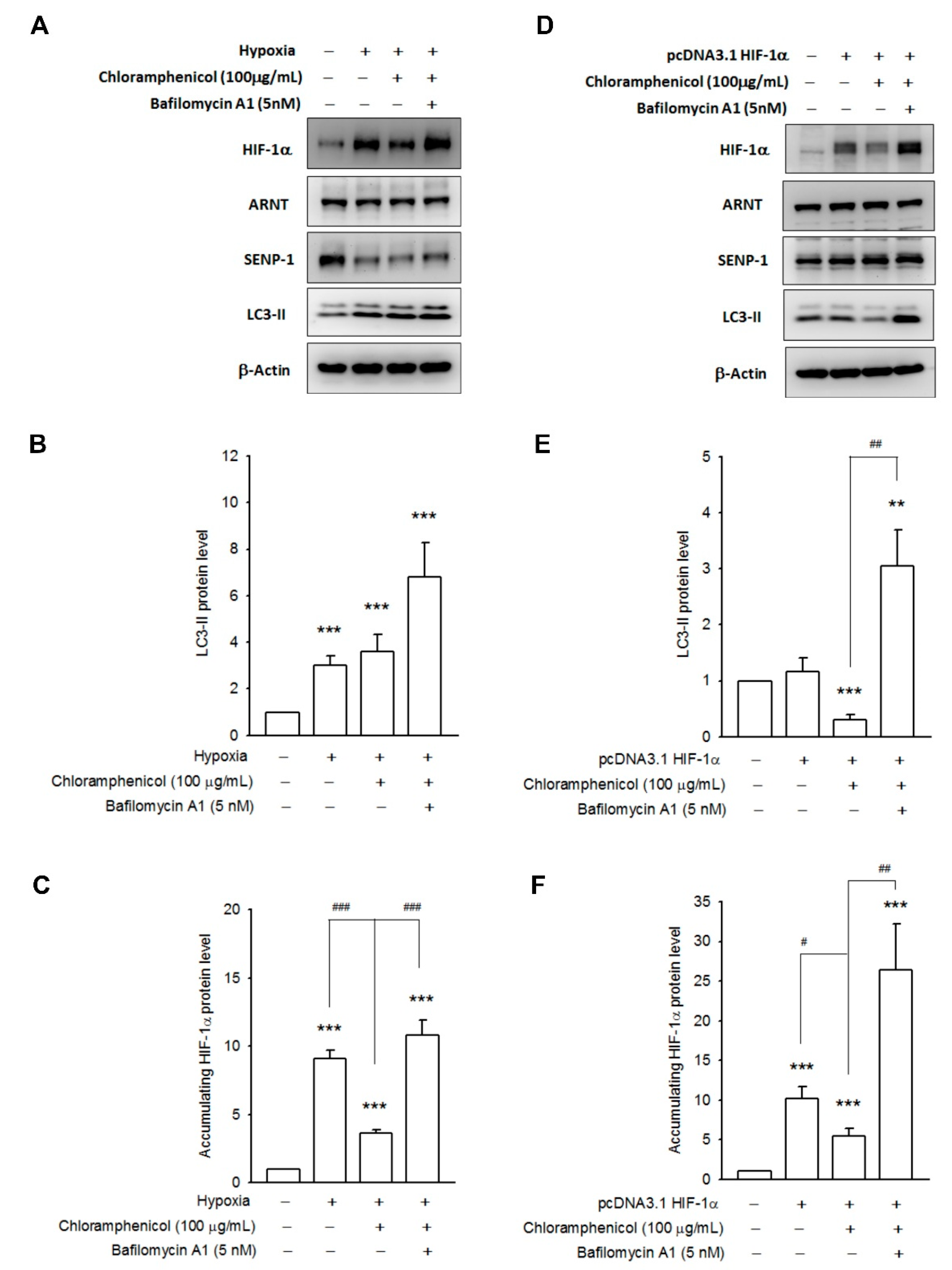
2.4. Chloramphenicol Promoted HIF-1α Protein Degradation Via Autophagy-Dependent Pathway
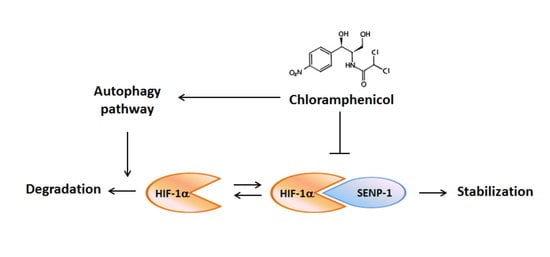
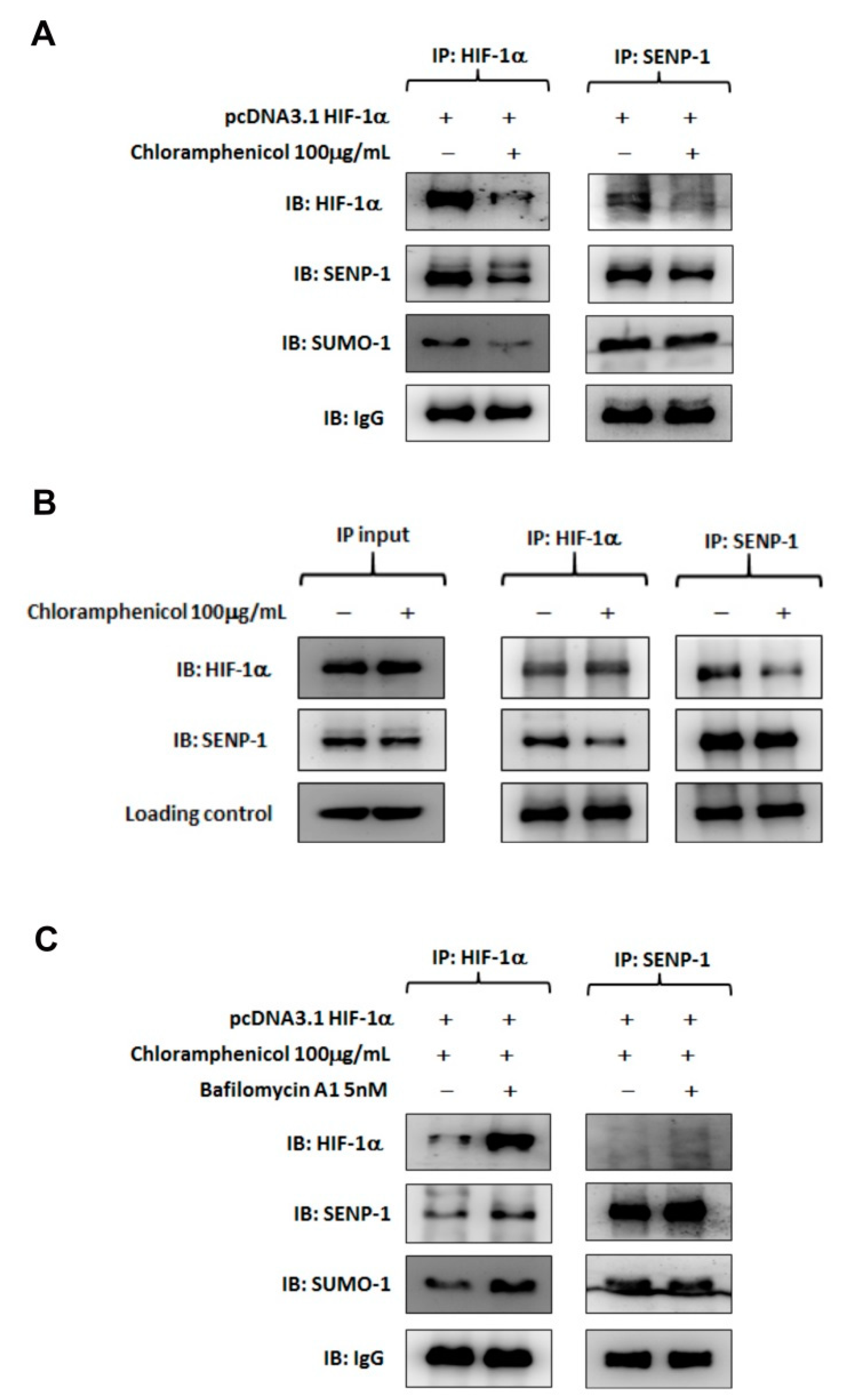
2.5. Chloramphenicol Interfered with Protein Interaction between SENP-1 and HIF-1α to Promote HIF-1α Degradation via Autophagy Pathway
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Hypoxia Condition
4.3. Cell Viability Assay
4.4. Western Blot Analysis
4.5. Reverse Transcription and Quantitative Polymerase Chain Reaction (RT-qPCR)
4.6. Enzyme Linked Immunosorbent Assay (ELISA)
4.7. Transfection
4.8. Luciferase Reporter Assay
4.9. Co-Immunoprecipitation
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HIF-1α | Hypoxia inducible factor-1 alpha |
| NSCLC | Non-small cell lung cancer |
| VEGF | Vascular endothelial growth factor |
| PHD | Prolyl hydroxylase |
| pVHL | von Hippel–Lindau protein |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| HREs | Hypoxia response elements |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| GLUT-1 | Glucose transporter 1 |
| PDGF | Platelet-derived growth factor |
| UPS | Ubiquitin-proteasome system |
| SUMO | Small ubiquitin-like modifier |
| SENP-1 | Sentrin/SUMO-specific protease 1 |
References
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Firth, J.D.; Ratcliffe, P.J. Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences. J. Biol. Chem. 1995, 270, 29083–29089. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Sakata, M.; Takeda, T.; Yamamoto, T.; Okamoto, Y.; Sawada, K.; Kimura, A.; Minekawa, R.; Tahara, M.; Tasaka, K.; et al. Induction of glucose transporter 1 expression through hypoxia-inducible factor 1alpha under hypoxic conditions in trophoblast-derived cells. J. Endocrinol. 2004, 183, 145–154. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.A.D.; Andrade, D.R., Jr. HIF-1alpha and infectious diseases: A new frontier for the development of new therapies. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e92. [Google Scholar] [CrossRef]
- Liu, X.D.; Yao, J.; Tripathi, D.N.; Ding, Z.; Xu, Y.; Sun, M.; Zhang, J.; Bai, S.; German, P.; Hoang, A.; et al. Autophagy mediates HIF2α degradation and suppresses renal tumorigenesis. Oncogene 2015, 34, 2450–2460. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox. Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Kirkin, V.; Dikic, I.; Johansen, T. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 2009, 8, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- IARC. Chloramphenicol. In Pharmaceutical Drugs. IARC Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Humans; International Agency for Research on Cancer: Lyon, France, 1990; Volume 50, pp. 169–193. ISBN 92 832 1250 9. [Google Scholar]
- Eliakim-Raz, N.; Lador, A.; Leibovici-Weissman, Y.; Elbaz, M.; Paul, M.; Leibovici, L. Efficacy and safety of chloramphenicol: Joining the revival of old antibiotics? Systematic review and meta-analysis of randomized controlled trials. J. Antimicrob. Chemother. 2015, 70, 979–996. [Google Scholar] [CrossRef]
- Hanekamp, J.C.; Bast, A. Antibiotics exposure and health risks: Chloramphenicol. Environ. Toxicol. Pharmacol. 2015, 39, 213–220. [Google Scholar] [CrossRef]
- Nitzan, O.; Kennes, Y.; Colodner, R.; Saliba, W.; EdelsteinI, H.; Raz, R.; Chazan, B. Chloramphenicol use and susceptibility patterns in Israel: A national survey. Isr. Med. Assoc. J. 2015, 17, 27–31. [Google Scholar] [PubMed]
- Čivljak, R.; Giannella, M.; Di Bella, S.; Petrosillo, N. Could chloramphenicol be used against ESKAPE pathogens? A review of in vitro data in the literature from the 21st century. Expert Rev. Anti. Infect. Ther. 2014, 12, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Grammatikos, A.P.; Michalopoulos, A. Potential of old-generation antibiotics to address current need for new antibiotics. Expert Rev. Anti. Infect. Ther. 2008, 6, 593–600. [Google Scholar] [CrossRef]
- Ingebrigtsen, S.G.; Didriksen, A.; Johannessen, M.; Škalko-Basnet, N.; Holsæter, A.M. Old drug, new wrapping—A possible comeback for chloramphenicol? Int. J. Pharm. 2017, 526, 538–546. [Google Scholar] [CrossRef]
- Bae, S.; Jeong, H.J.; Cha, H.J.; Kim, K.; Choi, Y.M.; An, I.S.; Koh, H.J.; Lim, D.J.; Lee, S.J.; An, S. The hypoxia-mimetic agent cobalt chloride induces cell cycle arrest and alters gene expression in u266 multiple myeloma cells. Int. J. Mol. Med. 2012, 30, 1180–1186. [Google Scholar] [CrossRef]
- Yamanaka-Tatematsu, M.; Nakashima, A.; Fujita, N.; Shima, T.; Yoshimori, T.; Saito, S. Autophagy induced by HIF1α overexpression supports trophoblast invasion by supplying cellular energy. PLoS ONE 2013, 8, e76605. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, V.O.; Piskunova, T.; Zborovskaya, I.B.; Tchevkina, E.M.; Zhivotovsky, B. Suppression of basal autophagy reduces lung cancer cell proliferation and enhances caspase-dependent and -independent apoptosis by stimulating ROS formation. Autophagy 2012, 8, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Kang, X.; Zhang, S.; Yeh, E.T. SUMO-specific protease 1 is essential for stabilization of HIF1alpha during hypoxia. Cell 2007, 131, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Mermis, J.; Gu, H.; Xue, B.; Li, F.; Tawfik, O.; Buch, S.; Bartolome, S.; O’Brien-Ladner, A.; Dhillon, N.K. Hypoxia-inducible factor-1a/platelet derived growth factor axis in HIV-associated pulmonary vascular remodeling. Respir. Res. 2011, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, X.; Simet, S.M.; Hu, G.; Cai, Y.; Niu, F.; Kook, Y.; Buch, S.J. Reactive oxygen species/hypoxia-inducible factor-1α/platelet-derived growth factor-BB autocrine loop contributes to cocaine-mediated alveolar epithelial barrier damage. Am. J. Respir. Cell Mol. Biol. 2016, 55, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Choi, Y.J.; Park, J.W. SIRT1 and AMPK mediate hypoxia-induced resistance of non-small cell lung cancers to cisplatin and doxorubicin. Cancer Res. 2014, 74, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, G.; Yang, Z.; Wang, L.; Zhang, L.; Wang, T.; Zhang, Y.; Zhang, S.; Han, Y.; Jia, L. Uncoupling protein 2 downregulation by hypoxia through repression of peroxisome proliferator-activated receptor γ promotes chemoresistance of non-small cell lung cancer. Oncotarget 2017, 8, 8083–8094. [Google Scholar] [CrossRef]
- Lu, Y.; Yu, L.Q.; Zhu, L.; Zhao, N.; Zhou, X.J.; Lu, X. Expression of HIF-1α and P-gp in non-small cell lung cancer and the relationship with HPV infection. Oncol. Lett. 2016, 12, 1455–1459. [Google Scholar] [CrossRef]
- Polke, M.; Seiler, F.; Lepper, P.M.; Kamyschnikow, A.; Langer, F.; Monz, D.; Herr, C.; Bals, R.; Beisswenger, C. Hypoxia and the hypoxia-regulated transcription factor HIF-1α suppress the host defense of airway epithelial cells. Innate Immun. 2017, 23, 373–380. [Google Scholar] [CrossRef]
- Chen, L.W.; Egan, L.; Li, Z.W.; Greten, F.R.; Kagnoff, M.F.; Karin, M. The two faces of IKK and NFkappaB inhibition: Prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat. Med. 2003, 9, 575–581. [Google Scholar] [CrossRef]
- Belton, M.; Brilha, S.; Manavaki, R.; Mauri, F.; Nijran, K.; Hong, Y.T.; Patel, N.H.; Dembek, M.; Tezera, L.; Green, J.; et al. Hypoxia and tissue destruction in pulmonary TB. Thorax 2016, 71, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Peyssonnaux, C.; Cejudo-Martin, P.; Doedens, A.; Zinkernagel, A.S.; Johnson, R.S.; Nizet, V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J. Immunol. 2007, 178, 7516–7519. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Hilliard, G.; Ferguson, T.; Millhorn, D.E. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von hippel-lindau protein by direct binding to hypoxia-inducible factor-alpha. J. Biol. Chem. 2003, 278, 15911–15916. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, K.; Kamitani, T. pVHL-independent ubiquitination of HIF1alpha and its stabilization by cobalt ion. Biochem. Biophys. Res. Commun. 2003, 306, 750–755. [Google Scholar] [CrossRef]
- Triantafyllou, A.; Liakos, P.; Tsakalof, A.; Georgatsou, E.; Simos, G.; Bonanou, S. Cobalt induces hypoxia-inducible factor-1alpha (HIF-1alpha) in HeLa cells by an iron-independent, but ROS-, PI-3K- and MAPK-dependent mechanism. Free Radic. Res. 2006, 40, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Maruyama, M.; Inoue, A.; Fujita, T.; Kawahara, J.; Sassa, K.; Hayashi, R.; Kawagishi, Y.; Yamashita, N.; Sugiyama, E.; et al. Inhibition of proteasome activity is involved in cobalt-induced apoptosis of human alveolar macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L849–L858. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Lapham, A.; Brimmell, M.; Wilkinson, H.; Packham, G. Inhibition of proteasomal degradation of Mcl-1 by cobalt chloride suppresses cobalt chloride-induced apoptosis in HCT116 colorectal cancer cells. Apoptosis 2008, 13, 972–982. [Google Scholar] [CrossRef]
- Tomco, D.; Schmitt, S.; Ksebati, B.; Heeg, M.J.; Dou, Q.P.; Verani, C.N. Effects of tethered ligands and of metal oxidation state on the interactions of cobalt complexes with the 26S proteasome. J. Inorg. Biochem. 2011, 105, 1759–1766. [Google Scholar] [CrossRef]
- Liu, X.W.; Cai, T.Y.; Zhu, H.; Cao, J.; Su, Y.; Hu, Y.Z.; He, Q.J.; Yang, B. Q6, a novel hypoxia-targeted drug, regulates hypoxia-inducible factor signaling via an autophagy-dependent mechanism in hepatocellular carcinoma. Autophagy 2014, 10, 111–122. [Google Scholar] [CrossRef]
- Wu, F.Q.; Fang, T.; Yu, L.X.; Lv, G.S.; Lv, H.W.; Liang, D.; Li, T.; Wang, C.Z.; Tan, Y.X.; Ding, J.; et al. ADRB2 signaling promotes HCC progression and sorafenib resistance by inhibiting autophagic degradation of HIF1α. J. Hepatol. 2016, 65, 314–324. [Google Scholar] [CrossRef]
- Zheng, B.; Mao, J.H.; Qian, L.; Zhu, H.; Gu, D.H.; Pan, X.D.; Yi, F.; Ji, D.M. Pre-clinical evaluation of AZD-2014, a novel mTORC1/2 dual inhibitor, against renal cell carcinoma. Cancer Lett. 2015, 357, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.W.; Hou, J.K.; He, W.; Fan, L.; Huang, Y. Chloroquine enhances cobalt chloride-induced leukemic cell differentiation via the suppression of autophagy at the late phase. Biochem. Biophys. Res. Commun. 2013, 430, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, C.; Mazure, N.; Pouysségur, J. Signalling via the hypoxia-inducible factor-1alpha requires multiple posttranslational modifications. Cell Signal. 2005, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.T. SUMOylation and De-SUMOylation: Wrestling with life’s processes. J. Biol. Chem. 2009, 284, 8223–8227. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Baek, S.H. Emerging roles of desumoylating enzymes. Biochim. Biophys. Acta 2009, 1792, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.P.; Wong, C.C.; Kai, A.K.; Ho, D.W.; Lau, E.Y.; Tsui, Y.M.; Chan, L.K.; Cheung, T.T.; Chok, K.S.; Chan, A.C.Y.; et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 2017, 66, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Dai, A.; Jiang, Y.; Tan, X.; Zhang, X. SENP-1 enhances hypoxia-induced proliferation of rat pulmonary artery smooth muscle cells by regulating hypoxia-inducible factor-1α. Mol. Med. Rep. 2016, 13, 3482–3490. [Google Scholar] [CrossRef]
- Li, C.H.; Liao, P.L.; Yang, Y.T.; Huang, S.H.; Lin, C.H.; Cheng, Y.W.; Kang, J.J. Minocycline accelerates hypoxia-inducible factor-1 alpha degradation and inhibits hypoxia-induced neovasculogenesis through prolyl hydroxylase, von Hippel-Lindau-dependent pathway. Arch. Toxicol. 2014, 88, 659–671. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Chloramphenicol (g/mL), 3 h Incubation | Chloramphenicol (g/mL), 24 h Incubation | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 10 | 100 | 0 | 1 | 10 | 100 | ||
| Cell viability (%) | A549 | 100.0 ± 1.0 | 103.6 ± 2.3 | 95.6 ± 1.5 | 97.0 ± 3.9 | 100.0 ± 0.8 | 96.0 ± 1.1 * | 92.4 ± 1.4 *** | 89.3 ± 1.4 *** |
| H1299 | 100.0 ± 0.9 | 101.1 ± 1.3 | 98.3 ± 1.5 | 98.1 ± 5.0 | 100.0 ± 2.4 | 97.5 ± 1.8 ** | 93.8 ± 2.1 *** | 92.4 ± 2.5 *** | |
| Chloramphenicol treatment followed by an 8 h hypoxia condition | |||||||||
| Cell viability (%) | A549 | 100.0 ± 0.8 | 102.0 ± 3.6 | 99.1 ± 1.6 | 102.9 ± 1.3 | 100.0 ± 0.4 | 92.4 ± 1.0 *** | 85.6 ± 1.0 *** | 81.0 ± 0.8 *** |
| H1299 | 100.0 ± 1.0 | 101.3 ± 0.9 | 98.7 ± 0.9 | 103.3 ± 1.9 | 100.0 ± 1.7 | 98.5 ± 0.8 | 98.1 ± 1.6 | 93.4 ± 1.6 *** | |
| Chloramphenicol treatment followed by an 8 h CoCl2 (250 M) incubation | |||||||||
| Cell viability (%) | A549 | 100.0 ± 1.5 | 101.1 ± 1.9 | 97.4 ± 1.2 | 99.2 ± 0.9 | 100.0 ± 1.1 | 98.4 ± 1.7 | 93.0 ± 2.2 * | 77.0 ± 1.1 *** |
| H1299 | 100.0 ± 0.9 | 101.8 ± 1.1 | 95.3 ± 1.6 | 93.8 ± 4.5 | 100.0 ± 1.2 | 100.6 ± 2.5 | 99.8 ± 2.0 | 88.5 ± 4.1 *** | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, H.-L.; Liao, P.-L.; Cheng, Y.-W.; Huang, S.-H.; Wu, C.-H.; Li, C.-H.; Kang, J.-J. Chloramphenicol Induces Autophagy and Inhibits the Hypoxia Inducible Factor-1 Alpha Pathway in Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2019, 20, 157. https://doi.org/10.3390/ijms20010157
Hsu H-L, Liao P-L, Cheng Y-W, Huang S-H, Wu C-H, Li C-H, Kang J-J. Chloramphenicol Induces Autophagy and Inhibits the Hypoxia Inducible Factor-1 Alpha Pathway in Non-Small Cell Lung Cancer Cells. International Journal of Molecular Sciences. 2019; 20(1):157. https://doi.org/10.3390/ijms20010157
Chicago/Turabian StyleHsu, Han-Lin, Po-Lin Liao, Yu-Wen Cheng, Shih-Hsuan Huang, Chien-Hua Wu, Ching-Hao Li, and Jaw-Jou Kang. 2019. "Chloramphenicol Induces Autophagy and Inhibits the Hypoxia Inducible Factor-1 Alpha Pathway in Non-Small Cell Lung Cancer Cells" International Journal of Molecular Sciences 20, no. 1: 157. https://doi.org/10.3390/ijms20010157
APA StyleHsu, H.-L., Liao, P.-L., Cheng, Y.-W., Huang, S.-H., Wu, C.-H., Li, C.-H., & Kang, J.-J. (2019). Chloramphenicol Induces Autophagy and Inhibits the Hypoxia Inducible Factor-1 Alpha Pathway in Non-Small Cell Lung Cancer Cells. International Journal of Molecular Sciences, 20(1), 157. https://doi.org/10.3390/ijms20010157