Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte

{kind=link}
{kind=link}
Abstract
1. Introduction
2. STAT3 as a Conditional Oncogene
3. STAT3 Multifaceted Functions: How, Where, and When?
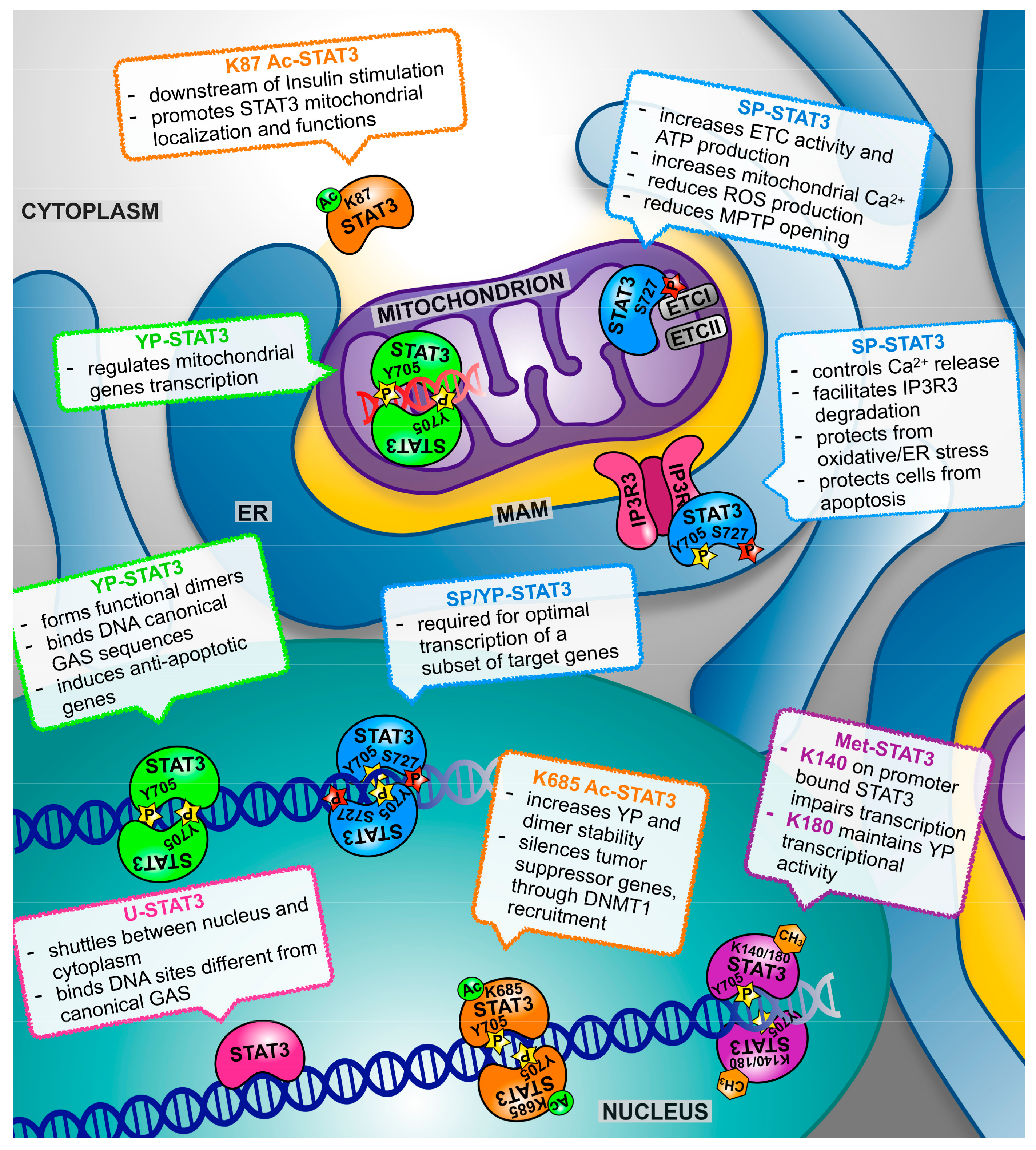
3.1. STAT3α and STAT3β
3.2. Tyrosine Phosphorylation
3.3. Serine Phosphorylation (SP)
3.4. Acetylation
3.5. Methylation
3.6. Oxidation and Glutathionylation
4. Both Nuclear and Mitochondrial STAT3 Affect Energy Metabolism and Cellular Respiration
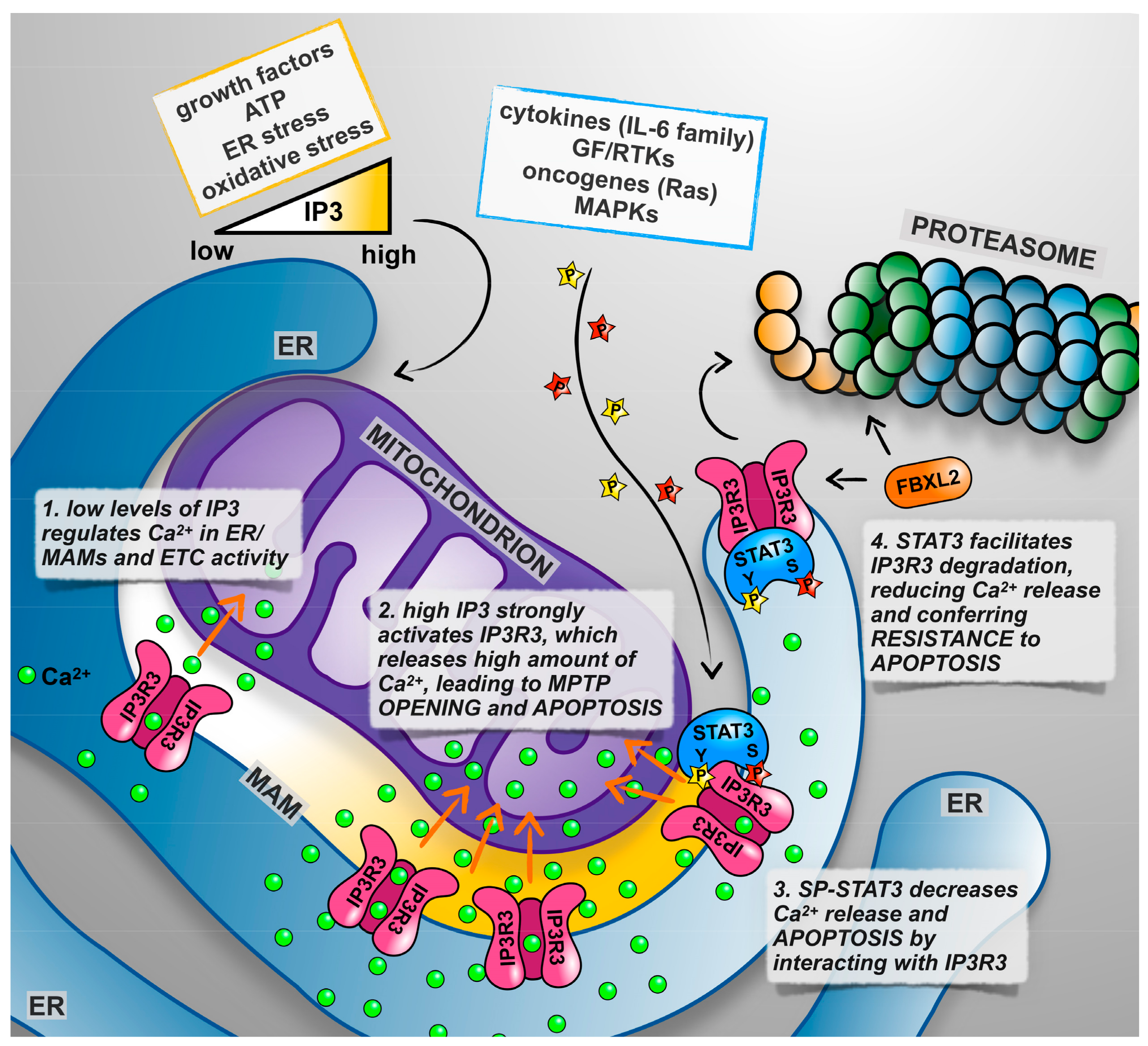
5. Not only Nucleus and Mitochondrion: The Endoplasmic Reticulum Takes Centre Stage
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| G-CSF | Granulocyte colony-stimulating factor |
| EGF | Epidermal growth factor |
| PDGF | Platelet-derived growth factor |
| K-RAS | Kirsten rat sarcoma viral oncogene homolog |
| TPA | 12-O-Tetradecanoylphorbol-13-acetate |
| DSS | Dextran Sodium Sulfate |
| DMBA | 7,12-Dimethylbenz[a]-anthracene |
| AOM | Azoxymethane |
| GAS | Gamma interferon activation site |
| FAK | Focal adhesion kinase |
| NAD | Nicotinamide adenine dinucleotide |
| ROS | Reactive oxygen species |
| IGF1 | Insulin-like growth factor 1 |
| HIF1α | Hypoxia-inducible factor 1, α subunit |
| LIF | Leukemia inhibitory factor |
| TFAM | Mitochondrial transcription factor A |
| H-RAS | Harvey rat sarcoma virus oncogene |
References
- Turkson, J.; Jove, R. Stat proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef] [PubMed]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.R.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Levy, D.E.; Decker, T. Jak-stat signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. Stats in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Dalton, W.S.; Jove, R. Stat proteins as novel targets for cancer therapy. Signal transducer an activator of transcription. Curr. Opin. Oncol. 1999, 11, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, F.; Niu, R. Multiple regulation pathways and pivotal biological functions of stat3 in cancer. Sci. Rep. 2015, 5, 17663. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting stat3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liang, X.; Kellendonk, C.; Poli, V.; Taub, R. Stat3 contributes to the mitogenic response of hepatocytes during liver regeneration. J. Biol. Chem. 2002, 277, 28411–28417. [Google Scholar] [CrossRef] [PubMed]
- Alonzi, T.; Maritano, D.; Gorgoni, B.; Rizzuto, G.; Libert, C.; Poli, V. Essential role of stat3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol. Cell. Biol. 2001, 21, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Fornek, J.L.; Tygrett, L.T.; Waldschmidt, T.J.; Poli, V.; Rickert, R.C.; Kansas, G.S. Critical role for stat3 in t-dependent terminal differentiation of igg b cells. Blood 2006, 107, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.J.; Grosso, J.F.; Yen, H.R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for stat3 signaling in th17 development and th17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mule, J.; Kerr, W.G.; et al. Inhibiting stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D. Hyperactivation of stat3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, M.; Ogura, H.; Ueda, N.; Tsuruoka, M.; Kitabayashi, C.; Tsuji, F.; Aono, H.; Ishihara, K.; Huseby, E.; Betz, U.A.; et al. Il-6-gp130-stat3 in t cells directs the development of il-17+ th with a minimum effect on that of treg in the steady state. Int. Immunol. 2007, 19, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Theurich, S.; Tsaousidou, E.; Hanssen, R.; Lempradl, A.M.; Mauer, J.; Timper, K.; Schilbach, K.; Folz-Donahue, K.; Heilinger, C.; Sexl, V.; et al. Il-6/stat3-dependent induction of a distinct, obesity-associated nk cell subpopulation deteriorates energy and glucose homeostasis. Cell Metab. 2017, 26, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Kreuzaler, P.A.; Staniszewska, A.D.; Li, W.; Omidvar, N.; Kedjouar, B.; Turkson, J.; Poli, V.; Flavell, R.A.; Clarkson, R.W.; Watson, C.J. Stat3 controls lysosomal-mediated cell death in vivo. Nat. Cell Biol. 2011, 13, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Fuchs, M.; Kaminski, K.; Schaefer, A.; Schieffer, B.; Hillmer, A.; Schmiedl, A.; Ding, Z.; Podewski, E.; et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004, 95, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. Stat3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Guadagnin, E.; Mazala, D.; Chen, Y.W. Stat3 in skeletal muscle function and disorders. Int. J. Mol. Sci. 2018, 19, 2265. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.; Watson, C.J. The multifaceted role of stat3 in mammary gland involution and breast cancer. Int. J. Mol. Sci. 2018, 19, 1695. [Google Scholar] [CrossRef] [PubMed]
- Kasembeli, M.M.; Bharadwaj, U.; Robinson, P.; Tweardy, D.J. Contribution of stat3 to inflammatory and fibrotic diseases and prospects for its targeting for treatment. Int. J. Mol. Sci. 2018, 19, 2299. [Google Scholar] [CrossRef] [PubMed]
- Laudisi, F.; Cherubini, F.; Monteleone, G.; Stolfi, C. Stat3 interactors as potential therapeutic targets for cancer treatment. Int. J. Mol. Sci. 2018, 19, 1787. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M.; Pereira, F.V. A new perspective: Mitochondrial stat3 as a regulator for lymphocyte function. Int. J. Mol. Sci 2018, 19, 1656. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.L.; Banerjee, S.; White, S.V.; Kortylewski, M. Stat3 in tumor-associated myeloid cells: Multitasking to disrupt immunity. Int. J. Mol. Sci. 2018, 19, 1803. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the jak/stat pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. Socs: Physiological suppressors of cytokine signaling. J. Cell Sci. 2000, 113, 2813–2819. [Google Scholar] [PubMed]
- Shuai, K. Modulation of stat signaling by stat-interacting proteins. Oncogene 2000, 19, 2638–2644. [Google Scholar] [CrossRef] [PubMed]
- Krebs, D.L.; Hilton, D.J. Socs proteins: Negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of stat3 signal transduction by pias3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef] [PubMed]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; Leblanc, F.; Peng Ng, K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. Stat3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of nk cells and t-cell large granular lymphocyte leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Ma, L.; Merker, J.D.; Martinez, B.; Zehnder, J.L.; Arber, D.A. Stat3 mutations are frequent in cd30+ t-cell lymphomas and t-cell large granular lymphocytic leukemia. Leukemia 2013, 27, 2244–2247. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Ma, L.; Monabati, A.; Zehnder, J.L.; Arber, D.A. Stat3 mutations are present in aggressive b-cell lymphomas including a subset of diffuse large b-cell lymphomas with cd30 expression. Haematologica 2014, 99, e105–107. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial stat3 supports ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.M.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Regis, G.; Poli, V. Universal and Specific Functions of Stat3 in Solid Tumors. In Jak-Stat Signaling: From Basics to Disease; Springer: Wien, Austria, 2012; pp. 305–333. [Google Scholar]
- Demaria, M.; Camporeale, A.; Poli, V. Stat3 and metabolism: How many ways to use a single molecule? Int. J. Cancer 2014, 135, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Laklai, H.; Miroshnikova, Y.A.; Pickup, M.W.; Collisson, E.A.; Kim, G.E.; Barrett, A.S.; Hill, R.C.; Lakins, J.N.; Schlaepfer, D.D.; Mouw, J.K.; et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 2016, 22, 497–505. [Google Scholar] [CrossRef] [PubMed]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; Bonni, A. Identification of a pten-regulated stat3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Schlederer, M.; Gruber, W.; Unger, C.; Walker, S.M.; Chalaris, A.; Marié, I.J.; Hassler, M.R.; Javaheri, T.; Aksoy, O.; et al. Stat3 regulated arf expression suppresses prostate cancer metastasis. Nat. Commun. 2015, 6, 7736. [Google Scholar] [CrossRef] [PubMed]
- Grabner, B.; Schramek, D.; Mueller, K.M.; Moll, H.P.; Svinka, J.; Hoffmann, T.; Bauer, E.; Blaas, L.; Hruschka, N.; Zboray, K.; et al. Disruption of stat3 signalling promotes kras-induced lung tumorigenesis. Nat. Commun. 2015, 6, 6285. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. Il-6 and stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, O.; Schwitalla, S.; et al. Gp130-mediated stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 is a negative regulator of intestinal tumor progression in apcmin mice. Gastroenterology 2010, 138, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, J.C.K.; Lee, S.E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (stat3) protein suppresses adenoma-to-carcinoma transition in apcmin/+ mice via regulation of snail-1 (snai) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar] [CrossRef] [PubMed]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. Stat3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Vallania, F.; Schiavone, D.; Dewilde, S.; Pupo, E.; Garbay, S.; Calogero, R.; Pontoglio, M.; Provero, P.; Poli, V. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: The case of stat3. Proc. Natl. Acad. Sci. USA 2009, 106, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.E.; Frank, D.A. Stat3-interacting proteins as modulators of transcription factor function: Implications to targeted cancer therapy. ChemMedChem 2016, 11, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Huso, D.L.; Nathans, D.; Desiderio, S. Specific ablation of stat3β distorts the pattern of stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell 2002, 108, 331–344. [Google Scholar] [CrossRef]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.; Murray-Tait, V.; Chiarle, R.; Poli, V. The stat3 isoforms α and β have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Dewilde, S.; Vercelli, A.; Chiarle, R.; Poli, V. Of αs and βs: Distinct and overlapping functions of stat3 isoforms. Front. Biosci. 2008, 13, 6501–6514. [Google Scholar] [CrossRef] [PubMed]
- Caldenhoven, E.; van Dijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; de Groot, R.P. Stat3β, a splice variant of transcription factor stat3, is a dominant negative regulator of transcription. J. Biol. Chem. 1996, 271, 13221–13227. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, T.S.; Sanders, L.K.; Nathans, D. Cooperative transcriptional activity of jun and stat3 β, a short form of stat3. Proc. Natl. Acad. Sci. USA 1995, 92, 9097–9101. [Google Scholar] [CrossRef] [PubMed]
- Aigner, P.; Just, V.; Stoiber, D. Stat3 isoforms: Alternative fates in cancer? Cytokine 2018. [Google Scholar] [CrossRef] [PubMed]
- Ecker, A.; Simma, O.; Hoelbl, A.; Kenner, L.; Beug, H.; Moriggl, R.; Sexl, V. The dark and the bright side of stat3: Proto-oncogene and tumor-suppressor. Front. Biosci. (Landmark Ed.) 2009, 14, 2944–2958. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Baldwin, W.M., III; Lee, C.Y.; Desiderio, S. Stat3β mitigates development of atherosclerosis in apolipoprotein e-deficient mice. J. Mol. Med. (Berl.) 2013, 91, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Marino, F.; Orecchia, V.; Regis, G.; Musteanu, M.; Tassone, B.; Jon, C.; Forni, M.; Calautti, E.; Chiarle, R.; Eferl, R.; et al. Stat3β controls inflammatory responses and early tumor onset in skin and colon experimental cancer models. Am. J. Cancer Res. 2014, 4, 484–494. [Google Scholar] [PubMed]
- Cimica, V.; Chen, H.C.; Iyer, J.K.; Reich, N.C. Dynamics of the stat3 transcription factor: Nuclear import dependent on ran and importin-β1. PLoS ONE 2011, 6, e20188. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated stat3 accumulates in response to il-6 and activates transcription by binding to nf b. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, O.A.; Chasovskikh, S.; Lonskaya, I.; Tarasova, N.I.; Khavrutskii, L.; Tarasov, S.G.; Zhang, X.; Korostyshevskiy, V.R.; Cheema, A.; Zhang, L.; et al. Mechanisms of unphosphorylated stat3 transcription factor binding to DNA. J. Biol. Chem. 2012, 287, 14192–14200. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, A.; Kugimiya, N.; Hosoyama, T.; Enoki, T.; Li, T.S.; Hamano, K. Jab1 regulates unphosphorylated stat3 DNA-binding activity through protein-protein interaction in human colon cancer cells. Biochem. Biophys. Res. Commun. 2013, 438, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Lopes, M.H.; Bleggi-Torres, L.F.; Suzuki, S.; Cunha, I.W.; Sanematsu, P.; Martins, V.R. Nuclear unphosphorylated stat3 correlates with a worse prognosis in human glioblastoma. Pathol. Res. Pract. 2016, 212, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Inghirami, G. Stat3: A multifaceted oncogene. Proc. Natl. Acad. Sci. USA 2006, 103, 10151–10152. [Google Scholar] [CrossRef] [PubMed]
- Silver, D.L.; Naora, H.; Liu, J.; Cheng, W.; Montell, D.J. Activated signal transducer and activator of transcription (stat) 3: Localization in focal adhesions and function in ovarian cancer cell motility. Cancer Res. 2004, 64, 3550–3558. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.C.; Lin, B.H.; Lim, C.P.; Huang, G.; Zhang, T.; Poli, V.; Cao, X. Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell. Biol. 2006, 172, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Marino, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic stat3 represses autophagy by inhibiting pkr activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Niso-Santano, M.; Shen, S.; Adjemian, S.; Malik, S.A.; Marino, G.; Lachkar, S.; Senovilla, L.; Kepp, O.; Galluzzi, L.; Maiuri, M.C.; et al. Direct interaction between stat3 and eif2ak2 controls fatty acid-induced autophagy. Autophagy 2013, 9, 415–417. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Uchida, E.; Grammer, T.C.; Blenis, J. Stat3 serine phosphorylation by erk-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 1997, 17, 6508–6516. [Google Scholar] [CrossRef] [PubMed]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of stat3 during cntf signaling is mediated by the rapamycin target mtor. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef]
- Aznar, S.; Valeron, P.F.; del Rincon, S.V.; Perez, L.F.; Perona, R.; Lacal, J.C. Simultaneous tyrosine and serine phosphorylation of stat3 transcription factor is involved in rho a gtpase oncogenic transformation. Mol. Biol. Cell 2001, 12, 3282–3294. [Google Scholar] [CrossRef] [PubMed]
- Garama, D.J.; White, C.L.; Balic, J.J.; Gough, D.J. Mitochondrial stat3: Powering up a potent factor. Cytokine 2016, 87, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Guan, Y.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Zhang, P.; Herrmann, A.; Yang, C.; Xin, H.; Wang, Z.; Hoon, D.S.B.; Forman, S.J.; Jove, R.; Riggs, A.D.; et al. Acetylated stat3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA 2012, 109, 7765–7769. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. Stat3 undergoes acetylation-dependent mitochondrial translocation to regulate pyruvate metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. Stat3 inhibition of gluconeogenesis is downregulated by sirt1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative regulation of stat3 protein-mediated cellular respiration by sirt1 protein. J. Biol. Chem. 2011, 286, 19270–19279. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound stat3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, M.; Woo, D.H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of ezh2 activates stat3 signaling via stat3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Booz, G.W. Evidence that il-6-type cytokine signaling in cardiomyocytes is inhibited by oxidative stress: Parthenolide targets jak1 activation by generating ros. J. Cell. Physiol. 2007, 212, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kole, S.; Precht, P.; Pazin, M.J.; Bernier, M. S-glutathionylation impairs signal transducer and activator of transcription 3 activation and signaling. Endocrinology 2009, 150, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cheung, S.H.; Evans, E.L.; Shaw, P.E. Modulation of gene expression and tumor cell growth by redox modification of stat3. Cancer Res. 2010, 70, 8222–8232. [Google Scholar] [CrossRef] [PubMed]
- Sobotta, M.C.; Liou, W.; Stocker, S.; Talwar, D.; Oehler, M.; Ruppert, T.; Scharf, A.N.; Dick, T.P. Peroxiredoxin-2 and stat3 form a redox relay for h2o2 signaling. Nat. Chem. Biol. 2015, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Simon, A.R.; Rai, U.; Fanburg, B.L.; Cochran, B.H. Activation of the jak-stat pathway by reactive oxygen species. Am. J. Physiol. 1998, 275, C1640–C1652. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Edderkaoui, M.; Truong, P.; Ohno, I.; Jang, K.T.; Berti, A.; Pandol, S.J.; Gukovskaya, A.S. Nadph oxidase promotes pancreatic cancer cell survival via inhibiting jak2 dephosphorylation by tyrosine phosphatases. Gastroenterology 2007, 133, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Choi, M.J.; Jeong, K.J.; Kim, J.J.; Hwang, M.H.; Shin, S.C.; Park, C.G.; Lee, H.Y. A ros/stat3/hif-1α signaling cascade mediates egf-induced twist1 expression and prostate cancer cell invasion. Prostate 2014, 74, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.; Bak, Y.; Park, Y.H.; Jang, G.B.; Nam, J.S.; Yoo, J.E.; Park, Y.N.; Bak, I.S.; Kim, J.M.; Yoon, D.Y.; et al. Peroxiredoxin ii is essential for maintaining stemness by redox regulation in liver cancer cells. Stem Cells 2016, 34, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Linher-Melville, K.; Singh, G. The complex roles of stat3 and stat5 in maintaining redox balance: Lessons from stat-mediated xct expression in cancer cells. Mol. Cell. Endocrinol. 2017, 451, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Tumor metabolism: Cancer cells give and take lactate. J. Clin. Investig. 2008, 118, 3835–3837. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A stat3-mediated metabolic switch is involved in tumour transformation and stat3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Misale, S.; Giorgi, C.; Miano, V.; Camporeale, A.; Campisi, J.; Pinton, P.; Poli, V. Stat3 can serve as a hit in the process of malignant transformation of primary cells. Cell Death Differ. 2012, 19, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Sarafian, T.A.; Montes, C.; Imura, T.; Qi, J.; Coppola, G.; Geschwind, D.H.; Sofroniew, M.V. Disruption of astrocyte stat3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS ONE 2010, 5, e9532. [Google Scholar] [CrossRef] [PubMed]
- Mantel, C.; Messina-Graham, S.; Moh, A.; Cooper, S.; Hangoc, G.; Fu, X.Y.; Broxmeyer, H.E. Mouse hematopoietic cell-targeted stat3 deletion: Stem/progenitor cell defects, mitochondrial dysfunction, ros overproduction, and a rapid aging-like phenotype. Blood 2012, 120, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Lirussi, D.; Thornton, T.M.; Jelley-Gibbs, D.M.; Diehl, S.A.; Case, L.K.; Madesh, M.; Taatjes, D.J.; Teuscher, C.; Haynes, L.; et al. Mitochondrial ca(2)(+) and membrane potential, an alternative pathway for interleukin 6 to regulate cd4 cell effector function. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor stat3 into mitochondria depends on grim-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Lizano, P.; Laure, L.; Sui, X.; Rashed, E.; Park, J.Y.; Hong, C.; Gao, S.; Holle, E.; Morin, D.; et al. H11 kinase/heat shock protein 22 deletion impairs both nuclear and mitochondrial functions of stat3 and accelerates the transition into heart failure on cardiac overload. Circulation 2011, 124, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial stat3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Marie, I.J.; Lobry, C.; Aifantis, I.; Levy, D.E. Stat3 supports experimental k-rasg12d-induced murine myeloproliferative neoplasms dependent on serine phosphorylation. Blood 2014, 124, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Szczepanek, K.; Chen, Q.; Derecka, M.; Salloum, F.N.; Zhang, Q.; Szelag, M.; Cichy, J.; Kukreja, R.C.; Dulak, J.; Lesnefsky, E.J.; et al. Mitochondrial-targeted signal transducer and activator of transcription 3 (stat3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 2011, 286, 29610–29620. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., III. The expression of the receptor for advanced glycation endproducts (rage) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef] [PubMed]
- Carbognin, E.; Betto, R.M.; Soriano, M.E.; Smith, A.G.; Martello, G. Stat3 promotes mitochondrial transcription and oxidative respiration during maintenance and induction of naive pluripotency. EMBO J. 2016, 35, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Garama, D.J.; Harris, T.J.; White, C.L.; Rossello, F.J.; Abdul-Hay, M.; Gough, D.J.; Levy, D.E. A synthetic lethal interaction between glutathione synthesis and mitochondrial reactive oxygen species provides a tumor-specific vulnerability dependent on stat3. Mol. Cell. Biol. 2015, 35, 3646–3656. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Koetz, L.; Levy, D.E. The mek-erk pathway is necessary for serine phosphorylation of mitochondrial stat3 and ras-mediated transformation. PLoS ONE 2013, 8, e83395. [Google Scholar] [CrossRef]
- Macias, E.; Rao, D.; Carbajal, S.; Kiguchi, K.; DiGiovanni, J. Stat3 binds to mtdna and regulates mitochondrial gene expression in keratinocytes. J. Investig. Dermatol. 2014, 134, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Chong, S.J.; Ooi, V.Z.; Vali, S.; Kumar, A.; Kapoor, S.; Abbasi, T.; Hirpara, J.L.; Loh, T.; Goh, B.C.; et al. Overexpression of bcl-2 induces stat-3 activation via an increase in mitochondrial superoxide. Oncotarget 2015, 6, 34191–34205. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Huang, L.; Alston, N.; Ouyang, N.; Vrankova, K.; Mattheolabakis, G.; Constantinides, P.P.; Rigas, B. Targeting mitochondrial stat3 with the novel phospho-valproic acid (mdc-1112) inhibits pancreatic cancer growth in mice. PLoS ONE 2013, 8, e61532. [Google Scholar] [CrossRef] [PubMed]
- Tateno, T.; Asa, S.L.; Zheng, L.; Mayr, T.; Ullrich, A.; Ezzat, S. The fgfr4-g388r polymorphism promotes mitochondrial stat3 serine phosphorylation to facilitate pituitary growth hormone cell tumorigenesis. PLoS Genet. 2011, 7, e1002400. [Google Scholar] [CrossRef] [PubMed]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (mams). Biochim. Biophys. Acta 2017, 1858, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Mak, D.O.; Foskett, J.K. Inositol 1,4,5-trisphosphate receptors in the endoplasmic reticulum: A single-channel point of view. Cell Calcium 2015, 58, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, M.; Parys, J.B.; Pinton, P.; Bultynck, G. Er functions of oncogenes and tumor suppressors: Modulators of intracellular ca(2+) signaling. Biochim. Biophys. Acta 2016, 1863, 1364–1378. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial permeability transition involves dissociation of f1fo atp synthase dimers and c-ring conformation. EMBO Rep. 2017, 18, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Mendes, C.C.; Gomes, D.A.; Thompson, M.; Souto, N.C.; Goes, T.S.; Goes, A.M.; Rodrigues, M.A.; Gomez, M.V.; Nathanson, M.H.; Leite, M.F. The type iii inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic ca2+ signals into mitochondria. J. Biol. Chem. 2005, 280, 40892–40900. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Ito, K.; Lin, H.K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R.; et al. Pml regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Bonora, M.; Marchi, S.; Missiroli, S.; Poletti, F.; Giorgi, C.; Pandolfi, P.P.; Pinton, P. Identification of pten at the er and mams and its regulation of ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 2013, 20, 1631–1643. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. Bap1 regulates ip3r3-mediated ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. Pten counteracts fbxl2 to promote ip3r3- and ca2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Morciano, G.; Caroccia, N.; Ghetti, E.; Orecchia, V.; Viavattene, D.; Giorgi, C.; Pinton, P.; Poli, V. Stat3 localizes to the er, acting as a gatekeeper for er-mitochondrion ca(2+) fluxes and apoptotic responses. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int. J. Mol. Sci. 2018, 19, 2820. https://doi.org/10.3390/ijms19092820
Avalle L, Poli V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. International Journal of Molecular Sciences. 2018; 19(9):2820. https://doi.org/10.3390/ijms19092820
Chicago/Turabian StyleAvalle, Lidia, and Valeria Poli. 2018. "Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte" International Journal of Molecular Sciences 19, no. 9: 2820. https://doi.org/10.3390/ijms19092820
APA StyleAvalle, L., & Poli, V. (2018). Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. International Journal of Molecular Sciences, 19(9), 2820. https://doi.org/10.3390/ijms19092820