The Dynamic Changes of Gut Microbiota in Muc2 Deficient Mice


Abstract
1. Introduction
2. Results
2.1. Histopathology of the Muc2 Mouse Models
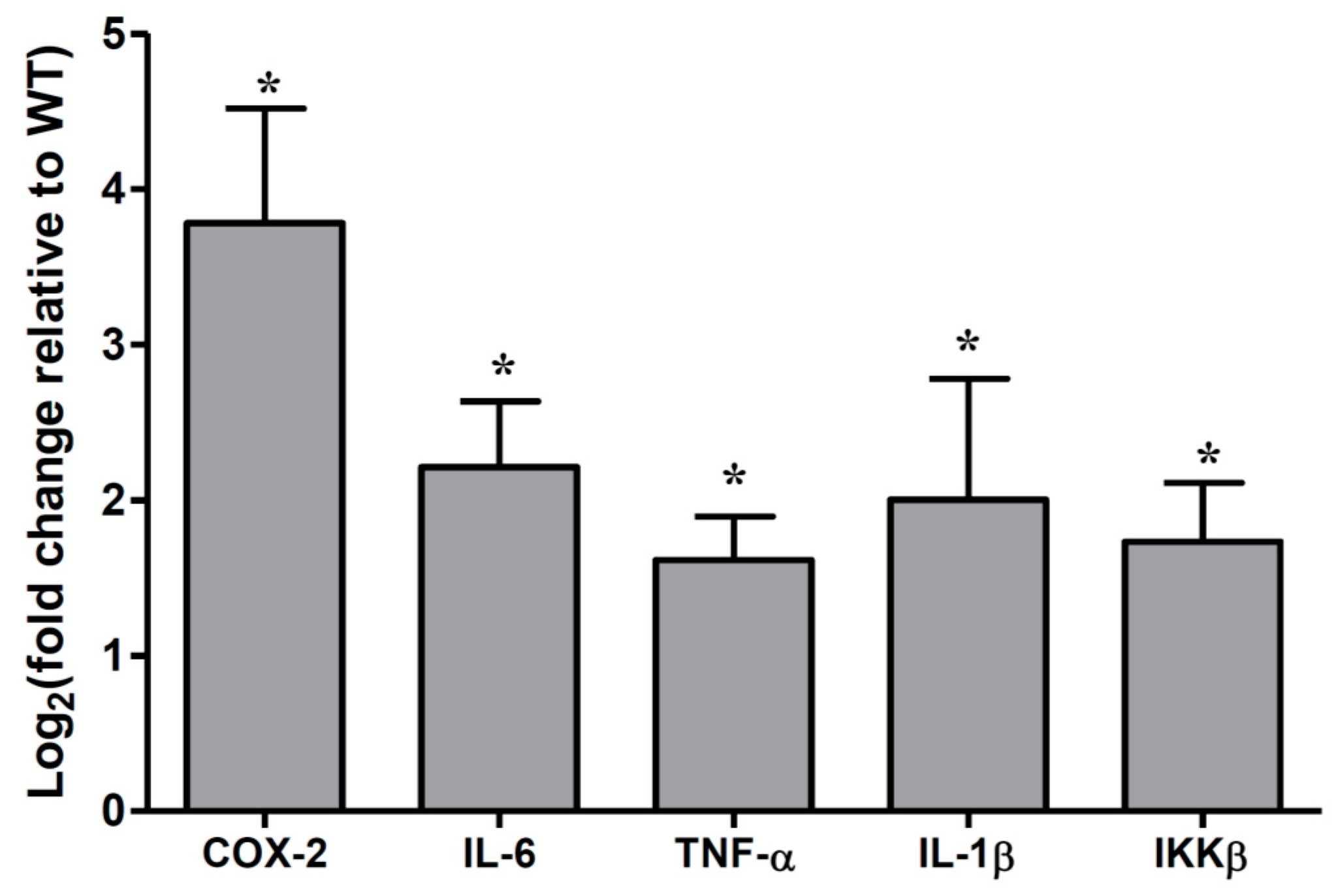
2.2. Cytokines Abundances in the Muc2 Mouse Colonic Epithelial Cells
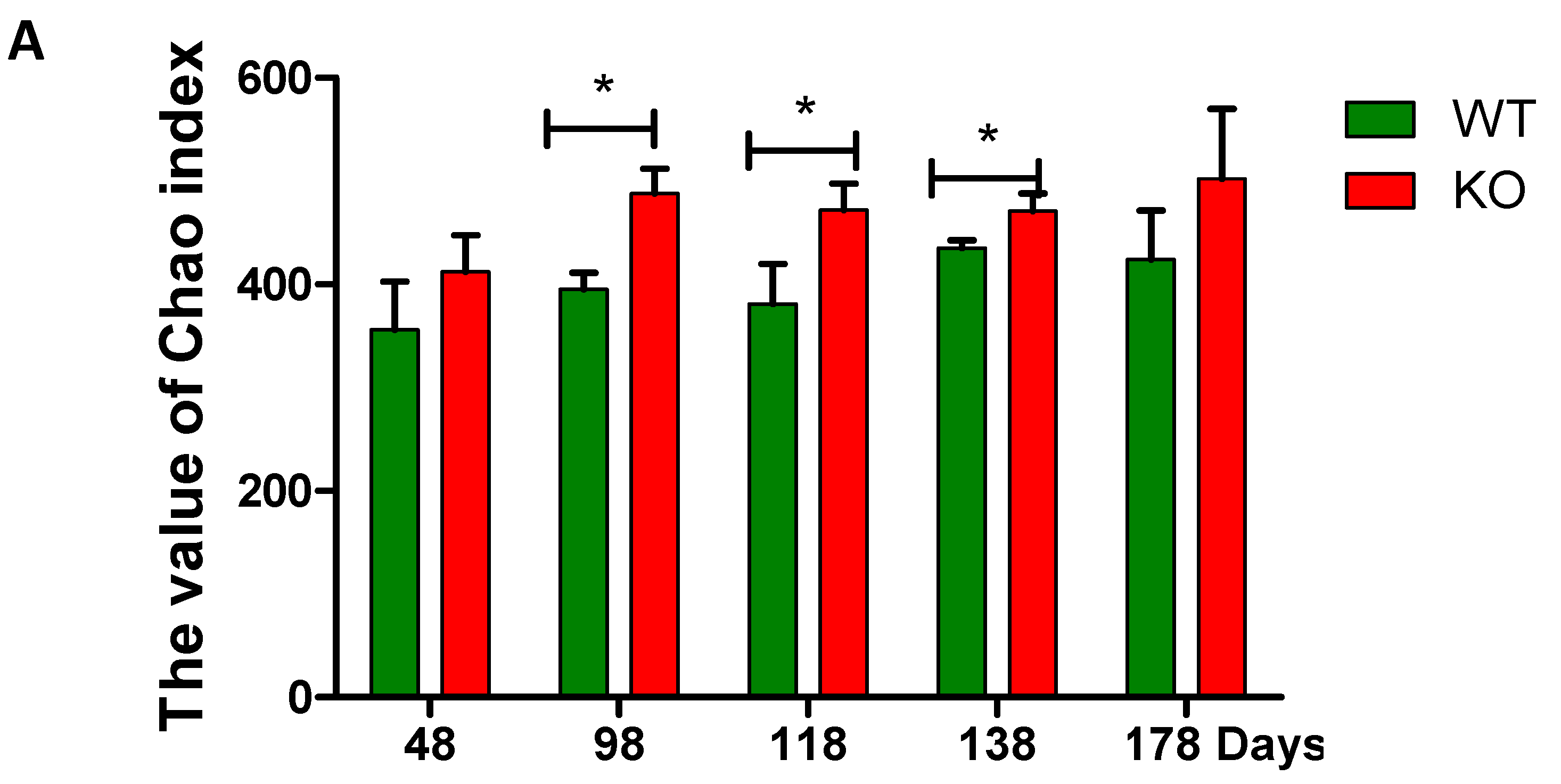
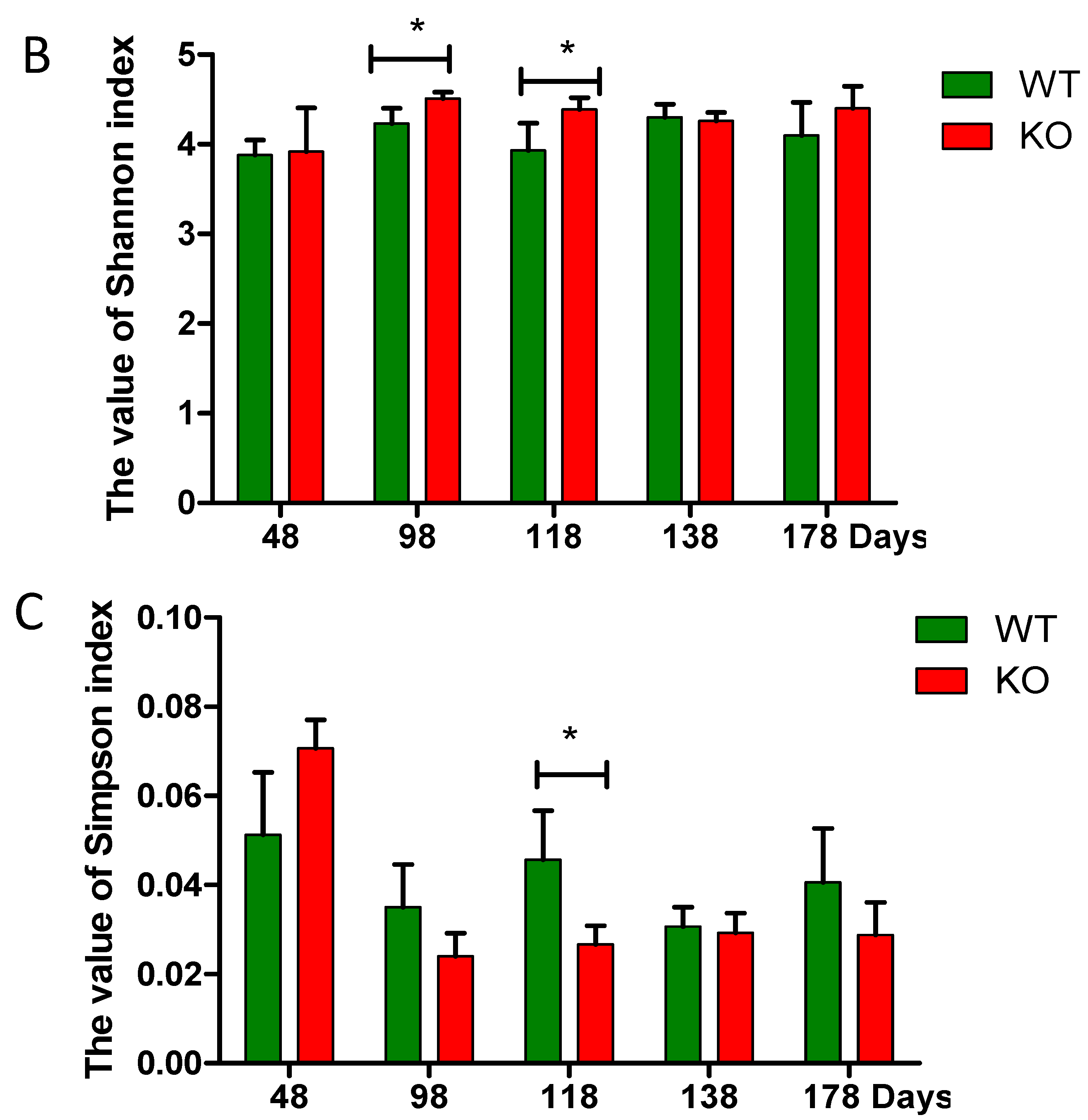
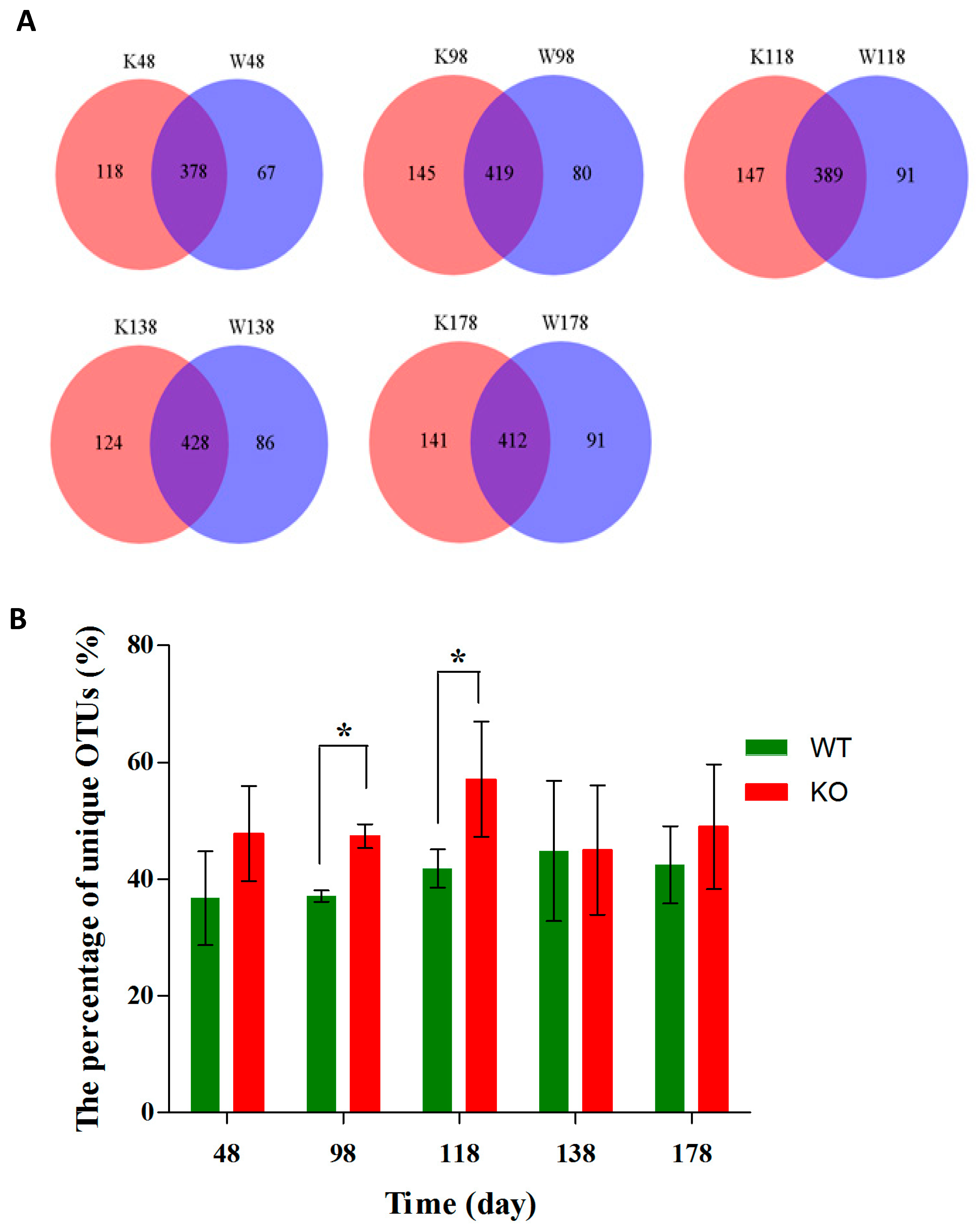
2.3. Dynamic Changes of the Diversity Index in Muc2 Mouse Models
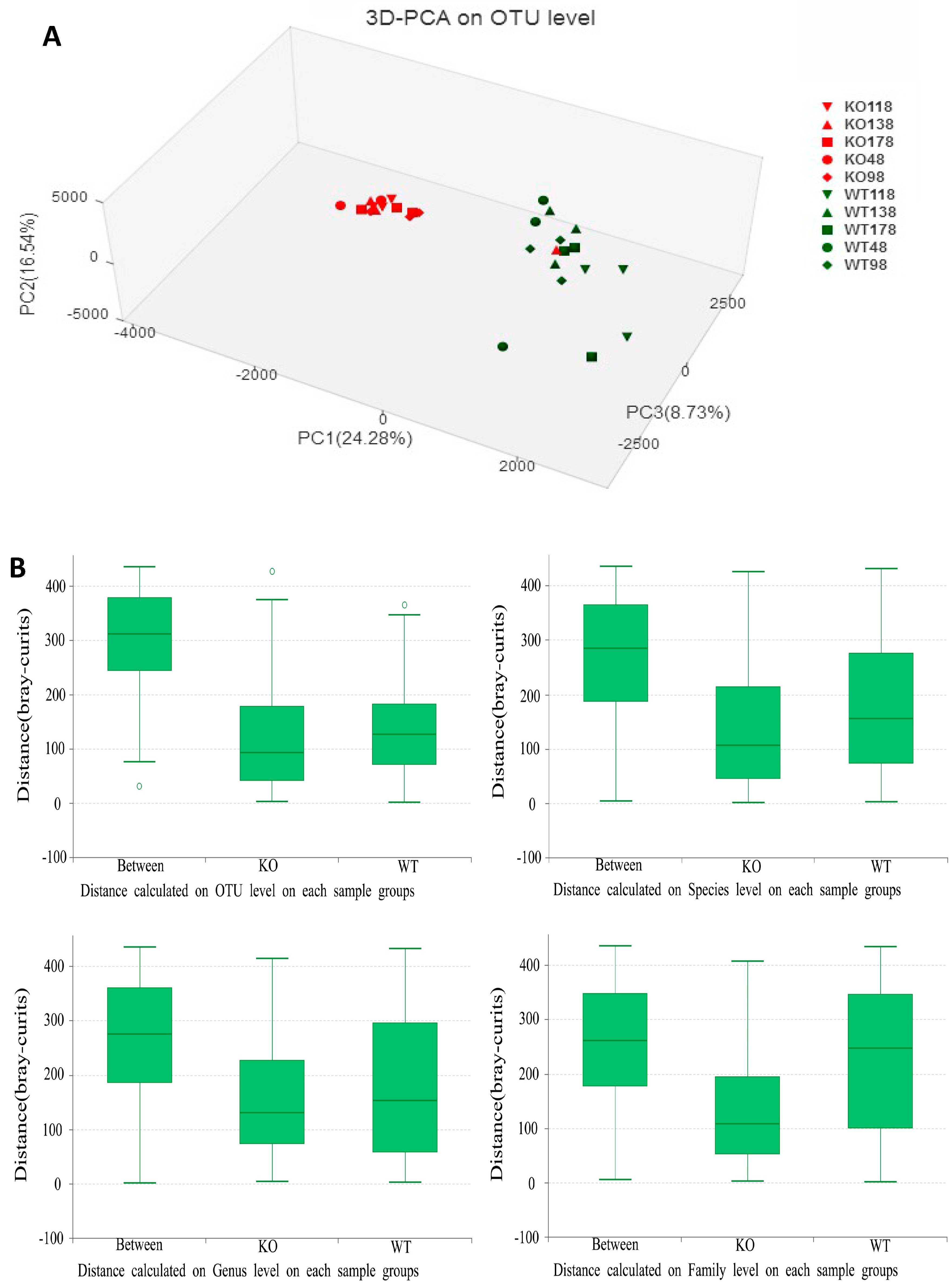
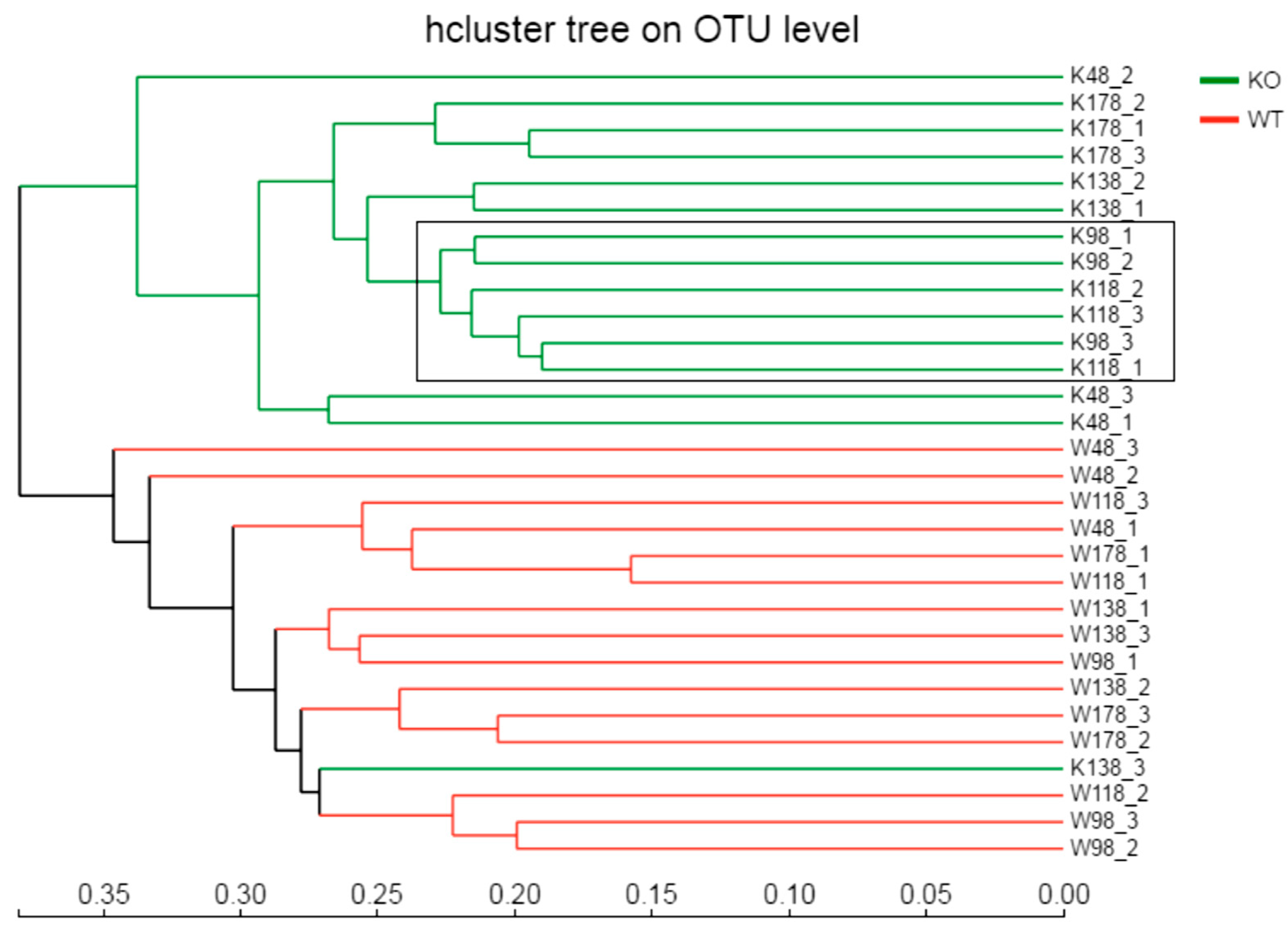
2.4. Clustering of Bacterial Communities in Muc2−/− Mice
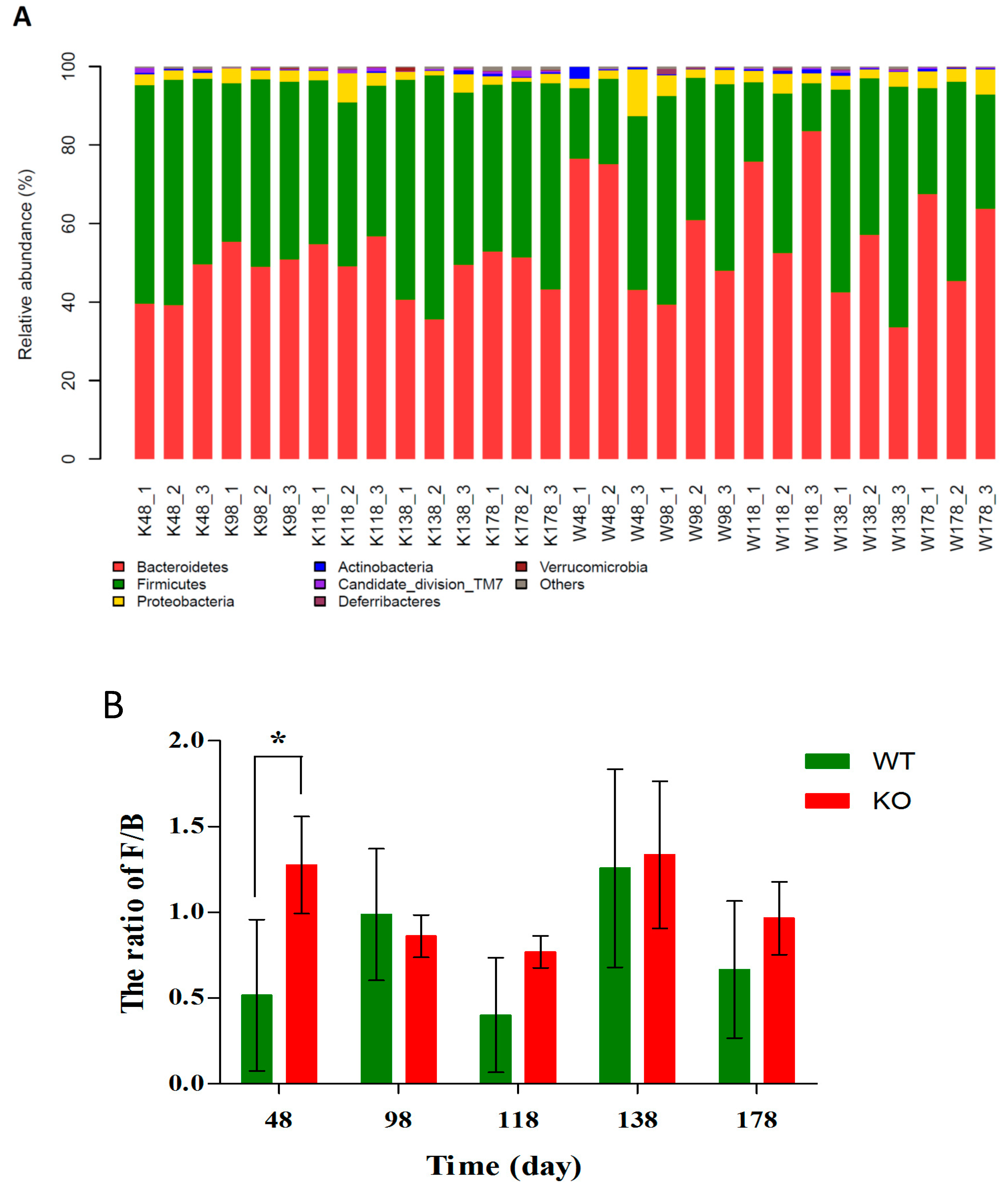
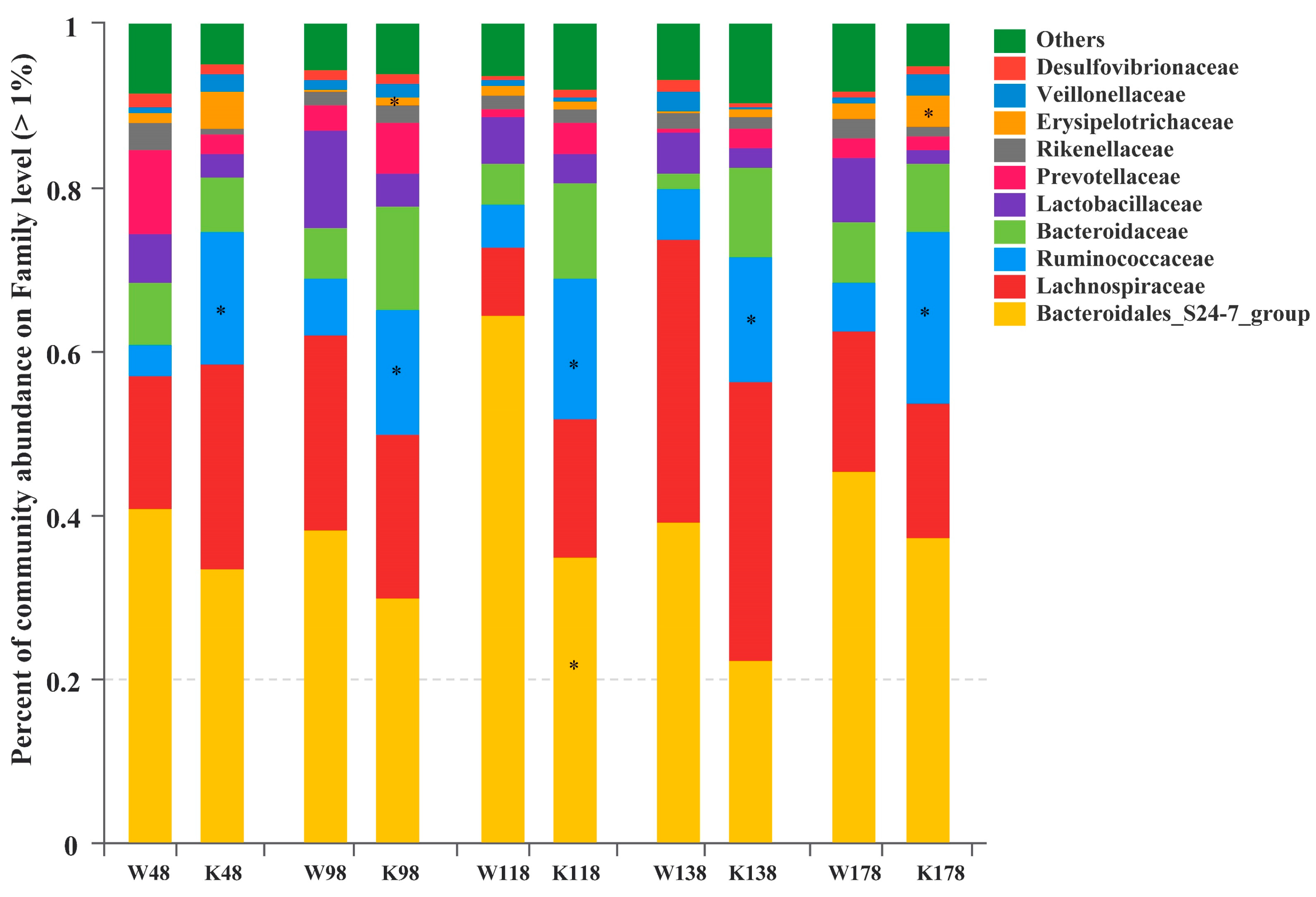
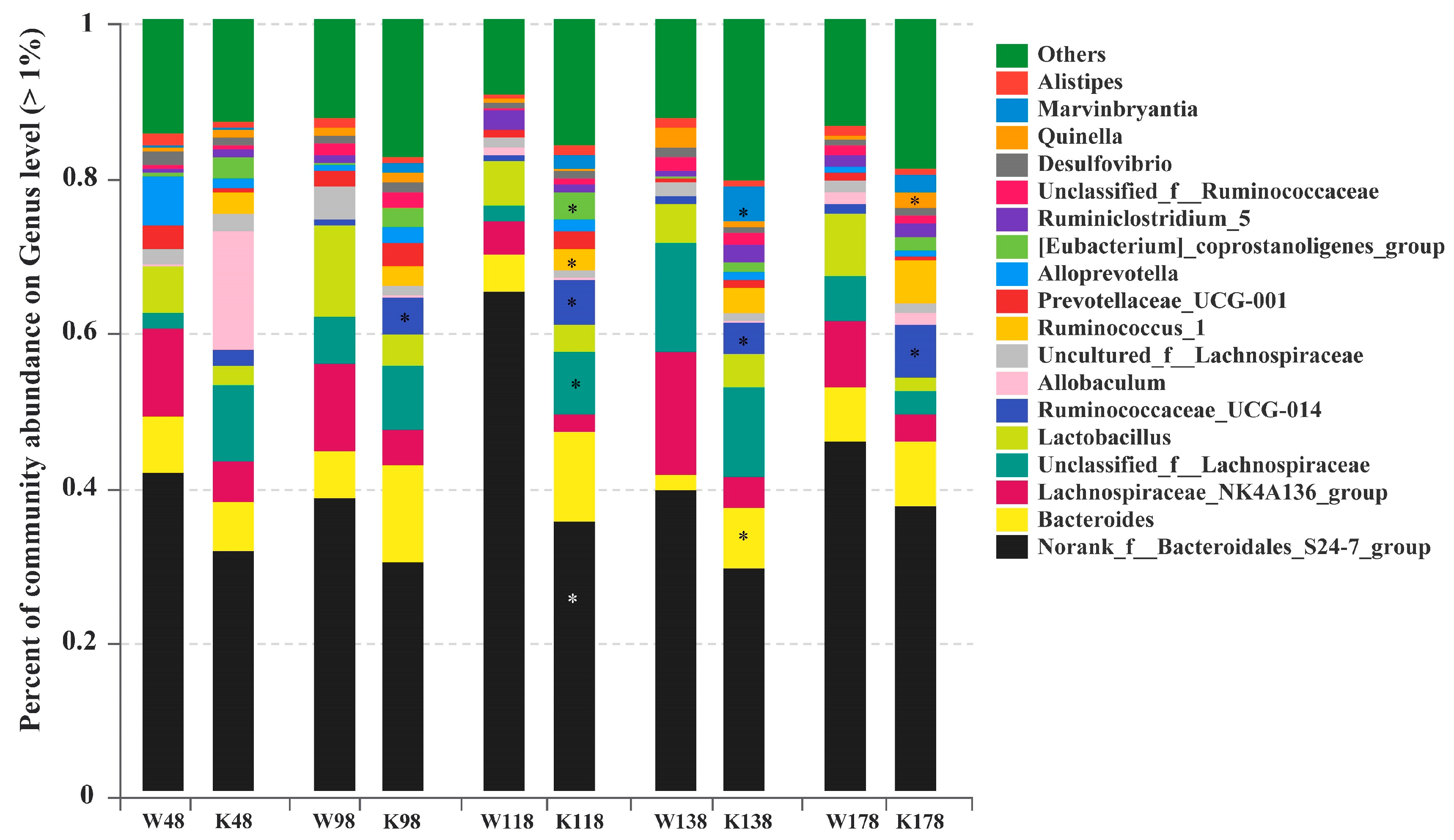
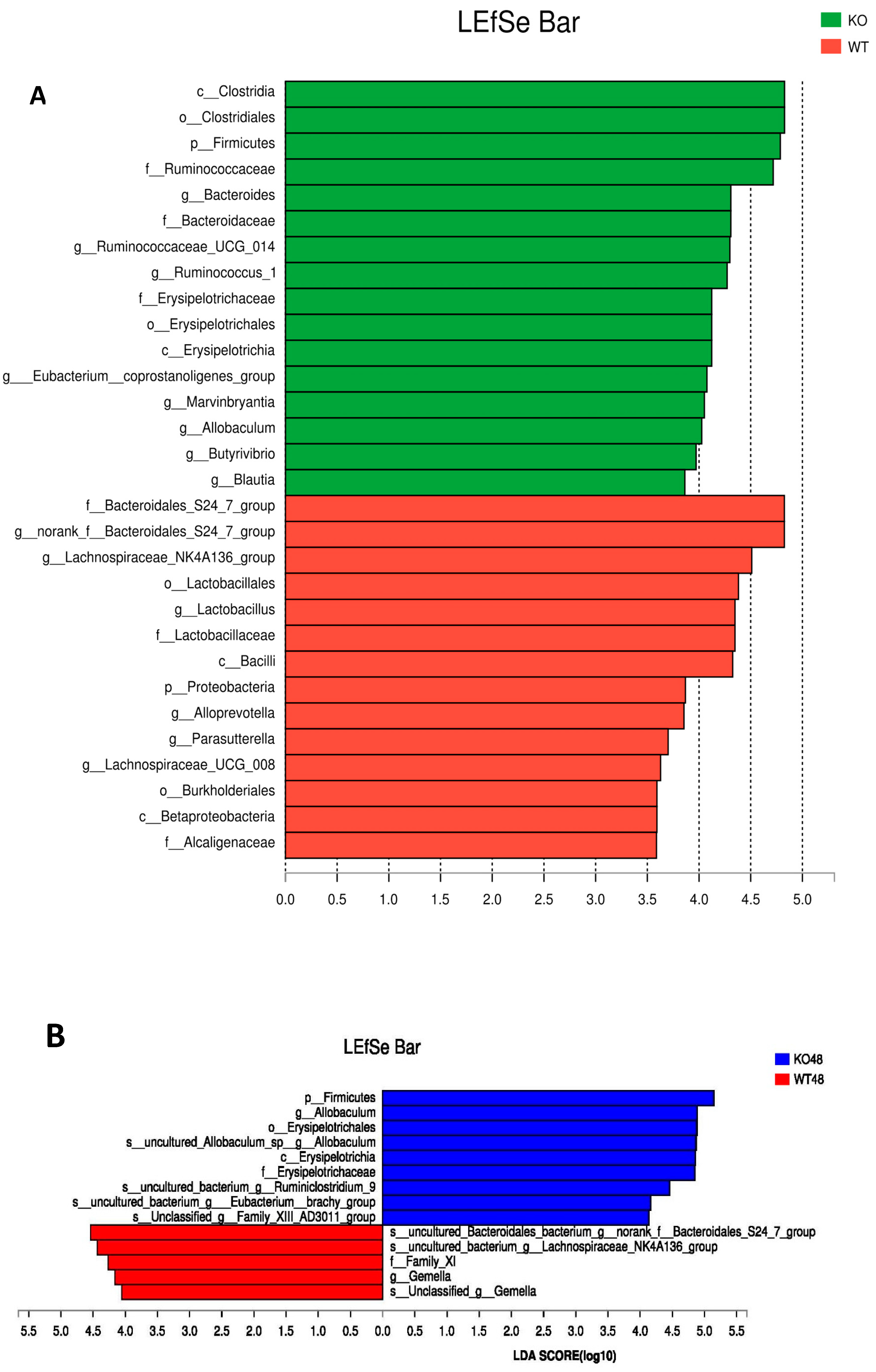
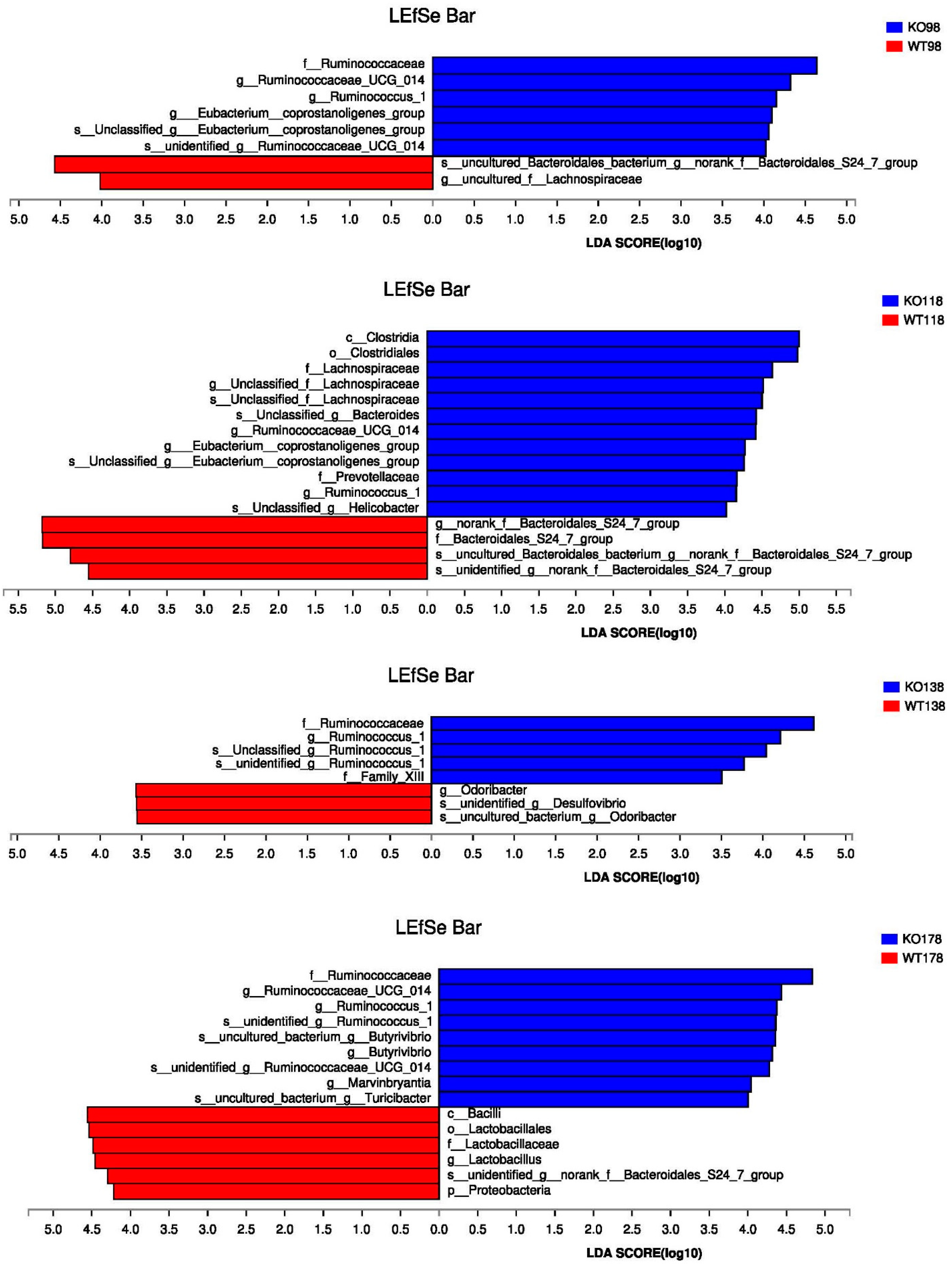
2.5. Changes of Gut Microbiota at Various Levels in Muc2 Mice
3. Discussion
4. Materials and Methods
4.1. Mouse Models, Fecal Samples Collection, and Histopathology
4.2. The Analysis of Cytokines in Colonic Epithelial Cells by Quantitative RT-PCR
4.3. High-throughput Sequencing and Bioinformatics Analysis
4.4. Statistical Analysis
4.5. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.K.; Weiderpass, E. Infection and cancer: Global distribution and burden of diseases. Ann. Glob. Health 2014, 80, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.; Trinchieri, G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat. Rev. Immunol. 2011, 11, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Martin, A. Microbiome and colorectal cancer: Unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin. Immunol. 2017, 32, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Nowarski, R.; Thaiss, C.A.; Hu, B.; Jin, C.; Flavell, R.A. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Hold, G.L.; Garrett, W.S. Gut microbiota. Microbiota organization—A key to understanding CRC development. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Weng, W.; Peng, J.; Hong, L.; Yang, L.; Toiyama, Y.; Gao, R.; Liu, M.; Yin, M.; Pan, C.; et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-kappaB, and up-regulating expression of microRNA-21. Gastroenterology 2017, 152, 851–866. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Li, H.; Tian, G.; Li, S. Dynamic microbe and molecule networks in a mouse model of colitis-associated colorectal cancer. Sci. Rep. 2014, 4, 4985. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Jin, Z.; Wu, W.; Gao, R.; Guo, B.; Gao, Z.; Yang, Y.; Qin, H. Analysis of the intestinal lumen microbiota in an animal model of colorectal cancer. PLoS ONE 2014, 9, e90849. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Jesus, E.C.; Lopes, A.; Aguiar, S., Jr.; Begnami, M.D.; Rocha, R.M.; Carpinetti, P.A.; Camargo, A.A.; Hoffmann, C.; Freitas, H.C.; et al. Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling. Front. Cell. Infect. Microbiol. 2016, 6, 179. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Lynch, D.B.; Brown, J.M.; Jeffery, I.B.; Ryan, F.J.; Claesson, M.J.; O’Riordain, M.; Shanahan, F.; O’Toole, P.W. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut 2017, 66, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Dharmani, P.; Leung, P.; Chadee, K. Tumor necrosis factor-alpha and Muc2 mucin play major roles in disease onset and progression in dextran sodium sulphate-induced colitis. PLoS ONE 2011, 6, e25058. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Guo, Y.; Li, Z.; Fang, W.; Yang, Y.; Li, X.; Li, Z.; Xiong, B.; Chen, Z.; Wang, J.; et al. MicroRNA profiling in Muc2 knockout mice of colitis-associated cancer model reveals epigenetic alterations during chronic colitis malignant transformation. PLoS ONE 2014, 9, e99132. [Google Scholar] [CrossRef] [PubMed]
- Velcich, A.; Yang, W.; Heyer, J.; Fragale, A.; Nicholas, C.; Viani, S.; Kucherlapati, R.; Lipkin, M.; Yang, K.; Augenlicht, L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 2002, 295, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Van, A.P.; Van, W.T.; Verstraete, W.; Possemiers, S. The host selects mucosal and luminal associations of coevolved gut microorganisms: A novel concept. FEMS Microbiol. Rev. 2011, 35, 681–704. [Google Scholar]
- Morampudi, V.; Dalwadi, U.; Bhinder, G.; Sham, H.P.; Gill, S.K.; Chan, J.; Bergstrom, K.S.; Huang, T.; Ma, C.; Jacobson, K.; et al. The goblet cell-derived mediator RELM-beta drives spontaneous colitis in Muc2-deficient mice by promoting commensal microbial dysbiosis. Mucosal. Immunol. 2016, 9, 1218–1233. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.Y.; Inoue, T.; Leone, V.A.; Dalal, S.; Touw, K.; Wang, Y.; Musch, M.W.; Theriault, B.; Higuchi, K.; Donovan, S.; et al. Using corticosteroids to reshape the gut microbiome: Implications for inflammatory bowel diseases. Inflamm. Bowel. Dis. 2015, 21, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Velcich, A.; Lozonschi, I.; Liang, J.; Nicholas, C.; Zhuang, M.; Bancroft, L.; Augenlicht, L.H. Inactivation of p21WAF1/cip1 enhances intestinal tumor formation in Muc2−/− mice. Am. J. Pathol. 2005, 166, 1239–1246. [Google Scholar] [CrossRef]
- Baxter, N.T.; Zackular, J.P.; Chen, G.Y.; Schloss, P.D. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, A.C.; Akter, S.; Hanson, M.M.; Busi, S.B.; Parker, T.W.; Schehr, R.J.; Hankins, M.A.; Ahner, C.E.; Davis, J.W.; Franklin, C.L.; et al. Differential susceptibility to colorectal cancer due to naturally occurring gut microbiota. Oncotarget 2015, 6, 33689–33704. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Huycke, M.M. Commensal bacteria drive endogenous transformation and tumour stem cell marker expression through a bystander effect. Gut 2015, 64, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Gagniere, J.; Bonnin, V.; Jarrousse, A.S.; Cardamone, E.; Agus, A.; Uhrhammer, N.; Sauvanet, P.; Dechelotte, P.; Barnich, N.; Bonnet, R.; et al. Interactions between microsatellite instability and human gut colonization by Escherichia coli in colorectal cancer. Clin. Sci. 2017, 131, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wu, Y.; Deng, B.; Li, J.; Cao, H.; Qu, Y.; Qian, X.; Zhong, G. Isoliquiritigenin decreases the incidence of colitis-associated colorectal cancer by modulating the intestinal microbiota. Oncotarget 2016, 7, 85318–85331. [Google Scholar] [CrossRef] [PubMed]
- Klimesova, K.; Kverka, M.; Zakostelska, Z.; Hudcovic, T.; Hrncir, T.; Stepankova, R.; Rossmann, P.; Ridl, J.; Kostovcik, M.; Mrazek, J.; et al. Altered gut microbiota promotes colitis-associated cancer in IL-1 receptor-associated kinase M-deficient mice. Inflamm. Bowel. Dis. 2013, 19, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, K.; Liu, X.; Zhao, Y.; Gao, N.; Wu, Q.; Song, K.; Cui, Y.; Li, Y.; McDaniel, J.M.; McGee, S.; et al. Defective intestinal mucin-type O-glycosylation causes spontaneous colitis-associated cancer in mice. Gastroenterology 2016, 151, 152–164.e11. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z.; et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chen, J.; Zheng, J.; Hu, G.; Wang, J.; Huang, C.; Lou, L.; Wang, X.; Zeng, Y. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci. Rep. 2016, 6, 26337. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Yang, X.; Xu, S.; Wu, C.; Song, Y.; Qin, N.; Chen, S.; Xiao, Q. Alteration of the fecal microbiota in Chinese patients with Parkinson’s disease. Brain. Behav. Immun. 2018, 70, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; Stams, A.J. The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 2008, 6, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gallego, C.; Pohl, S.; Salminen, S.; De Vos, W.M.; Kneifel, W. Akkermansia muciniphila: A novel functional microbe with probiotic properties. Benef. Microbes. 2016, 7, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huycke, M.M. Colorectal cancer: Role of commensal bacteria and bystander effects. Gut Microbes. 2015, 6, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.; Montgomery, S.A.; Truax, A.D.; Brickey, W.J.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. The intracellular innate immune sensor NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 2017, 18, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.; Schwab, C.; Milinovich, G.; Reichert, J.; Ben Mahfoudh, K.; Decker, T.; Engel, M.; Hai, B.; Hainzl, E.; Heider, S.; et al. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 2012, 6, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Liu, S.; Zhou, Y.; Yao, Z.; Zhang, D.; Cao, S.; Wei, Z.; Tan, B.; Li, Y.; Lian, Z.; et al. Evolutionary biologic changes of gut microbiota in an ’adenoma-carcinoma sequence’ mouse colorectal cancer model induced by 1, 2-Dimethylhydrazine. Oncotarget 2017, 8, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Cocconi, D.; van Sinderen, D.; Ventura, M. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis. FEMS Microbiol. Ecol. 2017, 93, 153. [Google Scholar] [CrossRef] [PubMed]
- Darnaud, M.; Santos, A.D.; Gonzalez, P.; Augui, S.; Lacoste, C.; Desterke, C.; De Hertogh, G.; Valentino, E.; Braun, E.; Zheng, J.; et al. Enteric delivery of regenerating family member 3 alpha alters the intestinal microbiota and controls inflammation in mice with colitis. Gastroenterology 2018, 154, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE 2013, 8, e70803. [Google Scholar] [CrossRef] [PubMed]
- Lupton, J.R. Microbial degradation products influence colon cancer risk: The butyrate controversy. J. Nutr. 2004, 134, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Muir, J.G.; Gibson, P.R. Does butyrate protect from colorectal cancer? J. Gastroenterol. Hepatol. 2006, 21, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, P.; Martel, F. Butyrate and colorectal cancer: The role of butyrate transport. Curr. Drug Metab. 2013, 14, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Yang, W.; Koeffler, H.P. Mouse models of colorectal cancer. Chin. J. Cancer 2011, 30, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Dennis, P.G.; Guo, K.; Imelfort, M.; Jensen, P.; Tyson, G.W.; Rabaey, K. Spatial uniformity of microbial diversity in a continuous bioelectrochemical system. Bioresour. Technol. 2013, 129, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome. Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative Abundance (%) | Genus | |||||||
|---|---|---|---|---|---|---|---|---|
| Roseburia (×10−2) | Butyricimonas (×10−2) | Coprococcus (×10−2) | Clostridium (×10−2) | Ruminococcus | Eubacterium | Total | ||
| D48 | WT | 5.5 ± 1.12 | 1.9 ± 0.35 | 0.5 ± 0.14 | 0 | 0.83 ± 0.13 | 0.54 ± 0.27 | 1.45 ± 0.031 |
| KO | 85.3 ± 19.80 | 5.3 ± 1.33 | 8.8 ± 0.97 | 30.3 ± 0.22 | 3.23 ± 0.57 | 2.97 ± 1.17 | 7.50 ± 1.39 * | |
| D98 | WT | 50.4 ± 9.79 | 0.5 ± 0.08 | 6.7 ± 0.91 | 0 | 0.090 ± 0.031 | 0.14 ± 0.092 | 0.77 ± 0.15 |
| KO | 216.8 ± 3.72 | 9.8 ± 2.22 | 43 ± 3.59 | 52.8 ± 1.51 | 3.30 ± 0.98 | 4.12 ± 1.34 | 10.65 ± 1.36 * | |
| D118 | WT | 38.1 ± 7.33 | 0 | 4.1 ± 0.72 | 0 | 0.01 ± 0.008 | 0.14 ± 0.014 | 0.57 ± 0.14 |
| KO | 58.3 ± 12.08 | 7.9 ± 1.71 | 13 ± 3.34 | 3.8 ± 0.79 | 3.06 ± 0.82 | 3.70 ± 0.67 | 7.58 ± 1.28 * | |
| D138 | WT | 202.9 ± 30.85 | 0 | 6.7 ± 1.47 | 0 | 0.02 ± 0.0056 | 0.08 ± 0.04 | 2.93 ± 0.44 |
| KO | 24.2 ± 3.91 | 4.5 ± 0.88 | 12 ± 2.58 | 1.9 ± 0.19 | 3.30 ± 0.74 | 1.12 ± 0.47 | 4.85 ± 0.97 * | |
| D178 | WT | 33.3 ± 2.82 | 0 | 4.0 ± 0.81 | 0 | 0.02 ± 0.0069 | 0.17 ± 0.07 | 0.55 ± 0.058 |
| KO | 51.2 ± 11.40 | 4.1 ± 0.76 | 7.2 ± 0.2 | 0.9 ± 0.00 | 5.89 ± 0.93 | 1.52 ± 0.81 | 8.04 ± 1.34 * | |
| Relative Abundance (%) | Genus | |||||
|---|---|---|---|---|---|---|
| Akkermansia (×10−3) [23,24] | Desulfovibrio (×10−3) [12,14] | Enterococcus (×10−24) [13,25] | Escherichia (×10−4) [9,14,26] | Turicibacter (×10−2) [27] | ||
| D48 | WT | 0 | 0.47 ± 0.35 | 4.7 ± 1.42 | 100.1 ± 61.3 * | 0.38 ± 0.06 |
| KO | 0 | 8.0 ± 1.91 * | 3.5 ± 0.27 | 0 | 12.4 ± 5.21 * | |
| D98 | WT | 0 | 0.12 ± 0.09 | 1.2 ± 0.89 | 0.3 ± 0.18 | 0 |
| KO | 0.95 ± 0.37 * | 13.2 ± 5.7 * | 5.2 ± 3.59 | 15.4 ± 0.011 * | 5.7 ± 0.98 * | |
| D118 | WT | 0 | 7.1 ± 3.28 | 5.2 ± 3.66 | 1.4 ± 0.98 | 0 |
| KO | 5.2 ± 0.13 * | 14.8 ± 2.1 * | 1.7 ± 1.26 | 18.0 ± 7.88 * | 7.8 ± 5.26 * | |
| D138 | WT | 0 | 8.4 ± 3.60 | 6.9 ± 3.47 | 2.9 ± 1.47 | 0 |
| KO | 3.5 ± 0.39 * | 14 ± 2.47 * | 1.8 ± 1.55 | 3.4 ± 2.51 | 0.7 ± 0.28 * | |
| D178 | WT | 0 | 6.7 ± 2.06 | 1.9 ± 0.81 | 6.6 ± 3.58 | 0 |
| KO | 0.95 ± 0.11 * | 11.3 ± 0.81 * | 1.5 ± 0.19 | 1.7 ± 1.30 | 2.2 ± 0.92 * | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.; Wu, Y.; Li, J.; Bao, Y.; Guo, Y.; Yang, W. The Dynamic Changes of Gut Microbiota in Muc2 Deficient Mice. Int. J. Mol. Sci. 2018, 19, 2809. https://doi.org/10.3390/ijms19092809
Wu M, Wu Y, Li J, Bao Y, Guo Y, Yang W. The Dynamic Changes of Gut Microbiota in Muc2 Deficient Mice. International Journal of Molecular Sciences. 2018; 19(9):2809. https://doi.org/10.3390/ijms19092809
Chicago/Turabian StyleWu, Minna, Yaqi Wu, Jianmin Li, Yonghua Bao, Yongchen Guo, and Wancai Yang. 2018. "The Dynamic Changes of Gut Microbiota in Muc2 Deficient Mice" International Journal of Molecular Sciences 19, no. 9: 2809. https://doi.org/10.3390/ijms19092809
APA StyleWu, M., Wu, Y., Li, J., Bao, Y., Guo, Y., & Yang, W. (2018). The Dynamic Changes of Gut Microbiota in Muc2 Deficient Mice. International Journal of Molecular Sciences, 19(9), 2809. https://doi.org/10.3390/ijms19092809