Papillomaviruses and Endocytic Trafficking

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
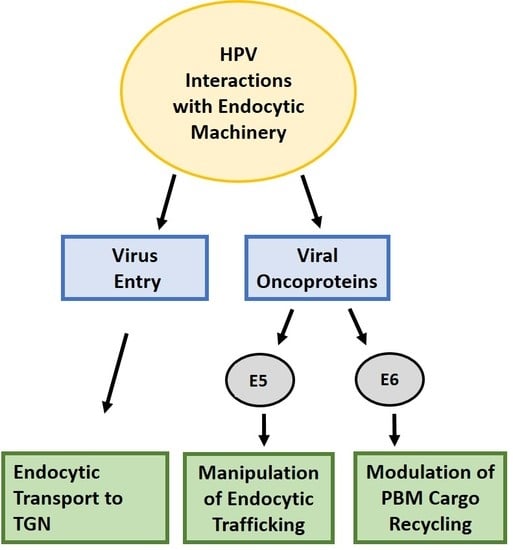
1. Introduction
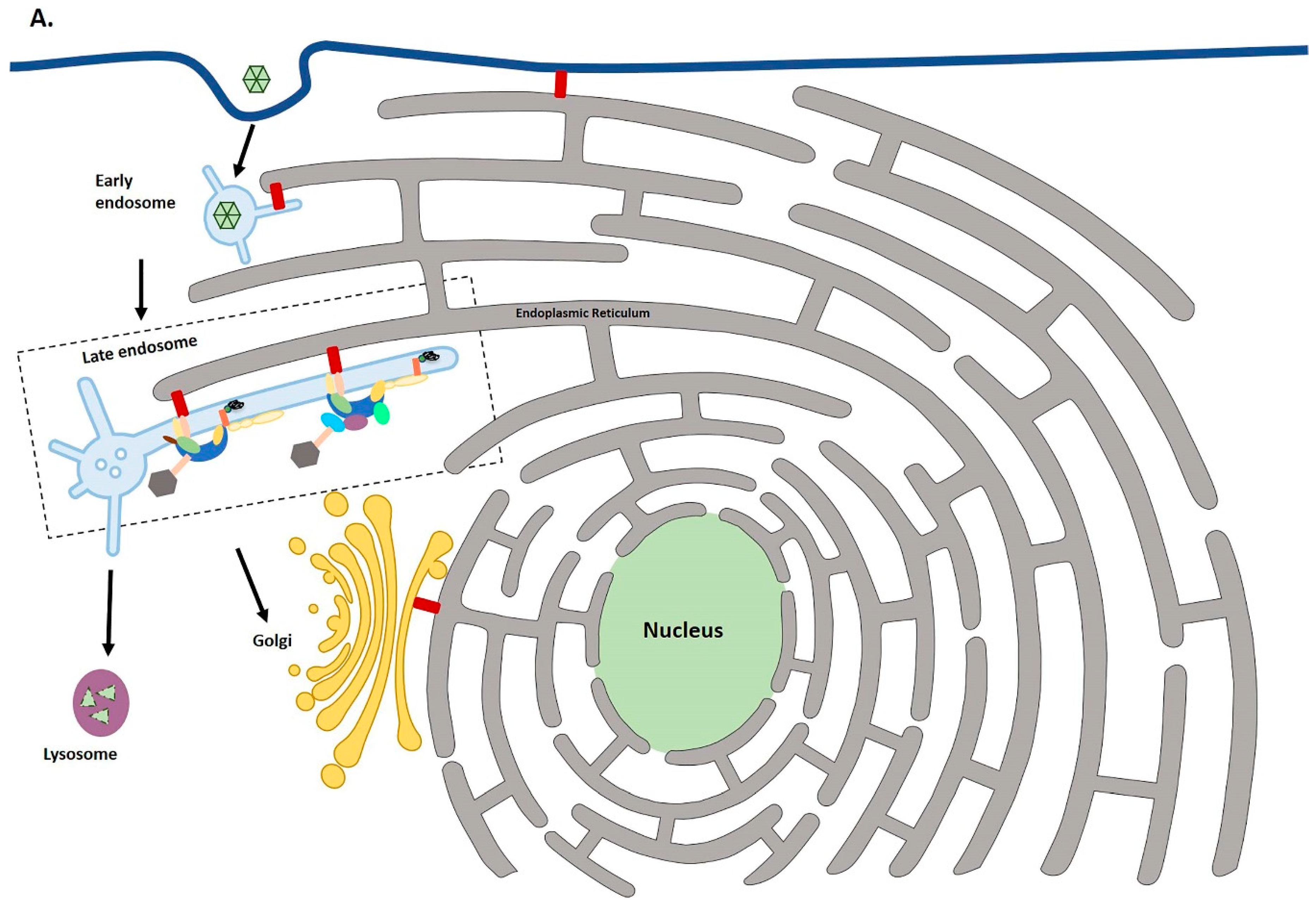
2. Virus Entry
3. The Early Endosome
4. Endosomal Maturation and Lysosomal Degradation
5. Egress from the Late Endosome and Entry into the TGN
6. Endosomal Tubulation
7. ER-Endosome Contact Points
8. L2 Membrane Spanning
9. Post-TGN Transport
10. Do Capsid Proteins Have a Role in Endocytic Transport in Late Infection?
11. The Papillomavirus Oncoproteins and Endocytic Trafficking
12. The Role of Viral Oncoproteins in HPV-Induced Carcinogenesis
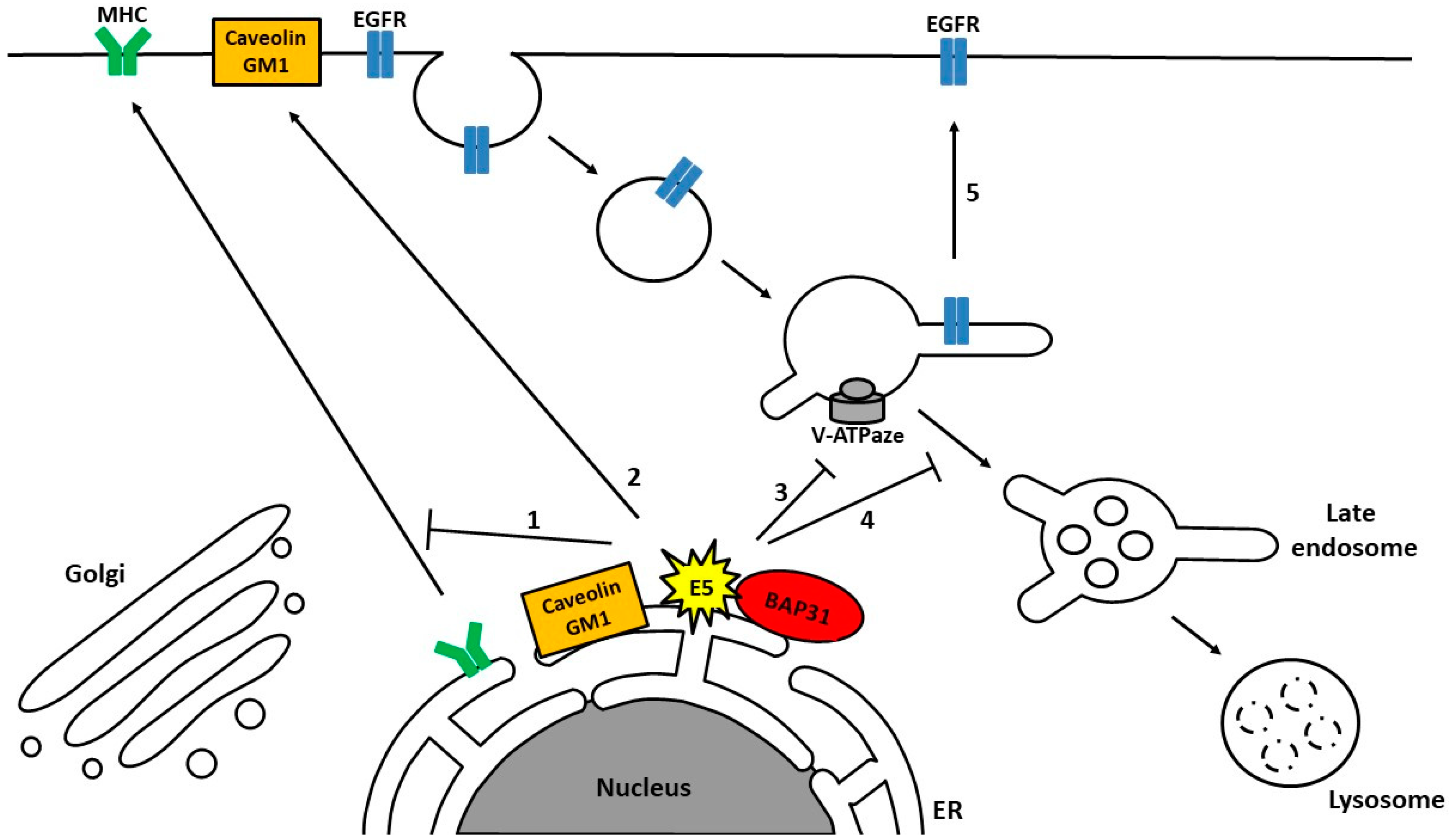
13. E5: Manipulation of Trafficking Pathways and Cancer Development
14. Novel and Unexpected Role of E6 in the Regulation of Endocytic Transport
15. Is There Any Link between E7 and the Endocytic Machinery?
16. Conclusion and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. (Lond) 2006, 110, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomavirus infections—A major cause of human cancers. Biochim. Biophys. Acta 1996, 1288, F55–F78. [Google Scholar] [CrossRef]
- Culp, T.D.; Budgeon, L.R.; Marinkovich, M.P.; Meneguzzi, G.; Christensen, N.D. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J. Virol. 2006, 80, 8940–8950. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Day, P.M.; Thompson, C.D.; Lubkowski, J.; Lu, W.; Lowy, D.R.; Schiller, J.T. Human alpha-defensins block papillomavirus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1516–1521. [Google Scholar] [CrossRef] [PubMed]
- Combita, A.L.; Touzé, A.; Bousarghin, L.; Sizaret, P.Y.; Muñoz, N.; Coursaget, P. Gene transfer using human papillomavirus pseudovirions varies according to virus genotype and requires cell surface heparan sulfate. FEMS Microbiol. Lett. 2001, 204, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Giroglou, T.; Florin, L.; Schafer, F.; Streeck, R.E.; Sapp, M. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 2001, 75, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009, 5, e1000524. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.F.; Bienkowska-Haba, M.; Dasgupta, J.; Chen, X.S.; Sapp, M. Multiple heparan sulfate binding site engagements are required for the infectious entry of human papillomavirus type 16. J. Virol. 2013, 87, 11426–11437. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, C.; Samperio Ventayol, P.; Vogeley, C.; Schelhaas, M. Kallikrein-8 Proteolytically Processes Human Papillomaviruses in the Extracellular Space to Facilitate Entry into Host Cells. J. Virol. 2015, 89, 7038–7052. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. USA 2006, 103, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J. Virol. 2008, 82, 12565–12568. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Gambhira, R.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. Mechanisms of human papillomavirus type 16 neutralization by l2 cross-neutralizing and l1 type-specific antibodies. J. Virol. 2008, 82, 4638–4646. [Google Scholar] [CrossRef] [PubMed]
- Cruz, L.; Biryukov, J.; Conway, M.J.; Meyers, C. Cleavage of the HPV16 Minor Capsid Protein L2 during Virion Morphogenesis Ablates the Requirement for Cellular Furin during De Novo Infection. Viruses 2015, 7, 5813–5830. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Selinka, H.C.; Florin, L.; Patel, H.D.; Freitag, K.; Schmidtke, M.; Makarov, V.A.; Sapp, M. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces non-infectious uptake of human papillomavirus. J. Virol. 2007, 81, 10970–10980. [Google Scholar] [CrossRef] [PubMed]
- Surviladze, Z.; Dziduszko, A.; Ozbun, M.A. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog. 2012, 8, e1002519. [Google Scholar] [CrossRef] [PubMed]
- Broniarczyk, J.; Ring, N.; Massimi, P.; Giacca, M.; Banks, L. HPV-16 virions can remain infectious for 2 weeks on senescent cells but require cell cycle re-activation to allow virus entry. Sci. Rep. 2018, 8, 811. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Guion, L.G.M.; Keiffer, T.R.; Sapp, M. Human papillomavirus major capsid protein L1 remains associated with the incoming viral genome throughout the entry process. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Weber, S.; Snijder, B.; Samperio Ventayol, P.; Kuhbacher, A.; Becker, M.; Day, P.M.; Schiller, J.T.; Kann, M.; Pelkmans, L.; et al. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014, 10, e1004162. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Luszczek, W.; Keiffer, T.R.; Bienkowska-Haba, M.; Guion, L.G.; Sapp, M.J. Incoming human papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. Proc. Natl. Acad. Sci. USA 2016, 113, 6289–6294. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Baker, C.C.; Lowy, D.R.; Schiller, J.T. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. USA 2004, 101, 14252–14257. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.F.; Porter, K.R. Yolk Protein Uptake in the Oocyte of the Mosquito Aedes Aegypti. L. J. Cell Biol. 1964, 20, 313–332. [Google Scholar] [CrossRef] [PubMed]
- Yamada, E. The fine structure of the gall bladder epithelium of the mouse. J. Biophys. Biochem. Cytol. 1955, 1, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Moya, M.; Dautry-Varsat, A.; Goud, B.; Louvard, D.; Boquet, P. Inhibition of coated pit formation in Hep2 cells blocks the cytotoxicity of diphtheria toxin but not that of ricin toxin. J. Cell Biol. 1985, 101, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Lamaze, C.; Dujeancourt, A.; Baba, T.; Lo, C.G.; Benmerah, A.; Dautry-Varsat, A. Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol. Cell 2001, 7, 661–671. [Google Scholar] [CrossRef]
- Kirkham, M.; Fujita, A.; Chadda, R.; Nixon, S.J.; Kurzchalia, T.V.; Sharma, D.K.; Pagano, R.E.; Hancock, J.F.; Mayor, S.; Parton, R.G. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell Biol. 2005, 168, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Bousarghin, L.; Touzé, A.; Sizaret, P.-Y.; Coursaget, P. Human Papillomavirus Types 16, 31, and 58 Use Different Endocytosis Pathways To Enter Cells. J. Virol. 2003, 77, 3846–3850. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Lowy, D.R.; Schiller, J.T. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 2003, 307, 1–11. [Google Scholar] [CrossRef]
- Hindmarsh, P.L.; Laimins, L.A. Mechanisms regulating expression of the HPV 31 L1 and L2 capsid proteins and pseudovirion entry. Virol. J. 2007, 4, 19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abban, C.Y.; Bradbury, N.A.; Meneses, P.I. HPV16 and BPV1 infection can be blocked by the dynamin inhibitor dynasore. Am. J. Ther. 2008, 15, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Campos, S.K.; Ozbun, M.A. Human papillomavirus type 31 uses a caveolin 1- and dynamin 2-mediated entry pathway for infection of human keratinocytes. J. Virol. 2007, 81, 9922–9931. [Google Scholar] [CrossRef] [PubMed]
- Spoden, G.; Freitag, K.; Husmann, M.; Boller, K.; Sapp, M.; Lambert, C.; Florin, L. Clathrin- and caveolin-independent entry of human papillomavirus type 16—Involvement of tetraspanin-enriched microdomains (TEMs). PLoS ONE 2008, 3, e3313. [Google Scholar] [CrossRef] [PubMed]
- Spoden, G.; Kuhling, L.; Cordes, N.; Frenzel, B.; Sapp, M.; Boller, K.; Florin, L.; Schelhaas, M. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. J. Virol. 2013, 87, 7765–7773. [Google Scholar] [CrossRef] [PubMed]
- Schelhaas, M.; Shah, B.; Holzer, M.; Blattmann, P.; Kuhling, L.; Day, P.M.; Schiller, J.T.; Helenius, A. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 2012, 8, e1002657. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Abban, C.Y.; Kiyashka, E.; Qiang, W.; Meneses, P.I. HPV16 infection of HaCaTs is dependent on β4 integrin, and α6 integrin processing. Virology 2014, 449, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Dziduszko, A.; Ozbun, M.A. Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J. Virol. 2013, 87, 7502–7515. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, K.D.; Gawlitza, A.; Spoden, G.A.; Zhang, X.A.; Lambert, C.; Berditchevski, F.; Florin, L. Tetraspanin CD151 mediates papillomavirus type 16 endocytosis. J. Virol. 2013, 87, 3435–3446. [Google Scholar] [CrossRef] [PubMed]
- Woodham, A.W.; Da Silva, D.M.; Skeate, J.G.; Raff, A.B.; Ambroso, M.R.; Brand, H.E.; Isas, J.M.; Langen, R.; Kast, W.M. The S100A10 subunit of the annexin A2 heterotetramer facilitates L2-mediated human papillomavirus infection. PLoS ONE 2012, 7, e43519. [Google Scholar] [CrossRef] [PubMed]
- Wüstenhagen, E.; Hampe, L.; Boukhallouk, F.; Schneider, M.A.; Spoden, G.A.; Negwer, I.; Koynov, K.; Kast, W.M.; Florin, L. The Cytoskeletal Adaptor Obscurin-Like 1 Interacts with the Human Papillomavirus 16 (HPV16) Capsid Protein L2 and Is Required for HPV16 Endocytosis. J. Virol. 2016, 90, 10629–10641. [Google Scholar] [CrossRef] [PubMed]
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: Intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Abban, C.Y.; Meneses, P.I. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 2010, 403, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Bergant Marusic, M.; Ozbun, M.A.; Campos, S.K.; Myers, M.P.; Banks, L. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 2012, 13, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Campos, S.K.; Wandinger-Ness, A.; Ozbun, M.A. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J. Virol. 2008, 82, 9505–9512. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, D.J.; Odorizzi, G.; Emr, S.D. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 2002, 3, 893–905. [Google Scholar] [CrossRef] [PubMed]
- Grassel, L.; Fast, L.A.; Scheffer, K.D.; Boukhallouk, F.; Spoden, G.A.; Tenzer, S.; Boller, K.; Bago, R.; Rajesh, S.; Overduin, M.; et al. The CD63-Syntenin-1 Complex Controls Post-Endocytic Trafficking of Oncogenic Human Papillomaviruses. Sci. Rep. 2016, 6, 32337. [Google Scholar] [CrossRef] [PubMed]
- Broniarczyk, J.; Bergant, M.; Gozdzicka-Jozefiak, A.; Banks, L. Human papillomavirus infection requires the TSG101 component of the ESCRT machinery. Virology 2014, 460–461, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Broniarczyk, J.; Pim, D.; Massimi, P.; Bergant, M.; Gozdzicka-Jozefiak, A.; Crump, C.; Banks, L. The VPS4 component of the ESCRT machinery plays an essential role in HPV infectious entry and capsid disassembly. Sci. Rep. 2017, 7, 45159. [Google Scholar] [CrossRef] [PubMed]
- Razi, M.; Futter, C.E. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol. Biol. Cell 2006, 17, 3469–3483. [Google Scholar] [CrossRef] [PubMed]
- Bache, K.G.; Stuffers, S.; Malerød, L.; Slagsvold, T.; Raiborg, C.; Lechardeur, D.; Wälchli, S.; Lukacs, G.L.; Brech, A.; Stenmark, H. The ESCRT-III subunit hVps24 is required for degradation but not silencing of the epidermal growth factor receptor. Mol. Biol. Cell. 2006, 17, 2513–2523. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.R.; Fernandez, D.J.; Thornton, S.M.; Skeate, J.G.; Luhen, K.P.; Da Silva, D.M.; Langen, R.; Kast, W.M. Heterotetrameric annexin A2/S100A10 (A2t) is essential for oncogenic human papillomavirus trafficking and capsid disassembly, and protects virions from lysosomal degradation. Sci. Rep. 2018, 8, 11642. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I. Endocytosis and molecular sorting. Annu. Rev. Cell Dev. Biol. 1996, 12, 575–625. [Google Scholar] [CrossRef] [PubMed]
- Selinka, H.C.; Giroglou, T.; Sapp, M. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 2002, 299, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Williams, C.; Kim, S.M.; Garcea, R.L.; Sapp, M. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J. Virol. 2012, 86, 9875–9887. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, E.Y.; Meneses, P.I. A Dual Role for the Nonreceptor Tyrosine Kinase Pyk2 during the Intracellular Trafficking of Human Papillomavirus 16. J. Virol. 2015, 89, 9103–9114. [Google Scholar] [CrossRef] [PubMed]
- Lipovsky, A.; Popa, A.; Pimienta, G.; Wyler, M.; Bhan, A.; Kuruvilla, L.; Guie, M.A.; Poffenberger, A.C.; Nelson, C.D.; Atwood, W.J.; et al. Genome-wide siRNA screen identifies the retromer as a cellular entry factor for human papillomavirus. Proc. Natl. Acad. Sci. USA 2013, 110, 7452–7457. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; Meneses, P.I. The Role of DCT in HPV16 Infection of HaCaTs. PLoS ONE 2017, 12, e0170158. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Hilbig, L.; Sapp, M. The nuclear retention signal of HPV16 L2 protein is essential for incoming viral genome to transverse the trans-Golgi network. Virology 2014, 458–459, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Schowalter, R.M.; Lowy, D.R.; Schiller, J.T. Identification of a role for the trans-Golgi network in human papillomavirus 16 pseudovirus infection. J. Virol. 2013, 87, 3862–3870. [Google Scholar] [CrossRef] [PubMed]
- Wiens, M.E.; Smith, J.G. alpha-Defensin HD5 Inhibits Human Papillomavirus 16 Infection via Capsid Stabilization and Redirection to the Lysosome. mBio 2017, 8, e02304-16. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T. Interferon Gamma Prevents Infectious Entry of Human Papillomavirus 16 via an L2-Dependent Mechanism. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Lipovsky, A.; Erden, A.; Kanaya, E.; Zhang, W.; Crite, M.; Bradfield, C.; MacMicking, J.; DiMaio, D.; Schoggins, J.W.; Iwasaki, A. The cellular endosomal protein stannin inhibits intracellular trafficking of human papillomavirus during virus entry. J. Gen. Virol. 2017, 98, 2821–2836. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, T. The gamma-secretase complex: Machinery for intramembrane proteolysis. Curr. Opin. Neurobiol. 2004, 14, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Saito, T.; Kondo, N.; Moriwaki, K.; Noda, K.; Ihara, S.; Takahashi, M.; Ide, Y.; Gu, J.; Inohara, H.; et al. A secreted type of beta1,6 N-acetylglucosaminyltransferase V (GnT-V), a novel angiogenesis inducer, is regulated by gamma-secretase. FASEB J. 2006, 20, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.L.; Strutz, N.; Gahring, L.C.; Rogers, S.W. Glutamate receptor subunit 3 is modified by site-specific limited proteolysis including cleavage by gamma-secretase. J. Biol. Chem. 2003, 278, 23786–23796. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.S.; Buck, C.B.; Lambert, P.F. Inhibition of gamma secretase blocks HPV infection. Virology 2010, 407, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Kazakov, T.; Popa, A.; DiMaio, D. Vesicular trafficking of incoming human papillomavirus 16 to the Golgi apparatus and endoplasmic reticulum requires gamma-secretase activity. mBio 2014, 5, e01777-14. [Google Scholar] [CrossRef] [PubMed]
- Karanam, B.; Peng, S.; Li, T.; Buck, C.; Day, P.M.; Roden, R.B. Papillomavirus infection requires gamma secretase. J. Virol. 2010, 84, 10661–10670. [Google Scholar] [CrossRef] [PubMed]
- Kwak, K.; Jiang, R.; Wang, J.W.; Jagu, S.; Kirnbauer, R.; Roden, R.B. Impact of inhibitors and L2 antibodies upon the infectivity of diverse alpha and beta human papillomavirus types. PLoS ONE 2014, 9, e97232. [Google Scholar] [CrossRef] [PubMed]
- Popa, A.; Zhang, W.; Harrison, M.S.; Goodner, K.; Kazakov, T.; Goodwin, E.C.; Lipovsky, A.; Burd, C.G.; DiMaio, D. Direct binding of retromer to human papillomavirus type 16 minor capsid protein L2 mediates endosome exit during viral infection. PLoS Pathog. 2015, 11, e1004699. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R.; McGraw, T.E. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 2004, 5, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Hsu, V.W.; Bai, M.; Li, J. Getting active: Protein sorting in endocytic recycling. Nat. Rev. Mol. Cell Biol. 2012, 13, 323–328. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Gallop, J.L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005, 438, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Rojas, R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 2006, 7, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.G. Physiology and pathology of endosome-to-Golgi retrograde sorting. Traffic 2011, 12, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Rocha, N.; Kuijl, C.; van der Kant, R.; Janssen, L.; Houben, D.; Janssen, H.; Zwart, W.; Neefjes, J. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol. 2009, 185, 1209–1225. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Chitwood, P.J.; Phillips, M.J.; Voeltz, G.K. ER contact sites define the position and timing of endosome fission. Cell 2014, 159, 1027–1041. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Dibenedetto, J.R.; West, M.; Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum–endosome contact increases as endosomes traffic and mature. Mol. Biol. Cell 2013, 24, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Saheki, Y.; Swarup, S.; Lucast, L.; Harper, J.W.; De Camilli, P. Endosome-ER Contacts Control Actin Nucleation and Retromer Function through VAP-Dependent Regulation of PI4P. Cell 2016, 166, 408–423. [Google Scholar] [CrossRef] [PubMed]
- Siddiqa, A.; Massimi, P.; Pim, D.; Broniarczyk, J.; Banks, L. Human Papillomavirus 16 Infection Induces VAP-Dependent Endosomal Tubulation. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Auvinen, E.; Kujari, H.; Arstila, P.; Hukkanen, V. Expression of the L2 and E7 genes of the human papillomavirus type 16 in female genital dysplasias. Am. J. Pathol. 1992, 141, 1217–1224. [Google Scholar] [PubMed]
- Hagensee, M.E.; Yaegashi, N.; Galloway, D.A. Self-assembly of human papillomavirus type 1 capsids by expression of the L1 protein alone or by coexpression of the L1 and L2 capsid proteins. J. Virol. 1993, 67, 315–322. [Google Scholar] [PubMed]
- Carlton, J.; Bujny, M.; Peter, B.J.; Oorschot, V.M.; Rutherford, A.; Mellor, H.; Klumperman, J.; McMahon, H.T.; Cullen, P.J. Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high- curvature membranes and 3-phosphoinositides. Curr. Biol. 2004, 14, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Haft, C.R.; de la Luz Sierra, M.; Bafford, R.; Lesniak, M.A.; Barr, V.A.; Taylor, S.I. Human orthologs of yeast vacuolar protein sorting proteins Vps26, 29, and 35: Assembly into multimeric complexes. Mol. Biol. Cell. 2000, 11, 4105–4116. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Lazar, C.S.; Tronchere, H.; Sato, T.; Meerloo, T.; Yeo, M.; Songyang, Z.; Emr, S.D.; Gill, G.N. Endosomal localization and function of sorting nexin 1. Proc. Natl. Acad. Sci. USA 2002, 99, 6767–6772. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.; Cullen, P.J. Retromer: A master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Balana, B.; Maslennikov, I.; Kwiatkowski, W.; Stern, K.M.; Bahima, L.; Choe, S.; Slesinger, P.A. Mechanism underlying selective regulation of G protein-gated inwardly rectifying potassium channels by the psychostimulant-sensitive sorting nexin 27. Proc. Natl. Acad. Sci. USA 2011, 108, 5831–5836. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, F.; Gallon, M.; Winfield, M.; Thomas, E.C.; Bell, A.J.; Heesom, K.J.; Tavare, J.M.; Cullen, P.J. A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat. Cell. Biol. 2013, 15, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Chang, J.; Blackstone, C. FAM21 directs SNX27-retromer cargoes to the plasma membrane by preventing transport to the Golgi apparatus. Nat. Commun. 2016, 7, 10939. [Google Scholar] [CrossRef] [PubMed]
- McNally, K.E.; Faulkner, R.; Steinberg, F.; Gallon, M.; Ghai, R.; Pim, D.; Langton, P.; Pearson, N.; Danson, C.M.; Nagele, H. Retriever is a multiprotein complex for retromer-independent endosomal cargo recycling. Nat. Cell Biol. 2017, 19, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Broniarczyk, J.; Bergant, M.; Playford, M.P.; Banks, L. A Novel Pdz Domain Interaction Mediates the Binding between Hpv-16 L2 and Sorting Nexin 27 and Modulates Virion Trafficking. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Liu, D.; Liu, N.; Xu, L.; Li, S.; Lin, S.; Shu, X.; Pei, D. SNX17 regulates Notch pathway and pancreas development through the retromer-dependent recycling of Jag1. Cell Regen. 2012, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Kämper, N.; Day, P.M.; Nowak, T.; Selinka, H.C.; Florin, L.; Bolscher, J.; Hilbig, L.; Schiller, J.T.; Sapp, M. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 2006, 80, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Monteiro da Silva, G.; Deatherage, C.; Burd, C.; DiMaio, D. Cell-Penetrating Peptide Mediates Intracellular Membrane Passage of Human Papillomavirus L2 Protein to Trigger Retrograde Trafficking. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Bronnimann, M.P.; Chapman, J.A.; Park, C.K.; Campos, S.K. A transmembrane domain and GxxxG motifs within L2 are essential for papillomavirus infection. J. Virol. 2013, 87, 464–473. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Keiffer, T.R.; Bienkowska-Haba, M.; Luszczek, W.; Guion, L.G.; Muller, M.; Sapp, M. Topography of the Human Papillomavirus Minor Capsid Protein L2 during Vesicular Trafficking of Infectious Entry. J. Virol. 2015, 89, 10442–10452. [Google Scholar] [CrossRef] [PubMed]
- Campos, S.K. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses 2017, 9, 370. [Google Scholar] [CrossRef] [PubMed]
- Calton, C.M.; Bronnimann, M.P.; Manson, A.R.; Li, S.; Chapman, J.A.; Suarez-Berumen, M.; Williamson, T.R.; Molugu, S.K.; Bernal, R.A.; Campos, S.K. Translocation of the papillomavirus L2/vDNA complex across the limiting membrane requires the onset of mitosis. PLoS Pathog. 2017, 13, e1006200. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.A.; Spoden, G.A.; Florin, L.; Lambert, C. Identification of the dynein light chains required for human papillomavirus infection. Cell. Microbiol. 2011, 13, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Aydin, I.; Villalonga-Planells, R.; Greune, L.; Bronnimann, M.P.; Calton, C.M.; Becker, M.; Lai, K.Y.; Campos, S.K.; Schmidt, M.A.; Schelhaas, M. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLoS Pathog. 2017, 13, e1006308. [Google Scholar] [CrossRef] [PubMed]
- Bund, T.; Spoden, G.A.; Koynov, K.; Hellmann, N.; Boukhallouk, F.; Arnold, P.; Hinderberger, D.; Florin, L. An L2 SUMO interacting motif is important for PML localization and infection of human papillomavirus type 16. Cell. Microbiol. 2014, 16, 1179–1200. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Luszczek, W.; Keiffer, T.R.; Guion, L.G.M.; DiGiuseppe, S.; Scott, R.S.; Sapp, M. Incoming human papillomavirus 16 genome is lost in PML protein-deficient HaCaT keratinocytes. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Stepp, W.H.; Meyers, J.M.; McBride, A.A. Sp100 provides intrinsic immunity against human papillomavirus infection. mBio 2013, 4, e00845-13. [Google Scholar] [CrossRef] [PubMed]
- Wustenhagen, E.; Boukhallouk, F.; Negwer, I.; Rajalingam, K.; Stubenrauch, F.; Florin, L. The Myb-related protein MYPOP is a novel intrinsic host restriction factor of oncogenic human papillomaviruses. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Thompson, C.D.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Involvement of Nucleophosmin (NPM1/B23) in Assembly of Infectious HPV16 Capsids. Papillomavirus Res. 2015, 1, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J. Virol. 1998, 72, 142–150. [Google Scholar] [PubMed]
- Becker, K.A.; Florin, L.; Sapp, C.; Sapp, M. Dissection of human papillomavirus type 33 L2 domains involved in nuclear domains (ND) 10 homing and reorganization. Virology 2003, 314, 161–167. [Google Scholar] [CrossRef]
- Becker, K.A.; Florin, L.; Sapp, C.; Maul, G.G.; Sapp, M. Nuclear localization but not PML protein is required for incorporation of the papillomavirus minor capsid protein L2 into virus-like particles. J. Virol. 2004, 78, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Sapp, C.; Streeck, R.E.; Sapp, M. Assembly and translocation of papillomavirus capsid proteins. J. Virol. 2002, 76, 10009–10014. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Becker, K.A.; Sapp, C.; Lambert, C.; Sirma, H.; Muller, M.; Streeck, R.E.; Sapp, M. Nuclear translocation of papillomavirus minor capsid protein L2 requires Hsc70. J. Virol. 2004, 78, 5546–5553. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Gao, B.; Prasad, K.; Greene, L.E.; Eisenberg, E. Hsc70 chaperones clathrin and primes it to interact with vesicle membranes. J. Boil. Chem. 2000, 275, 8439–8447. [Google Scholar] [CrossRef]
- Darshan, M.S.; Lucchi, J.; Harding, E.; Moroianu, J. The l2 minor capsid protein of human papillomavirus type 16 interacts with a network of nuclear import receptors. J. Virol. 2004, 78, 12179–12188. [Google Scholar] [CrossRef] [PubMed]
- Mamoor, S.; Onder, Z.; Karanam, B.; Kwak, K.; Bordeaux, J.; Crosby, L.; Roden, R.B.; Moroianu, J. The high risk HPV16 L2 minor capsid protein has multiple transport signals that mediate its nucleocytoplasmic traffic. Virology 2012, 422, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Goldenring, J.R. A central role for vesicle trafficking in epithelial neoplasia: Intracellular highways to carcinogenesis. Nat. Rev. Cancer 2013, 13, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Maufort, J.P.; Shai, A.; Pitot, H.C.; Lambert, P.F. A role for HPV16 E5 in cervical carcinogenesis. Cancer Res. 2010, 70, 2924–2931. [Google Scholar] [CrossRef] [PubMed]
- Stoppler, M.C.; Straight, S.W.; Tsao, G.; Schlegel, R.; McCance, D.J. The E5 gene of HPV-16 enhances keratinocyte immortalization by full-length DNA. Virology 1996, 223, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Matlashewski, G.; Gu, Z.M.; Storey, A.; Banks, L. The human papillomavirus type 16 E5 gene cooperates with the E7 gene to stimulate proliferation of primary cells and increases viral gene expression. Virology 1994, 203, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Valle, G.F.; Banks, L. The human papillomavirus (HPV)-6 and HPV-16 E5 proteins co-operate with HPV-16 E7 in the transformation of primary rodent cells. J. Gen. Virol. 1995, 76, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Petti, L.; Nilson, L.A.; DiMaio, D. Activation of the platelet-derived growth factor receptor by the bovine papillomavirus E5 transforming protein. EMBO J. 1991, 10, 845–855. [Google Scholar] [PubMed]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Yoshinouchi, M.; Yamada, T.; Kizaki, M.; Fen, J.; Koseki, T.; Ikeda, Y.; Nishihara, T.; Yamato, K. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol. Ther. 2003, 8, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Thompson, D.A.; Munger, K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 1997, 239, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Noya, F.; Chien, W.M.; Broker, T.R.; Chow, L.T. p21cip1 Degradation in differentiated keratinocytes is abrogated by costabilization with cyclin E induced by human papillomavirus E7. J. Virol. 2001, 75, 6121–6134. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Thomas, M.; Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Muller, M.; Wasson, C.W.; Bhatia, R.; Boxall, S.; Millan, D.; Goh, G.Y.; Haas, J.; Stonehouse, N.J.; Macdonald, A. YIP1 family member 4 (YIPF4) is a novel cellular binding partner of the papillomavirus E5 proteins. Sci. Rep. 2015, 5, 12523. [Google Scholar] [CrossRef] [PubMed]
- Bravo, I.G.; Alonso, A. Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. J. Virol. 2004, 78, 13613–13626. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, E.; Suprynowicz, F.A.; Hebert, J.D.; Kamonjoh, C.M.; Schlegel, R. The human papillomavirus type 16 E5 oncoprotein translocates calpactin I to the perinuclear region. J. Virol. 2011, 85, 10968–10975. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Bubb, V.J.; Schlegel, R. The human papillomavirus type 6 and 16 E5 proteins are membrane-associated proteins which associate with the 16-kilodalton pore-forming protein. J. Virol. 1993, 67, 6170–6178. [Google Scholar] [PubMed]
- Wetherill, L.F.; Holmes, K.K.; Verow, M.; Muller, M.; Howell, G.; Harris, M.; Fishwick, C.; Stonehouse, N.; Foster, R.; Blair, G.E.; et al. High-risk human papillomavirus E5 oncoprotein displays channel-forming activity sensitive to small-molecule inhibitors. J. Virol. 2012, 86, 5341–5351. [Google Scholar] [CrossRef] [PubMed]
- Royle, J.; Dobson, S.J.; Muller, M.; Macdonald, A. Emerging Roles of Viroporins Encoded by DNA Viruses: Novel Targets for Antivirals? Viruses 2015, 7, 5375–5387. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Leechanachai, P.; Banks, L.; Moreau, F.; Matlashewski, G. The E5 gene from human papillomavirus type 16 is an oncogene which enhances growth factor-mediated signal transduction to the nucleus. Oncogene 1992, 7, 19–25. [Google Scholar] [PubMed]
- Pim, D.; Collins, M.; Banks, L. Human papillomavirus type 16 E5 gene stimulates the transforming activity of the epidermal growth factor receptor. Oncogene 1992, 7, 27–32. [Google Scholar] [PubMed]
- Straight, S.W.; Hinkle, P.M.; Jewers, R.J.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 transforms fibroblasts and effects the downregulation of the epidermal growth factor receptor in keratinocytes. J. Virol. 1993, 67, 4521–4532. [Google Scholar] [PubMed]
- Disbrow, G.L.; Hanover, J.A.; Schlegel, R. Endoplasmic reticulum-localized human papillomavirus type 16 E5 protein alters endosomal pH but not trans-Golgi pH. J. Virol. 2005, 79, 5839–5846. [Google Scholar] [CrossRef] [PubMed]
- Straight, S.W.; Herman, B.; McCance, D.J. The E5 oncoprotein of human papillomavirus type 16 inhibits the acidification of endosomes in human keratinocytes. J. Virol. 1995, 69, 3185–3192. [Google Scholar] [PubMed]
- Suprynowicz, F.A.; Krawczyk, E.; Hebert, J.D.; Sudarshan, S.R.; Simic, V.; Kamonjoh, C.M.; Schlegel, R. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J. Virol. 2010, 84, 10619–10629. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, P.; van Deurs, B.; Norrild, B.; Kayser, L. The HPV16 E5 oncogene inhibits endocytic trafficking. Oncogene 2000, 19, 6023–6032. [Google Scholar] [CrossRef] [PubMed]
- Belleudi, F.; Leone, L.; Purpura, V.; Cannella, F.; Scrofani, C.; Torrisi, M.R. HPV16 E5 affects the KGFR/FGFR2b-mediated epithelial growth through alteration of the receptor expression, signaling and endocytic traffic. Oncogene 2011, 30, 4963–4976. [Google Scholar] [CrossRef] [PubMed]
- Purpura, V.; Belleudi, F.; Caputo, S.; Torrisi, M.R. HPV16 E5 and KGFR/FGFR2b interplay in differentiating epithelial cells. Oncotarget 2013, 4, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Haghshenas, M.; Marchetti, B.; Campo, M.S. E5 protein of human papillomavirus 16 downregulates HLA class I and interacts with the heavy chain via its first hydrophobic domain. Int. J. Cancer 2006, 119, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Brown, D.R.; Fife, K.H.; Campo, M.S. Down-regulation of MHC class I is a property common to papillomavirus E5 proteins. Virus Res. 2006, 120, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Haghshenas, M.R.; Marchetti, B.; O’Brien, P.M.; Campo, M.S. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int. J. Cancer 2005, 113, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.S.; Ashrafi, G.H.; Campo, M.S. All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int. J. Cancer 2010, 126, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Gruener, M.; Bravo, I.G.; Momburg, F.; Alonso, A.; Tomakidi, P. The E5 protein of the human papillomavirus type 16 down-regulates HLA-I surface expression in calnexin-expressing but not in calnexin-deficient cells. Virol. J. 2007, 4, 116. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.A.; Laimins, L.A. Bap31 is a novel target of the human papillomavirus E5 protein. J. Virol. 2008, 82, 10042–10051. [Google Scholar] [CrossRef] [PubMed]
- Kotnik Halavaty, K.; Regan, J.; Mehta, K.; Laimins, L. Human papillomavirus E5 oncoproteins bind the A4 endoplasmic reticulum protein to regulate proliferative ability upon differentiation. Virology 2014, 452–453, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, P.; Wang, E.; Brahmi, Z.; Dunn, K.W.; Blum, J.S.; Roman, A. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-gamma. Virology 2003, 310, 100–108. [Google Scholar] [CrossRef]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K.; et al. CD1d, asentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: A possible mechanism for immune evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.S.; Graham, S.V.; Cortese, M.S.; Ashrafi, G.H.; Araibi, E.H.; Dornan, E.S.; Miners, K.; Nunes, C.; Man, S. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 2010, 407, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Bravo, I.G.; Crusius, K.; Alonso, A. The E5 protein of the human papillomavirus type 16 modulates composition and dynamics of membrane lipids in keratinocytes. Arch. Virol. 2005, 150, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Suprynowicz, F.A.; Disbrow, G.L.; Krawczyk, E.; Simic, V.; Lantzky, K.; Schlegel, R. HPV-16 E5 oncoprotein upregulates lipid raft components caveolin-1 and ganglioside GM1 at the plasma membrane of cervical cells. Oncogene 2008, 27, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Tajima, O.; Furukawa, K.; Urano, T.; Furukawa, K. Over-expression of GM1 enhances cell proliferation with epidermal growth factor without affecting the receptor localization in the microdomain in PC12 cells. Int. J. Oncol. 2005, 26, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Breuza, L.; Halbeisen, R.; Jeno, P.; Otte, S.; Barlowe, C.; Hong, W.; Hauri, H.P. Proteomics of endoplasmic reticulum-Golgi intermediate compartment (ERGIC) membranes from brefeldin A-treated HepG2 cells identifies ERGIC-32, a new cycling protein that interacts with human Erv46. J. Biol. Chem. 2004, 279, 47242–47253. [Google Scholar] [CrossRef] [PubMed]
- Conchon, S.; Cao, X.; Barlowe, C.; Pelham, H.R. Got1p and Sft2p: Membrane proteins involved in traffic to the Golgi complex. EMBO J. 1999, 18, 3934–3946. [Google Scholar] [CrossRef] [PubMed]
- Geerts, D.; Wallick, C.J.; Koomoa, D.L.; Koster, J.; Versteeg, R.; Go, R.C.; Bachmann, A.S. Expression of prenylated Rab acceptor 1 domain family, member 2 (PRAF2) in neuroblastoma: Correlation with clinical features, cellular localization, and cerulenin-mediated apoptosis regulation. Clin. Cancer Res. 2007, 13, 6312–6319. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, S.; Ben-Tekaya, H.; Koegler, E.; Gruenberg, J.; Hauri, H.P. The cargo receptors Surf4, endoplasmic reticulum-Golgi intermediate compartment (ERGIC)-53, and p25 are required to maintain the architecture of ERGIC and Golgi. Mol. Biol.Cell 2008, 19, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.F.; Ghanekar, Y.; Frost, L.; Diao, A.; Morrison, D.; McKenzie, E.; Lowe, M. ZFPL1, a novel ring finger protein required for cis-Golgi integrity and efficient ER-to-Golgi transport. EMBO J. 2008, 27, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, A.; Hara, T.; Maejima, I.; Sato, M.; Sato, K.; Sato, K. Rer1p regulates the ER retention of immature rhodopsin and modulates its intracellular trafficking. Sci. Rep. 2014, 4, 5973. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.P.; Littleton, J.T. Vesicle trafficking: A Rab family profile. Curr. Biol. 2011, 21, R841–R843. [Google Scholar] [CrossRef] [PubMed]
- Howie, H.L.; Katzenellenbogen, R.A.; Galloway, D.A. Papillomavirus E6 proteins. Virology 2009, 384, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Mora, R.; Laimins, L.A. Intracellular localization and DNA-binding properties of human papillomavirus type 18 E6 protein expressed with a baculovirus vector. J. Virol. 1989, 63, 366–374. [Google Scholar] [PubMed]
- Guccione, E.; Massimi, P.; Bernat, A.; Banks, L. Comparative analysis of the intracellular location of the high- and low-risk human papillomavirus oncoproteins. Virology 2002, 293, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Tomaic, V.; Massimi, P.; Nicolaides, L.; Doorbar, J.; Banks, L. The high-risk HPV E6 target scribble (hScrib) is required for HPV E6 expression in cervical tumour-derived cell lines. Papillomavirus Res. 2016, 2, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Fanning, A.S.; Fu, C.; Xu, J.; Marfatia, S.M.; Chishti, A.H.; Crompton, A.; Chan, A.C.; Anderson, J.M.; Cantley, L.C. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 1997, 275, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A.; Thomas, M.; Banks, L.; Roberts, S. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J. Cell Sci. 2003, 116, 4925–4934. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Nguyen, M.M.; Lee, D.; Griep, A.E.; Lambert, P.F. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6’s induction of epithelial hyperplasia in vivo. J. Virol. 2003, 77, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Laimins, L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004, 78, 12366–12377. [Google Scholar] [CrossRef] [PubMed]
- Delury, C.P.; Marsh, E.K.; James, C.D.; Boon, S.S.; Banks, L.; Knight, G.L.; Roberts, S. The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J. Virol. 2013, 87, 9463–9472. [Google Scholar] [CrossRef] [PubMed]
- Banks, L.; Pim, D.; Thomas, M. Human tumour viruses and the deregulation of cell polarity in cancer. Nat. Rev. Cancer 2012, 12, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Myers, M.P.; Massimi, P.; Guarnaccia, C.; Banks, L. Analysis of multiple HPV E6 PDZ interactions defines type-specific PDZ fingerprints that predict oncogenic potential. PLoS Pathog. 2016, 12, e1005766. [Google Scholar] [CrossRef] [PubMed]
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaic, V.; et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef] [PubMed]
- Facciuto, F.; Cavatorta, A.L.; Valdano, M.B.; Marziali, F.; Gardiol, D. Differential expression of PDZ domain-containing proteins in human diseases-challenging topics and novel issues. FEBS J. 2012, 279, 3538–3548. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Huibregtse, J.M. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.; Delury, C.; Marsh, E. The PDZ protein discs-large (DLG): The ‘Jekyll and Hyde’ of the epithelial polarity proteins. FEBS J. 2012, 279, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
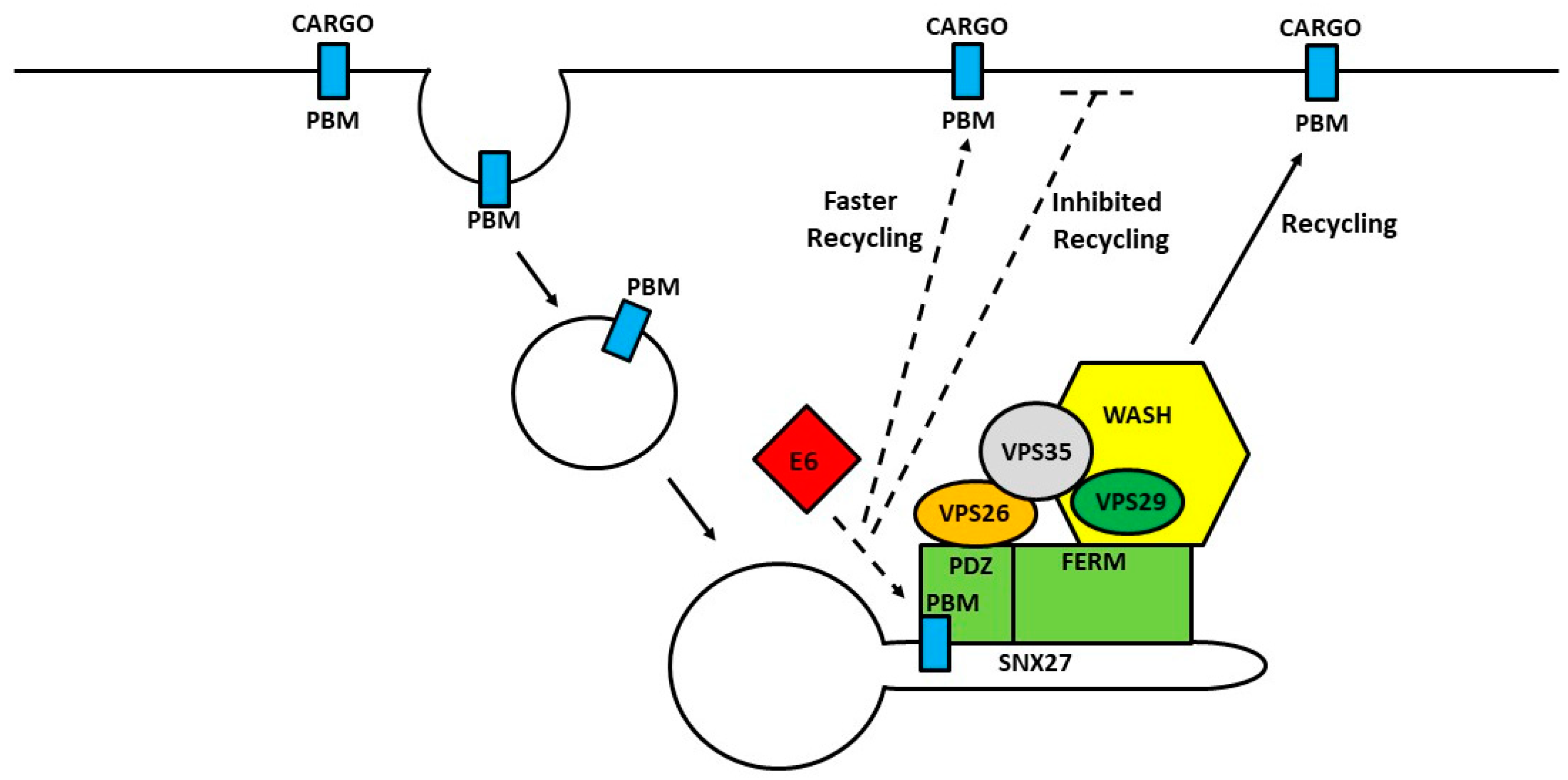
- Ganti, K.; Massimi, P.; Manzo-Merino, J.; Tomaic, V.; Pim, D.; Playford, M.P.; Lizano, M.; Roberts, S.; Kranjec, C.; Doorbar, J.; et al. Interaction of the Human Papillomavirus E6 Oncoprotein with Sorting Nexin 27 Modulates Endocytic Cargo Transport Pathways. PLoS Pathog. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I.; Nusrat, A.; Parkos, C.A. Endocytosis of the apical junctional complex: Mechanisms and possible roles in regulation of epithelial barriers. Bioessays 2005, 27, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Shivas, J.M.; Morrison, H.A.; Bilder, D.; Skop, A.R. Polarity and endocytosis: Reciprocal regulation. Trends Cell Biol. 2010, 20, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Belotti, E.; Polanowska, J.; Daulat, A.M.; Audebert, S.; Thome, V.; Lissitzky, J.C.; Lembo, F.; Blibek, K.; Omi, S.; Lenfant, N.; et al. The human PDZome: A gateway to PSD95-Disc large-zonula occludens (PDZ)-mediated functions. Mol. Cell. Proteom. 2013, 12, 2587–2603. [Google Scholar] [CrossRef] [PubMed]
- Joubert, L.; Hanson, B.; Barthet, G.; Sebben, M.; Claeysen, S.; Hong, W.; Marin, P.; Dumuis, A.; Bockaert, J. New sorting nexin (SNX27) and NHERF specifically interact with the 5-HT4a receptor splice variant: Roles in receptor targeting. J. Cell Sci. 2004, 117, 5367–5379. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, A.J.; Mansour, M.; Pohajdak, B. Sorting nexin 27 interacts with the Cytohesin associated scaffolding protein (CASP) in lymphocytes. Biochem. Biophys. Res. Commun. 2007, 359, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.P.; Hueschen, C.L.; Malide, D.; Milgram, S.L.; Playford, M.P. Sorting nexin 27 (SNX27) associates with zonula occludens-2 (ZO-2) and modulates the epithelial tight junction. Biochem. J. 2013, 455, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Lauffer, B.E.; Melero, C.; Temkin, P.; Lei, C.; Hong, W.; Kortemme, T.; von Zastrow, M. SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J. Cell Biol. 2010, 190, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Temkin, P.; Lauffer, B.; Jager, S.; Cimermancic, P.; Krogan, N.J.; von Zastrow, M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat. Cell Biol. 2011, 13, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Asahi, M. beta1-adrenergic receptor recycles via a membranous organelle, recycling endosome, by binding with sorting nexin27. J. Membr. Biol. 2013, 246, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Dreier, K.; Scheiden, R.; Lener, B.; Ehehalt, D.; Pircher, H.; Muller-Holzner, E.; Rostek, U.; Kaiser, A.; Fiedler, M.; Ressler, S. Subcellular localization of the human papillomavirus 16 E7 oncoprotein in CaSki cells and its detection in cervical adenocarcinoma and adenocarcinoma in situ. Virology 2011, 409, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Laurson, J.; Raj, K. Localisation of human papillomavirus 16 E7 oncoprotein changes with cell confluence. PLoS ONE 2011, 6, e21501. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Songock, W.K.; Kim, S.M.; Bodily, J.M. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res. 2017, 231, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Yarden, Y. Endocytosis and cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef] [PubMed]
- Corallino, S.; Malabarba, M.G.; Zobel, M.; Di Fiore, P.P.; Scita, G. Epithelial-to-Mesenchymal Plasticity Harnesses Endocytic Circuitries. Front. Oncol. 2015, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. The human papillomavirus E7 oncoprotein. Virology 2009, 384, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Szul, T.; Sztul, E. COPII and COPI traffic at the ER-Golgi interface. Physiology 2011, 26, 348–364. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siddiqa, A.; Broniarczyk, J.; Banks, L. Papillomaviruses and Endocytic Trafficking. Int. J. Mol. Sci. 2018, 19, 2619. https://doi.org/10.3390/ijms19092619
Siddiqa A, Broniarczyk J, Banks L. Papillomaviruses and Endocytic Trafficking. International Journal of Molecular Sciences. 2018; 19(9):2619. https://doi.org/10.3390/ijms19092619
Chicago/Turabian StyleSiddiqa, Abida, Justyna Broniarczyk, and Lawrence Banks. 2018. "Papillomaviruses and Endocytic Trafficking" International Journal of Molecular Sciences 19, no. 9: 2619. https://doi.org/10.3390/ijms19092619
APA StyleSiddiqa, A., Broniarczyk, J., & Banks, L. (2018). Papillomaviruses and Endocytic Trafficking. International Journal of Molecular Sciences, 19(9), 2619. https://doi.org/10.3390/ijms19092619