1. Introduction
Alcohol abuse causes alcoholic liver disease (ALD), which is a major cause of morbidity and mortality worldwide [
1]. Chronic alcohol consumption leads to socio-economic and public health problems [
2]. Hepatic steatosis, characterized by extensive triglyceride (TG) accumulation, occurs in the initial stages of ALD, and chronic alcohol intake causes hepatic steatosis to progress to the advanced stages, leading to steatohepatitis, fibrosis, cirrhosis, and liver cancer [
3]. Ethanol-induced hepatic steatosis is caused by increased de novo lipogenesis, decreased fatty acid oxidation, and the export of very low-density lipoprotein from the liver [
3]. In addition, alcohol exposure stimulates lipolysis in the adipose tissue, which increases the influx of fatty acid to the liver [
3].
Enhanced lipogenesis is one of the major reasons for the development of hepatic steatosis [
4]. Sterol regulatory element-binding protein-1c (SREBP-1c) is an essential transcription factor for de novo lipogenesis, which upregulates downstream lipogenesis genes, including fatty acid synthase (
FAS), stearoyl CoA desaturase 1 (
SCD1), acetyl-CoA carboxylase (
ACC), ATP-citrate lyase (
ACLY), and diacylglycerol acyltransferase (
DGAT) [
5]. Alcohol exposure stimulates the expression of
SREBP-1c and its target lipogenesis genes, leading to hepatic steatosis [
6]. Decreased fatty acid oxidation also plays an important role in the development of hepatic steatosis [
3]. Peroxisome proliferator-activated receptor-α (PPARα) is an important transcription factor that causes fatty acid oxidation by upregulating carnitine palmitoyltransferase 1 (
CPT1) and acyl-coenzyme A oxidase (
ACO) [
7]. Alcohol exposure downregulates the expression of fatty acid oxidation-related genes, including
PPARα,
CPT1, and
ACO [
7].
Reactive oxygen species (ROS) is also a major contributing factor to the pathogenesis of ALD [
3]. Alcohol exposure enhances the generation of ROS, including superoxide, hydroxyl radical, and hydrogen peroxide, and suppresses the cellular antioxidant defense system, which leads to oxidative stress in the liver. Several pathways are involved in alcohol-induced oxidative stress [
3]. The microsomal cytochrome P450 2E1 (CYP2E1) enzyme catalyzes the conversion of ethanol to acetaldehyde in the presence of iron, and is a major contributor to ROS generation [
8]. The expression and activity of CYP2E1 are upregulated due to alcohol exposure, which leads to liver injury [
9] and the suppression of CYP2E1 activity reduces the incidence of ethanol-induced fatty liver [
10]. Thus, CYP2E1 activation is critical to ALD pathogenesis. Furthermore, alcohol exposure impairs enzymatic and non-enzymatic antioxidant systems that protect hepatocytes against ROS damage [
11]. Endogenous antioxidant enzymes involved in ROS elimination include superoxide dismutase (SOD), catalase (CAT), glutathione (GSH)-S-transferase (GST), and glutathione peroxidase (GPX), which are involved in the primary defense against oxidative damage. Non-enzymatic antioxidants, such as GSH as well as vitamins C and E, also play an important role in protecting the cell against lipid peroxidation. Thus, the inhibition of CYP2E1 or the activation of antioxidant systems facilitates the elimination of ROS and protects against ALD.
The fruits of
Schisandra chinensis have been used for medicinal purposes in Asia [
12].
S. chinensis extract exhibits a wide spectrum of pharmacological activities, such as anti-aging, anti-melanogenic, anti-inflammatory, and anticancer activities [
12]. Previously, we reported that
S. chinensis extracts having protective effects against endoplasmic stress (ER) stress-induced hepatic steatosis [
13]. Numerous types of lignans have been isolated from
S. chinensis fruits, including gomisin N (GN), which are reported to have hepatoprotective, anticancer, anti-inflammatory and anti-melanogenic activities [
14]. Recently, we demonstrated that GN protects against nonalcoholic fatty liver disease (NAFLD) by inhibiting ER stress and activating AMP-activated protein kinase (AMPK) [
15,
16]. However, the protective effect of GN against ALD and its underlying mechanisms have still not been elucidated. As previously mentioned, TG accumulation, oxidative stress, and inflammation have been implicated in the pathogenesis of ALD, and thus, their inhibition may be a potential treatment strategy against ALD.
Therefore, the present study was designed to investigate the potential protective effects of GN against ethanol-induced liver injury and explore the related mechanisms in a chronic-binge alcohol mouse model and ethanol-treated HepG2 cells. Our findings suggest that GN exerts potential hepatoprotective effects against ethanol-induced liver injury by inhibiting hepatic steatosis, oxidative stress, and inflammation both in vivo and in vitro.
3. Discussion
Although ALD causes an economic burden and has serious effects on public health, there are few satisfactory therapeutic approaches to prevent or treat the progression of ALD [
4]. To date, numerous studies have attempted to discover bioactive components from herbal sources for the prevention and treatment of ALD [
20]. Previously, we reported that GN, a phytochemical derived from
S. chinensis, inhibits high-fat diet-induced hepatic steatosis by inhibiting endoplasmic reticulum stress and activating AMPK [
15,
16]. In the present study, we investigated whether GN also prevents ALD. We demonstrated that GN administration protected against ethanol-induced hepatic steatosis, oxidative stress, and inflammation in chronic-binge alcohol feeding mice. Consistent with in vivo results, GN downregulated lipogenesis genes and upregulated fatty acid oxidation genes in ethanol-treated HepG2 cells, thereby reducing intracellular TG accumulation. Furthermore, GN suppressed ethanol-induced ROS production and inflammation. GN reversed ethanol-mediated inhibition of SIRT1 and AMPK in both mice and HepG2 cells, and inhibited the expression and activity of CYP2E1.
The pathogenesis of ALD is associated with increased hepatic TG accumulation, oxidative stress, and inflammatory cytokines [
3]. Hepatic steatosis is the earliest stage of the broad spectrum of conditions that constitute ALD, and continuous alcohol intake causes a progression to the advanced disease stage due to oxidative stress and inflammation [
3]. Therefore, we hypothesized that the inhibition of TG accumulation, oxidative stress, and inflammation might be a potential strategy to prevent the progression of ALD. Hepatic TG accumulation is caused by enhanced de novo lipogenesis and impaired fatty acid oxidation [
3]. Alcohol intake stimulates lipogenesis by upregulating SREBP-1c and its downstream lipogenesis genes [
6], whereas it lowers fatty acid oxidation by downregulating PPARα-PGC-1α and its target fatty acid oxidation genes [
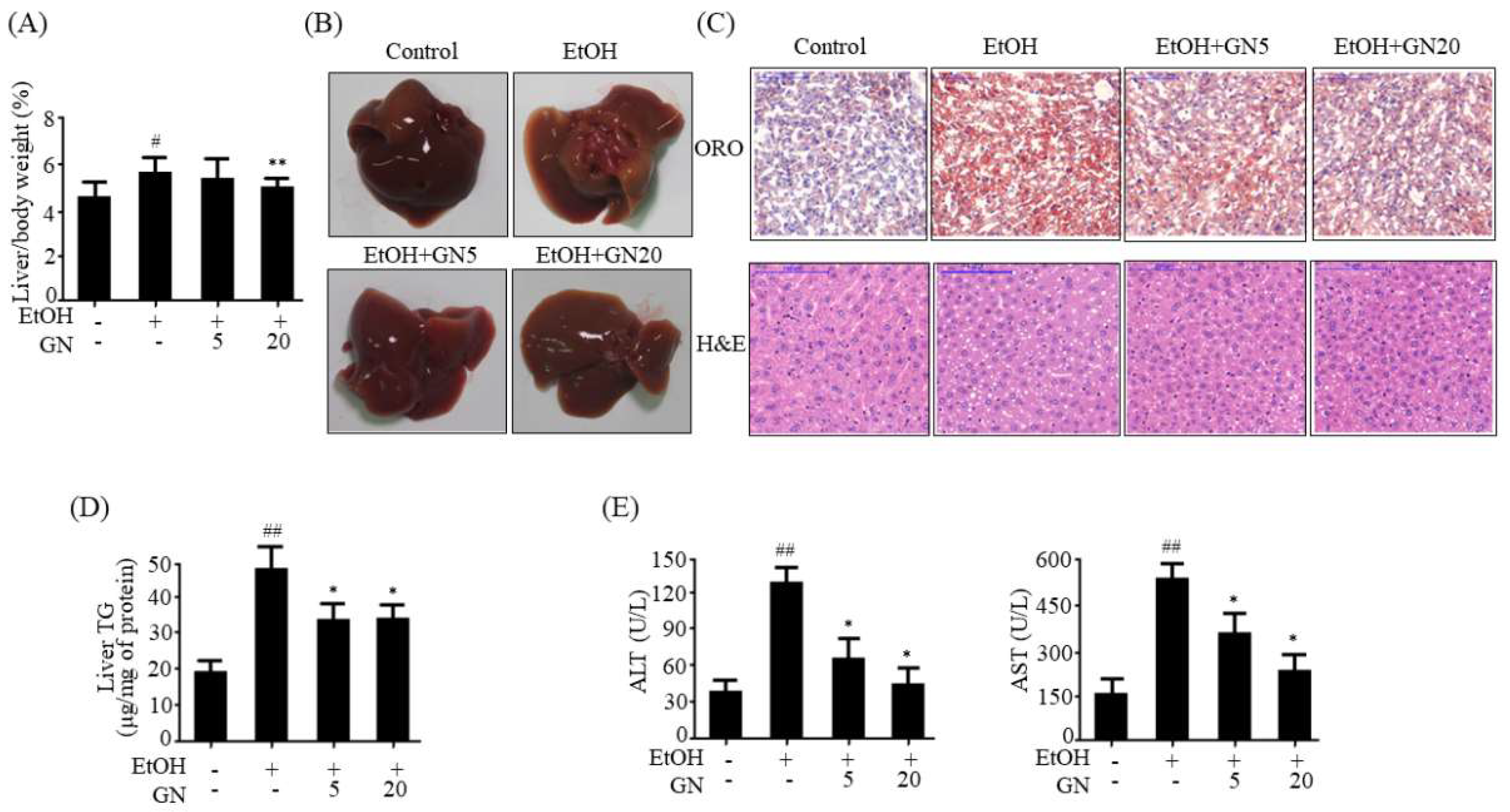
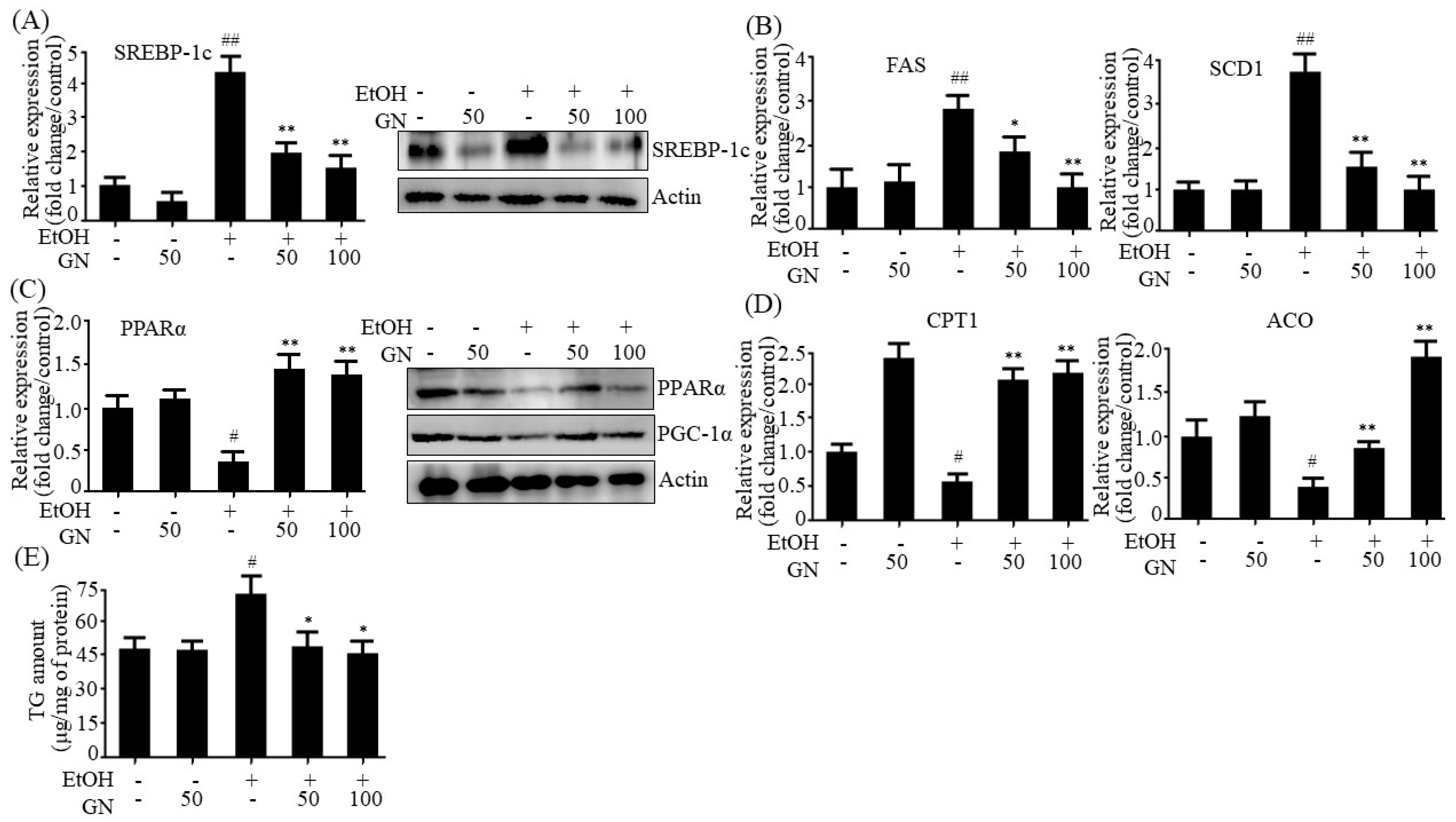
7]. Hence, we assessed the protective effects of GN against ethanol-induced hepatic steatosis in chronic-binge alcohol-fed mice and HepG2 cells. Our results showed that ethanol feeding induces hepatic steatosis, as observed by the measurement of hepatic TG accumulation and histological examination. However, GN administration efficiently blocked hepatic TG accumulation and converted the white-colored fatty liver into a relatively healthy liver. Moreover, GN administration reduced ethanol-induced serum levels of ALT and AST, indicating that GN ameliorates ethanol-induced hepatic injury and toxicity by attenuating hepatic steatosis. Furthermore, we observed that GN administration decreases the ethanol-induced expression of lipogenesis genes in ethanol-fed mice liver and HepG2 cells, but increases the expression of fatty acid oxidation genes, indicating that GN prevents ethanol-induced hepatic steatosis by decreasing lipogenesis and increasing fatty acid oxidation both in vivo and in vitro.
Sirtuins are a group of highly conserved NAD
+-dependent histone and protein deacetylases and/or ADP-ribosyl transferases that play important functions in numerous biological processes by regulating the transcriptional activation and/or gene expression of several crucial transcription factors and transcription co-activators that are involved in metabolic homeostasis [
17]. SIRT1 inhibits hepatic de novo lipogenesis by deacetylating lipogenesis transcription factors, such as SREBP-1c and carbohydrate-responsive element-binding protein (ChREBP), stimulates fatty acid β-oxidation by deacetylating the fatty acid oxidation transcription factor and coactivator PPARα/PGC-1α, inhibits hepatic oxidative stress by enhancing the antioxidant system, and reduces inflammation by deacetylating NF-κB in the liver. Recent study demonstrated that SIRT1 activation could protect against alcohol-induced liver diseases [
17]. Furthermore, SIRT1 and AMPK regulate each other’s functions. SIRT1 can stimulate AMPK by increasing cellular NAD
+ levels that is used as a substrate for SIRT1 activity [
18,
19]. In this regard, we investigated whether SIRT1-AMPK activation is associated with the beneficial protective effect of GN against ALD. Ethanol exposure significantly suppressed the levels of SIRT1 and phosphorylated AMPK in the liver of ethanol-fed mice and HepG2 cells, but GN administration blocked this reduction. In addition, we examined the effects of GN on cellular NAD
+ level in ethanol-treated HepG2 cells because NAD is a stimulator for SIRT1 activity. Ethanol treatment resulted in decreased NAD
+/NADH ratio; however, pretreatment with GN reversed the reduction. In consistent with this result, GN recovered NAD/NADH ratio in SK-Hep1 cells (
Figure S1). Moreover, the inhibition of SIRT1 using a SIRT1 inhibitor significantly prevented ethanol-induced cellular TG accumulation in HepG2 cells. Based on these results, although we have not measured the acetylated SREBP and PPARα/PGC-1α levels, GN-mediated SIRT1 activation might contribute to inhibition of lipogenesis and stimulation of fatty acid oxidation, thereby leading to decreased TG accumulation. To provide evidence for the involvement of SIRT1 in TG accumulation, the acetylated SREBP and PPARα/PGC-1α levels should be determined in further study. AMPK also plays a key role in the regulation of lipid and glucose metabolism [
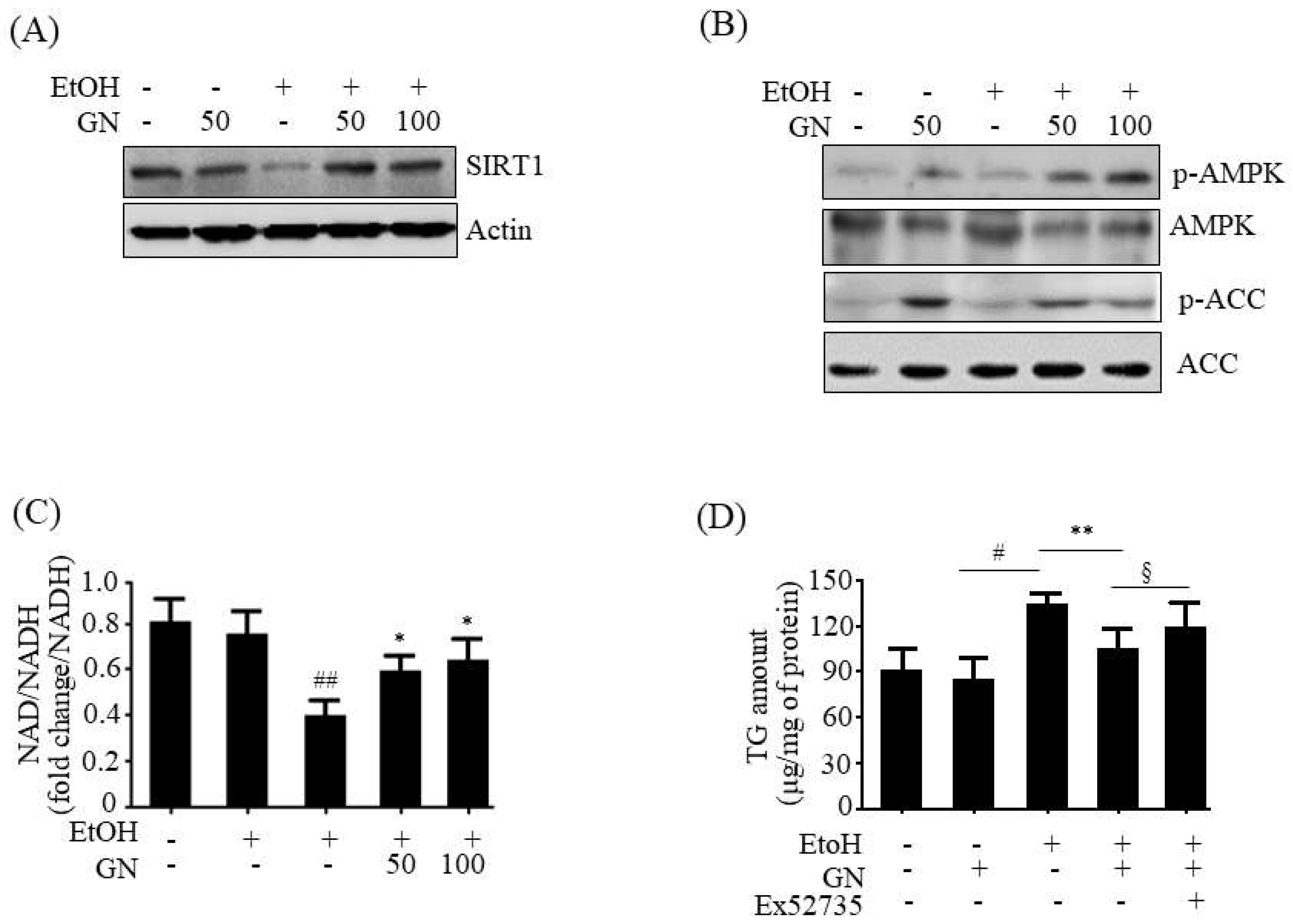
21]. AMPK can phosphorylate SREBP-1c and inhibit its transcriptional activity, thereby decreasing lipogenesis. In contrast, AMPK stimulates fatty acid oxidation by upregulating fatty acid oxidation genes and inactivating ACC1 that catalyzes the synthesis of malonyl-CoA, a potent inhibitor of CPT1. Our present results showed that GN treatment efficiently enhanced AMPK phosphorylation in the liver of chronic-binge alcohol feeding mice and ethanol-treated HepG2 cells, which was consistent with reduced lipogenesis genes expression and increased fatty acid oxidation genes expression, suggesting that AMPK activation might also participate in GN-mediated inhibition of TG accumulation. Taken together, these results suggest that the SIRT1-AMPK pathway might play a critical role in the protection of GN against ethanol-induced hepatic steatosis.
Oxidative stress is also a contributing factor to ethanol-induced ALD pathogenesis [
3]. Alcohol exposure increases the generation of ROS. Among the several pathways involved in ROS generation by alcohol, CYP2E1 is considered to be the major contributor to ethanol-induced ROS generation [
10]. The expression and activity of CYP2E1 are enhanced due to alcohol intake and cause liver injury. Therefore, the inhibition of CYP2E1 expression could effectively block the progression of ALD. In the present study, we evaluated the inhibitory effects of GN on ethanol-induced CYP2E1 expression and subsequent ROS generation in both chronic-binge alcohol feeding mice and ethanol-treated HepG2 cells. Alcohol feeding significantly elevated the expression and activity of CYP2E1 in the liver of chronic-binge alcohol-fed mice and subsequently, increased hepatic MDA levels. However, GN administration markedly reversed these ethanol-induced effects. Ethanol treatment also elevated CYP2E1 expression and increased ROS production in HepG2; however, GN treatment reduced these ethanol-induced effects in HepG2 cells. These results suggest that GN might exert protective effects against ethanol-induced oxidative stress through inhibition of CYP2E1. Furthermore, alcohol exposure impairs enzymatic and non-enzymatic antioxidant systems that protect hepatocytes against ROS damage [
11]. Therefore, we investigated whether GN exerts antioxidant effects by reversing the ethanol-mediated impairment of the antioxidant system. The present results showed that binge alcohol feeding in mice and ethanol treatment in HepG2 cells not only repressed the expression of antioxidant genes, but also reduced GSH levels. However, GN administration reversed these ethanol-mediated effects, indicating that GN might also protect against ethanol-induced ROS generation by upregulating antioxidant systems. SIRT1 stimulates antioxidant genes expression via nuclear factor E2-related factor 2 (Nrf-2) activation [
22] or PGC-1α deacetylation [
23], and inhibits oxidative stress. Based on the present results that GN treatment upregulated antioxidant genes expression in the liver of GN-treated mice and HepG2 cells concomitant with SIRT1 activation, SIRT1 might also play a role in GN-mediated inhibition of ROS generation through upregulation of antioxidant defense.
Furthermore, inflammation also plays a crucial role in the pathogenesis of ALD [
3]. Our present results showed that ethanol exposure significantly upregulates inflammation gene expression in the liver of chronic-binge alcohol feeding mice and in ethanol-treated HepG2 cells. However, these effects were efficiently reversed by GN administration through inhibiting NF-κB activation. ROS can activate transcription factors such as NF-κB and activator protein 1 (AP-1), and induce inflammatory gene expression [
24]. The present study revealed that GN inhibited ethanol-induced ROS generation and NF-κB activation consistent with repression of inflammatory genes expression. These results suggest that GN-mediated inhibition of ROS generation might lead to repress inflammation through attenuation of NF-κB. Furthermore, SIRT1 can deacetylate NF-κB and inhibit its transcriptional activity, thereby reducing inflammatory genes expression. Therefore, SIRT1 activation might also play a role in GN-mediated suppression of inflammation. In addition, GN reduced ethanol-induced inflammation gene expression in SK-Hep1 cells (
Figure S2). Taken together, our present study demonstrated that GN inhibits ethanol-induced inflammation, which might prevent the progression of ALD.
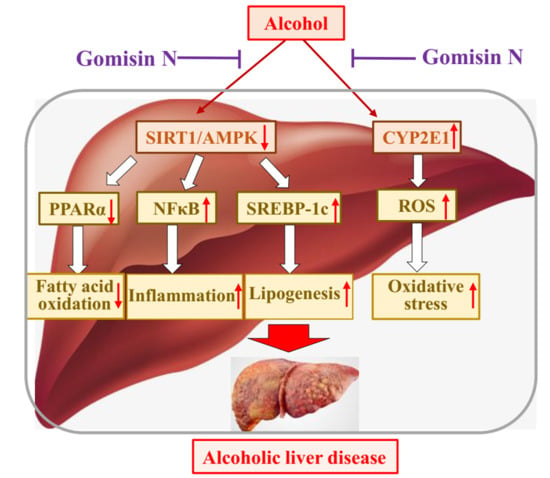
In conclusion, GN exerted a protective effect against ALD by inhibiting hepatic steatosis, oxidative stress, and inflammation both in vivo and in vitro. SIRT-AMPK activation and CYP2E1 inhibition might play a critical role in GN-mediated protection against ALD (
Figure 10). Our previous study also demonstrated the protective effect of GN against NAFLD [
10]. Altogether, GN could be a potential therapeutic agent for preventing both ALD and NAFLD, and conducting further clinical trials might be worthwhile.
4. Materials and Methods
4.1. Reagents
GN (≥98% purity) was obtained from ChemFaces (Wuhan, China). Antibodies against SREBP-1c (H-160; sc-8984), PPARα (H-2; sc-398394), PGC-1α (H-300; sc-13067), IκBα (H-4; sc-164), NF-κB p65 (F-6; sc-8008p65), SIRT1 (H-300; sc-15404), AMPK α1/2 (D-6; sc-74461), and β-actin (C4; sc-47778) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Specific primary antibodies against p-ACC (Ser79; #3661s) and ACC (C83B10; #3676s) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against CYP2E1 (ab28146) and p-AMPK α1 (Thr172; PA5-17831) were purchased from Abcam (Cambridge, MA, USA) and Invitrogen (Rockford, IL, USA), respectively. Ethanol and Ex52735 were obtained from Sigma (St. Louis, MO, USA).
4.2. Animal Experiments
All animal experiments were approved by Pusan National University’s Institutional Animal Care and Use Committee, in accordance with the established ethical and scientific care procedures (PNU-2017-1631: 28 July 2017). Male C57BL/6N mice (8-week-old, 20–22 g) were purchased from Doo Yeol Biotech (Seoul, South Korea). The mice were housed in a pathogen-free laboratory environment, maintained at 22 ± 2 °C temperature and 50–60% relative humidity, with a 12-h light-dark cycle throughout the experiment. All mice were randomly divided into 4 groups as follows: normal group (
n = 10), ethanol group (
n = 10), ethanol + GN-5 mg/kg (
n = 11) group, and ethanol + GN-20 mg/kg (
n = 11) group. Mice were fed according to the NIAAA model (chronic-binge) as described previously [
25]. The mice were initially fed the control Lieber-DeCarli liquid diet and ethanol Lieber-DeCarli diet (gradually increasing 0–5% ethanol concentrations) (Research Diets Inc., New Brunswick, NJ, USA) ad libitum for the first 5 days for acclimatization. Then, the feeding protocol was switched to chronic-binge feeding, in which mice were allowed free access to the ethanol Lieber-DeCarli diet containing 5% (
v/
v) ethanol for 10 days, and control-fed mice were pair-fed with an isocaloric control diet. GN-treated mice were orally administered daily 5 or 20 mg/kg of GN. On the 11th day, mice in ethanol groups were orally administered a single dose of ethanol (5 g/kg), and mice in control groups were orally administered dextrin maltose in the early morning. Further, the mice were euthanized, and blood and tissue samples were collected 9 h post oral administration. Isolated liver tissues were immediately frozen in liquid nitrogen and stored at −80 °C until further analyses.
4.3. Histopathological Analysis
Isolated liver tissues were dissected, fixed in 10% buffered formalin, embedded in paraffin, and then 5-μm sections were cut using a frozen microtome (HM560H, Microm International, Walldorf, Germany). The liver tissue sections were stained with hematoxylin and eosin (H&E) and then with oil red O (ORO); stained sections were then observed under a light microscope.
4.4. TG Measurement
HepG2 cell suspensions were mixed with 750 μL of chloroform/methanol/H2O (8:4:3, v/v/v) to extract TG. The cell suspensions were incubated at room temperature for 1 h and centrifuged at 800× g for 10 min. The bottom layer (organic phase) obtained was dried overnight, dissolved in ethanol, and then an enzyme reaction kit (Asan Pharmaceutical, Seoul, South Korea) was used to measure TG concentration, which was normalized to the protein concentration.
4.5. Biochemical Analysis
Blood samples were collected and centrifuged at 1000× g for 15 min at 4 °C to obtain serum samples, which were stored at −80 °C until analyses. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were determined by using FUJI DRI-CHEM 7000i (FUJI FILM, Tokyo, Japan).
4.6. Measurement of Hepatic Lipid Peroxidation
Hepatic lipid peroxidation was expressed in terms of malondialdehyde (MDA) concentration that was measured by the thiobarbituric acid reactive substances (TBARS) method using a commercially available kit (OxiSelect™ TBARS assay kit-MDA Quantitation, Cell Biolabs Inc., San Diego, CA, USA) according to the manufacturer’s instructions. Briefly, the liver tissue was homogenized in PBS containing 1× butylated hydroxytoluene solution on ice. After centrifugation at 10,000× g for 5 min at 4 °C, the supernatant was collected for the measurement of TBARS level. Absorbance was measured at 532 nm using a microplate reader.
4.7. Measurement of CYP2E1 Enzyme Activity
Mouse liver microsomes were prepared as described previously [
26]. Briefly, the liver tissues were homogenized in 125 mM KCl and 10 mM KPi buffer (pH 7.4), and centrifuged at 600×
g for 10 min. The supernatant was removed and centrifuged twice at 8500×
g for 10 min. Finally, the supernatant was centrifuged at 100,000×
g for 60 min and the pellet containing the microsomes was obtained. The pellet was resuspended in 125 mM KCl-10 mM KPi (pH 7.4) and the protein concentration was determined using the Bio-Rad DC Protein Assay Kit (Bio-Rad, Hercules, CA, USA).
Microsomal CYP2E1 (p-nitrophenol-2-hydroxylase) activity was assayed as described previously [
27]. Briefly, the microsomes were incubated in an assay buffer containing KPi buffer (50 mM, pH 7.4), NADPH (1 mM), and p-nitrophenol (0.1 mM) at 37 °C using a shaking water bath. After 30 min, the reaction was stopped by the addition of 500 μL of 0.75 N perchloride acid, and centrifuged at 5000×
g for 45 min. Subsequently, 250 μL of 2 M NaOH was added to the supernatant, and the absorbance was measured at 546 nM spectrophotometrically.
4.8. Measurement of Liver GSH Level
GSH level in the liver was measured with a commercially available kit (K261-100, BioVision, CA, USA) following the manufacturer’s protocol. Briefly, liver tissues were homogenized with cold GSH assay buffer, and then centrifuged for 10 min at 8000× g. The supernatant was collected and GSH level was measured based on the absorbance at 405 nm using a microplate reader.
4.9. Cell Culture
The HepG2 and SK-HEP-1 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and grown in DMEM supplemented with 10% heat-inactivated FBS, penicillin (100 units/mL), and streptomycin sulfate (100 μg/mL) at 37 °C in an atmosphere of 5% CO2.
4.10. Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was isolated from mouse livers or HepG2 cells using TRIzolTM (Invitrogen). Isolated RNA (1 µg) was reverse-transcribed using TOPScript RT DryMix (Enzynomics, Daejeon, Korea). Quantitative real-time PCR was performed using a SYBR Green premixed Taq reaction mixture with gene-specific primers as listed in
Table S1.
4.11. Western Blot Analysis
Protein lysates from liver tissues or HepG2 cells were prepared using Pro-Prep Protein Extraction Solution (Intron Biotechnology, Seoul, Korea) according to the manufacturer’s instructions. The protein lysates (50 μg) were subjected to SDS-PAGE and then transferred to polyvinylidene difluoride membranes (Amersham Pharmacia Biotech, Amersham, UK). The membranes were incubated with specific primary antibodies (1:1000) at 4 °C overnight followed by incubation with anti-rabbit or anti-mouse secondary antibodies (1:1000) (Santa Cruz Biotechnology) conjugated to peroxidase, and protein bands were visualized using an enhanced chemiluminescence system (ECL Advance, GE Healthcare, Hatfield, UK).
4.12. Determination of Intracellular ROS Levels
HepG2 cells were treated with ethanol (50 mM) for 3 h after incubating with GN (50 μM) for 3 h, and the ROS level was measured using the fluorescent dye, 2,7-dichlorofluorescein diacetate (DCFH-DA, Sigma) by flow cytometry with the FACSCantoII (BD Biosciences, San Jose, CA, USA). Briefly, HepG2 cells were suspended in PBS with DCFH-DA (10 μM) at 37 °C for 30 min. Then, the cells were harvested and fluorescence was determined using the FACS Diva 6.3.1 software (BD Biosciences).
4.13. NAD+/NADH Ratio Measurement
The NAD+/NADH ratio in HepG2 cells was measured with the NAD+/NADH quantification kit (K377-100; Biovision) based on an enzymatic cycling reaction, according to the manufacturer’s instructions.
4.14. Statistical Analysis
All data are presented as the mean ± SD. The data were analyzed using one-way ANOVA, and the differences between means were determined using the Tukey–Kramer post hoc test. Values were considered statistically significant at p < 0.05.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









