Combined Modulation of Tumor Metabolism by Metformin and Diclofenac in Glioma



 and
and
Abstract
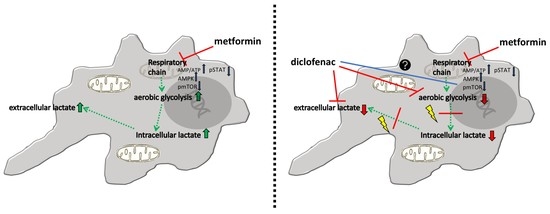
1. Introduction
2. Results
2.1. Stem Cell-Like BTICs Express Nestin and SOX
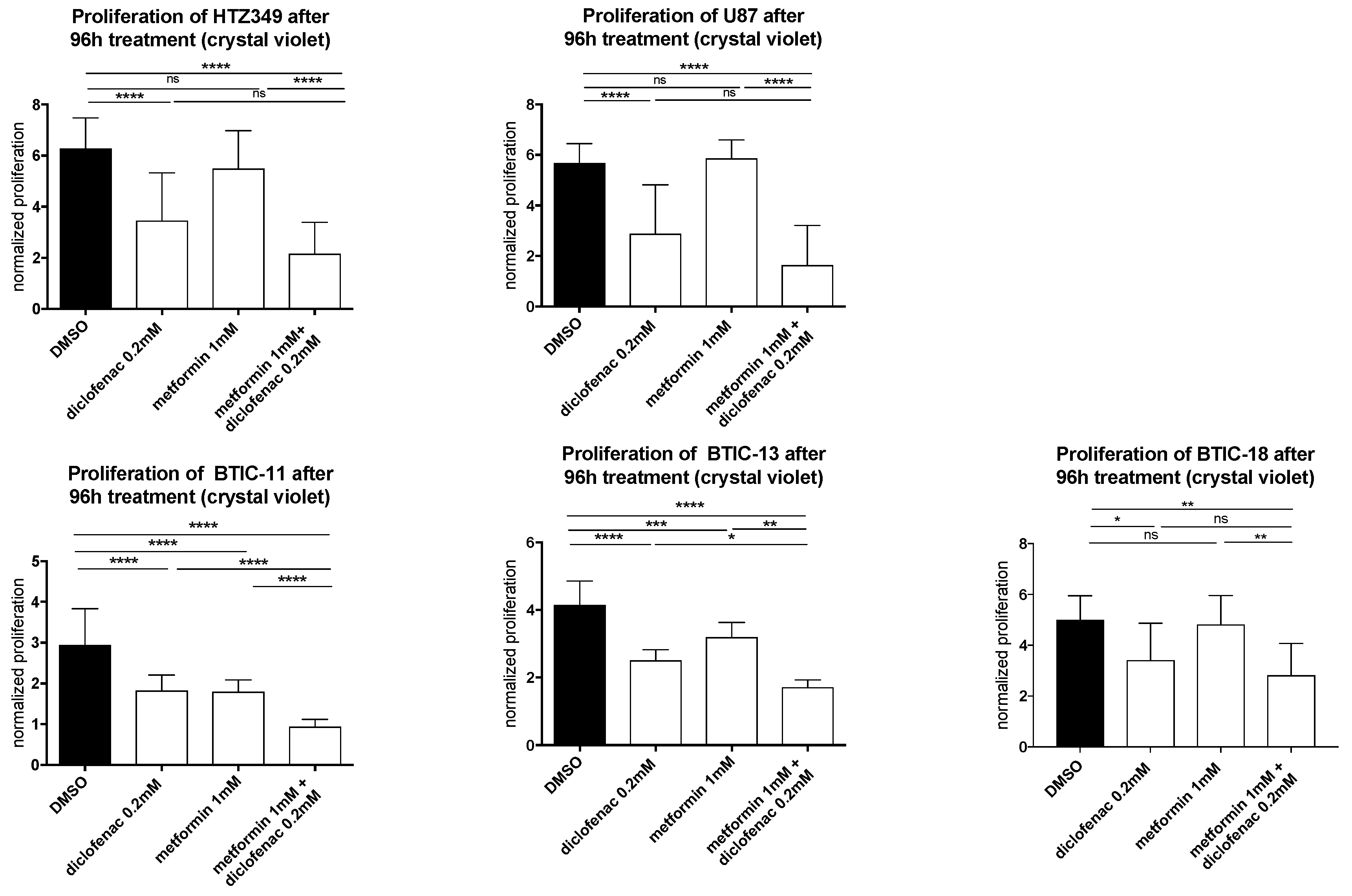
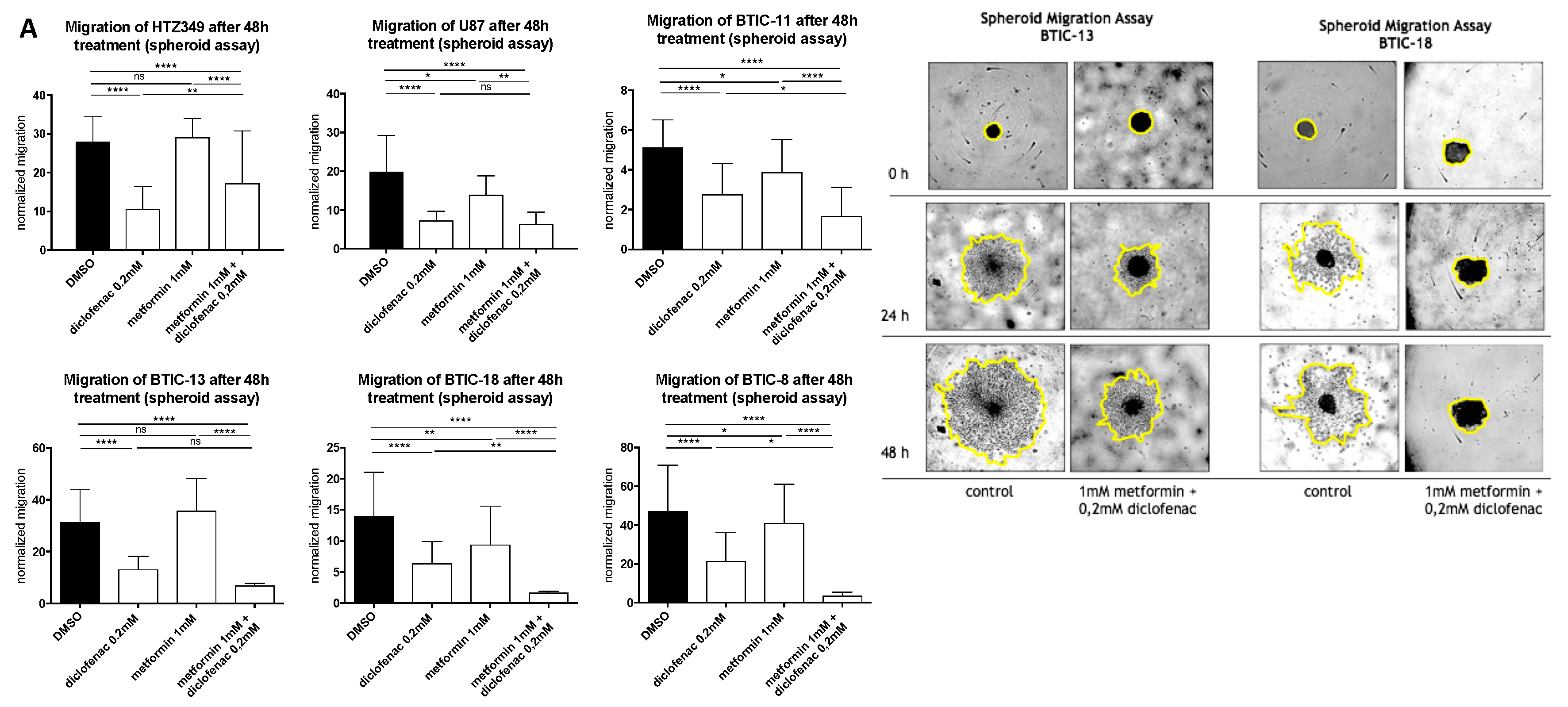
2.2. Combined Treatment of Metformin and Diclofenac Impairs Cell Proliferation and Migration
2.3. Metformin, Diclofenac, and Combined Treatment at Low Doses Do Not Increase Cell Death
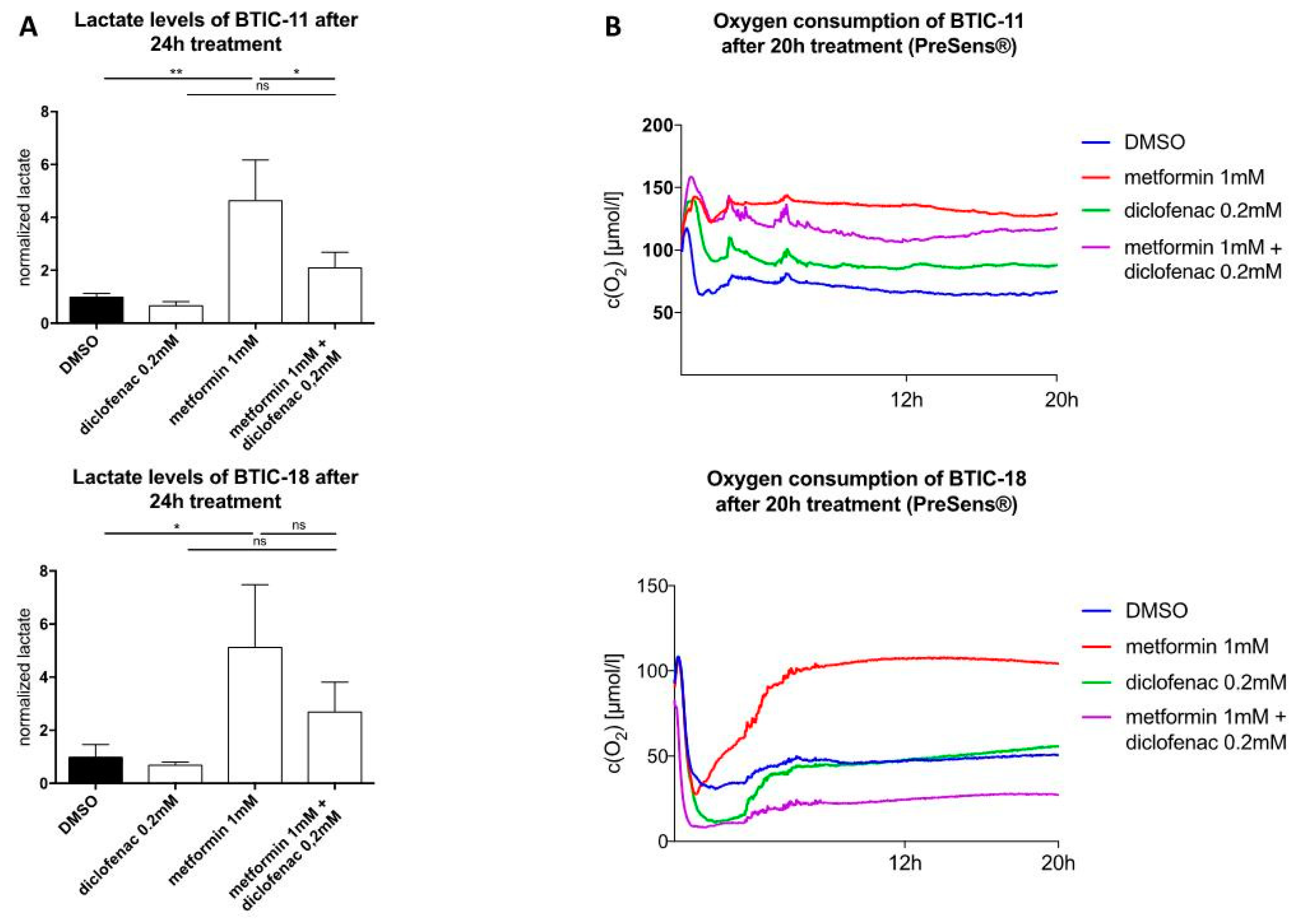
2.4. Combined Treatment of Metformin and Diclofenac Reduces Extracellular Lactate Levels Compared to Metformin Treatment. Effects on Oxygen Consumption Differ between Cell Lines
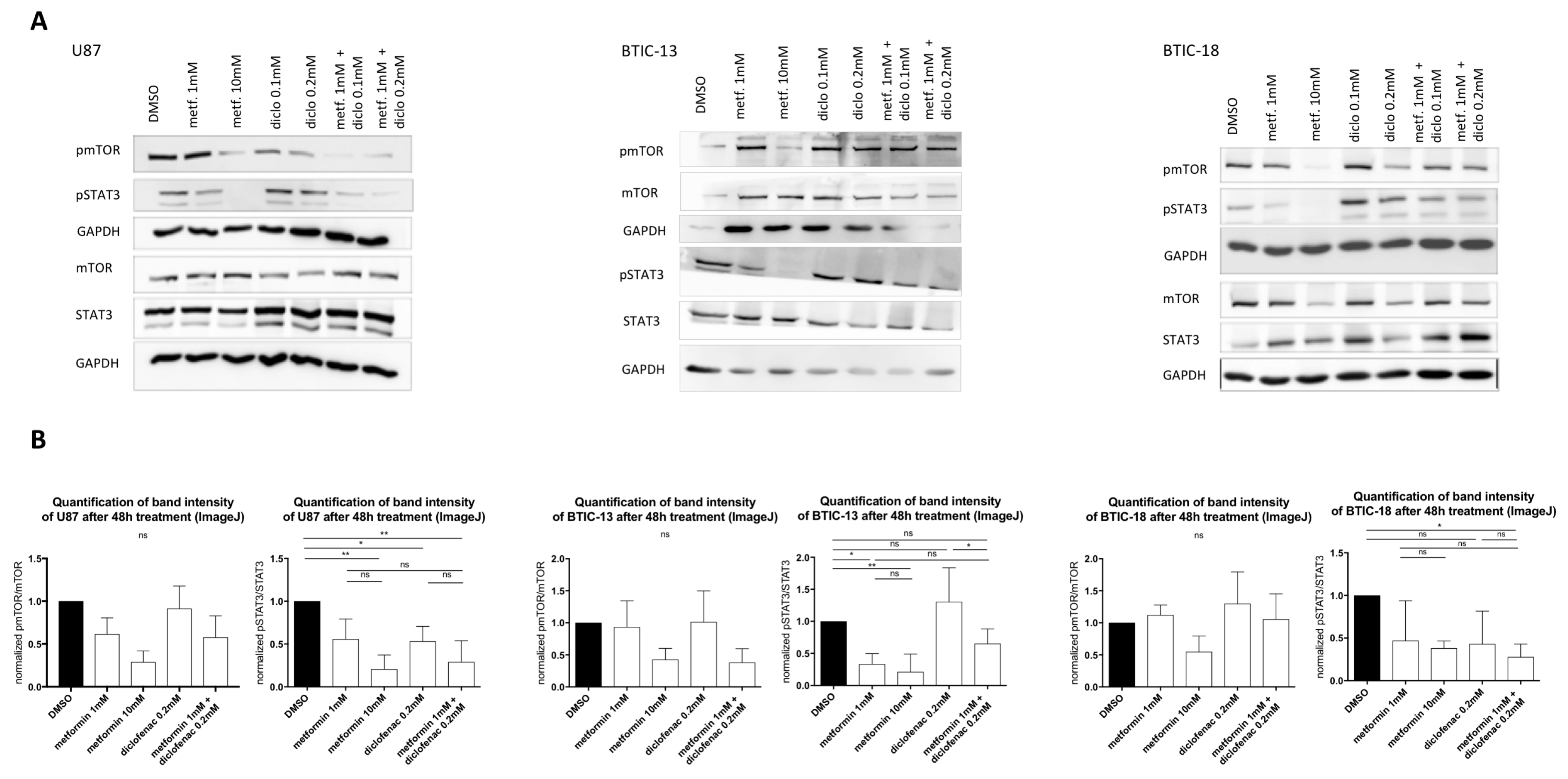
2.5. Combined Treatment Doesn’t Lead to Additive Effects on MTor or STAT3 (Signal Transducer and Activator of Transcription 3) Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Tumor Cell Lines
4.3. Proliferation Assay
4.4. Migration Assay
4.5. Protein Isolation and Western Blot
4.6. LDH Activity Measurement
4.7. Online Measurement of Oxygen Concentration and PH Levels in Cell Culture
4.8. Extracellular Lactate Levels
4.9. Combination Index
4.10. Immunocytochemistry
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. CENTRIC study team Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Das, S.; Srikanth, M.; Kessler, J.A. Cancer stem cells and glioma. Nat. Clin. Pract. Neurol. 2008, 4, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Goodarzi, M.O.; Bryer-Ash, M. Metformin revisited: Re-evaluation of its properties and role in the pharmacopoeia of modern antidiabetic agents. Diabetes Obes. Metab. 2005, 7, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.M.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS ONE 2013, 8, e71583. [Google Scholar] [CrossRef] [PubMed]
- Sesen, J.; Dahan, P.; Scotland, S.J.; Saland, E.; Dang, V.-T.; Lemarié, A.; Tyler, B.M.; Brem, H.; Toulas, C.; Moyal, E.C.-J.; et al. Metformin Inhibits Growth of Human Glioblastoma Cells and Enhances Therapeutic Response. PLoS ONE 2015, 10, e0123721. [Google Scholar] [CrossRef] [PubMed]
- Würth, R.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Corsaro, A.; Parodi, A.; Sirito, R.; Massollo, M.; Marini, C.; Zona, G.; et al. Metformin selectively affects human glioblastoma tumor-initiating cell viability. Cell Cycle 2013, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Seliger, C.; Meyer, A.-L.; Renner, K.; Leidgens, V.; Moeckel, S.; Jachnik, B.; Dettmer, K.; Tischler, U.; Gerthofer, V.; Rauer, L.; et al. Metformin inhibits proliferation and migration of glioblastoma cells independently of TGF-β2. Cell Cycle 2016, 15, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Sunayama, J.; Okada, M.; Watanabe, E.; Seino, S.; Shibuya, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem Cells Transl. Med. 2012, 1, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Gravel, S.-P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Seliger, C.; Renner, K.; Kreutz, M.; Meyer, A.; Jachnik, B.; Bogdahn, U.; Vollmann-Zwerenz, A.; Hau, P. P17.39turn Old into New: Metformin as Antineoplastic Agent in Glioblastoma. Neuro-Oncol. 2014, 16, ii96. [Google Scholar] [CrossRef]
- Seliger, C.; Ricci, C.; Meier, C.R.; Bodmer, M.; Jick, S.S.; Bogdahn, U.; Hau, P.; Leitzmann, M.F. Diabetes, use of antidiabetic drugs, and the risk of glioma. Neuro-Oncol. 2016, 18, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Chirasani, S.R.; Leukel, P.; Gottfried, E.; Hochrein, J.; Stadler, K.; Neumann, B.; Oefner, P.J.; Gronwald, W.; Bogdahn, U.; Hau, P.; et al. Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. Int. J. Cancer 2013, 132, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.I.; DuBois, R.N. NSAIDs and cancer prevention: Targets downstream of COX-2. Annu. Rev. Med. 2007, 58, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Taketo, M.M. Cyclooxygenase-2 Inhibitors in Tumorigenesis (Part I). J. Natl. Cancer Inst. 1998, 90, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Taketo, M.M. Cyclooxygenase-2 inhibitors in tumorigenesis (Part II). J. Natl. Cancer Inst. 1998, 90, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Beebe-Donk, J.; Alshafie, G.A. Reduction in the risk of human breast cancer by selective cyclooxygenase-2 (COX-2) inhibitors. BMC Cancer 2006, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Abnet, C.C.; Freedman, N.D.; Kamangar, F.; Leitzmann, M.F.; Hollenbeck, A.R.; Schatzkin, A. Non-steroidal anti-inflammatory drugs and risk of gastric and oesophageal adenocarcinomas: Results from a cohort study and a meta-analysis. Br. J. Cancer 2009, 100, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Lang, S.A.; Renner, K.; Bosserhoff, A.; Gronwald, W.; Rehli, M.; Einhell, S.; Gedig, I.; Singer, K.; Seilbeck, A.; et al. New aspects of an old drug—iclofenac targets MYC and glucose metabolism in tumor cells. PLoS ONE 2013, 8, e66987. [Google Scholar] [CrossRef] [PubMed]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: An in vivo study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Gonçalves, V.; Honavar, M.; Pinheiro, C.; Martinho, O.; Pires, M.M.; Pinheiro, C.; Cordeiro, M.; Bebiano, G.; Costa, P.; Palmeirim, I.; et al. Monocarboxylate transporters (MCTs) in gliomas: Expression and exploitation as therapeutic targets. Neuro-Oncol. 2013, 15, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Neradil, J.; Veselska, R. Nestin as a marker of cancer stem cells. Cancer Sci. 2015, 106, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Iacopino, F.; Angelucci, C.; Piacentini, R.; Biamonte, F.; Mangiola, A.; Maira, G.; Grassi, C.; Sica, G. Isolation of Cancer Stem Cells from Three Human Glioblastoma Cell Lines: Characterization of Two Selected Clones. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, V.; Seliger, C.; Jachnik, B.; Welz, T.; Leukel, P.; Vollmann-Zwerenz, A.; Bogdahn, U.; Kreutz, M.; Grauer, O.M.; Hau, P. Ibuprofen and Diclofenac Restrict Migration and Proliferation of Human Glioma Cells by Distinct Molecular Mechanisms. PLoS ONE 2015, 10, e0140613. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, V.; Proske, J.; Rauer, L.; Moeckel, S.; Renner, K.; Bogdahn, U.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Stattic and metformin inhibit brain tumor initiating cells by reducing STAT3-phosphorylation. Oncotarget 2016, 8, 8250–8263. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Rodin, S.; Simonson, O.E.; Hollande, F. Laminins and cancer stem cells: Partners in crime? Semin. Cancer Biol. 2017, 45, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Beckner, M.E.; Gobbel, G.T.; Abounader, R.; Burovic, F.; Agostino, N.R.; Laterra, J.; Pollack, I.F. Glycolytic glioma cells with active glycogen synthase are sensitive to PTEN and inhibitors of PI3K and gluconeogenesis. Lab. Investig. 2005, 85, 1457. [Google Scholar] [CrossRef] [PubMed]
- Isakovic, A.; Harhaji, L.; Stevanovic, D.; Markovic, Z.; Sumarac-Dumanovic, M.; Starcevic, V.; Micic, D.; Trajkovic, V. Dual antiglioma action of metformin: Cell cycle arrest and mitochondria-dependent apoptosis. Cell. Mol. Life Sci. 2007, 64, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Gritti, M.; Würth, R.; Angelini, M.; Barbieri, F.; Peretti, M.; Pizzi, E.; Pattarozzi, A.; Carra, E.; Sirito, R.; Daga, A.; et al. Metformin repositioning as antitumoral agent: Selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014, 5, 11252–11268. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Nerstedt, A.; Johansson, A.; Andersson, C.X.; Cansby, E.; Smith, U.; Mahlapuu, M. AMP-activated protein kinase inhibits IL-6-stimulated inflammatory response in human liver cells by suppressing phosphorylation of signal transducer and activator of transcription 3 (STAT3). Diabetologia 2010, 53, 2406–2416. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, M.; Lüscher, B. Proteins of the Myc network: Essential regulators of cell growth and differentiation. Adv. Cancer Res. 1996, 68, 109–182. [Google Scholar] [PubMed]
- Hirano, T.; Ishihara, K.; Hibi, M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 2000, 19, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.-L.; Bhattacharyya, K.K.; Dutta, S.K.; Bamlet, W.R.; Rabe, K.G.; Wang, E.; Smyrk, T.C.; Oberg, A.L.; Petersen, G.M.; Mukhopadhyay, D. Metformin suppresses pancreatic tumor growth with inhibition of NFκB/STAT3 inflammatory signaling. Pancreas 2015, 44, 636–647. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Yang, G.-L.; Yang, J.-H.; Lin, S.-L.; Liu, N.; Liu, S.-S.; Liu, M.-Y.; Zhang, L.-H.; Huang, Y.-R.; Shen, R.; et al. Metformin can block precancerous progression to invasive tumors of bladder through inhibiting STAT3-mediated signaling pathways. J. Exp. Clin. Cancer Res. 2015, 34, 77. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.-S.; Wang, S.; Deng, A.; Liu, B.; Edgerton, S.M.; Lind, S.E.; Wahdan-Alaswad, R.; Thor, A.D. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle 2012, 11, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Łabuzek, K.; Suchy, D.; Gabryel, B.; Bielecka, A.; Liber, S.; Okopień, B. Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol. Rep. 2010, 62, 956–965. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.-J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, S.; Dico, A.L.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [PubMed]
- Aldea, M.D.; Petrushev, B.; Soritau, O.; Tomuleasa, C.I.; Berindan-Neagoe, I.; Filip, A.G.; Chereches, G.; Cenariu, M.; Craciun, L.; Tatomir, C.; et al. Metformin plus sorafenib highly impacts temozolomide resistant glioblastoma stem-like cells. J. BUON Off. J. Balk. Union Oncol. 2014, 19, 502–511. [Google Scholar]
- Zhu, H.-Q.; Ma, J.-B.; Song, X.; Gao, H.-J.; Ma, C.-Q.; Chang, H.; Li, H.-G.; Liu, F.-F.; Lu, J.; Zhou, X. Metformin potentiates the anticancer activities of gemcitabine and cisplatin against cholangiocarcinoma cells in vitro and in vivo. Oncol. Rep. 2016, 36, 3488–3496. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Huang, Y.; Tao, T.; Peng, C.-Y.; Su, Q.; Xu, W.; Darko, K.O.; Tao, X.; Yang, X. Metformin and gefitinib cooperate to inhibit bladder cancer growth via both AMPK and EGFR pathways joining at Akt and Erk. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Ferdous, T.; Harada, T.; Ueyama, Y. Metformin in combination with 5-fluorouracil suppresses tumor growth by inhibiting the Warburg effect in human oral squamous cell carcinoma. Int. J. Oncol. 2016, 49, 276–284. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Bakhashab, S.; Ahmed, F.; Schulten, H.-J.; Ahmed, F.W.; Glanville, M.; Al-Qahtani, M.H.; Weaver, J.U. Proangiogenic Effect of Metformin in Endothelial Cells Is via Upregulation of VEGFR1/2 and Their Signaling under Hyperglycemia-Hypoxia. Int. J. Mol. Sci. 2018, 19, 293. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Saber, M.M.; Galal, M.A.; Ain-Shoka, A.A.; Shouman, S.A. Combination of metformin and 5-aminosalicylic acid cooperates to decrease proliferation and induce apoptosis in colorectal cancer cell lines. BMC Cancer 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Meena, A.S.; Bhat, M.K. Targeting metabolic flexibility by simultaneously inhibiting respiratory complex I and lactate generation retards melanoma progression. Oncotarget 2015, 6, 37281–37299. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.E.; Beebe-Donk, J.; Doss, H.; Burr Doss, D. Aspirin, ibuprofen, and other non-steroidal anti-inflammatory drugs in cancer prevention: A critical review of non-selective COX-2 blockade (review). Oncol. Rep. 2005, 13, 559–583. [Google Scholar] [CrossRef] [PubMed]
- Valle, B.L.; D’Souza, T.; Becker, K.G.; Wood, W.H.; Zhang, Y.; Wersto, R.P.; Morin, P.J. Non-steroidal anti-inflammatory drugs decrease E2F1 expression and inhibit cell growth in ovarian cancer cells. PLoS ONE 2013, 8, e61836. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Lindskog, M.; Ponthan, F.; Pettersen, I.; Elfman, L.; Orrego, A.; Sveinbjörnsson, B.; Kogner, P. NSAIDs in neuroblastoma therapy. Cancer Lett. 2005, 228, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Prostaglandins and cancer. Gut 2006, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J.; DuBois, R.N. COX-2: A target for colon cancer prevention. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 55–80. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Dubois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Futagi, Y.; Ideno, M.; Kobayashi, M.; Narumi, K.; Furugen, A.; Iseki, K. Effect of diclofenac on SLC16A3/MCT4 by the Caco-2 cell line. Drug Metab. Pharmacokinet. 2016, 31, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Vellonen, K.-S.; Häkli, M.; Merezhinskaya, N.; Tervo, T.; Honkakoski, P.; Urtti, A. Monocarboxylate transport in human corneal epithelium and cell lines. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2010, 39, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Bjerkvig, R.; Berens, M.E.; Westphal, M. Cost of Migration: Invasion of Malignant Gliomas and Implications for Treatment. J. Clin. Oncol. 2003, 21, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Lunetta, M.; DiMauro, M. Different effect of acute and chronic oral metformin administration on glucose and insulin response to bread and to pasta in non-insulin dependent diabetic patients. Diabetes Res. Clin. Pract. 1996, 33, 53–58. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Dietmaier, W.; Lorenz, J.; Riemenschneider, M.J. [Molecular diagnostics in neuropathology]. Pathol. 2015, 36, 171–180. [Google Scholar] [CrossRef]
- Bogdahn, U.; Apfel, R.; Hahn, M.; Gerlach, M.; Behl, C.; Hoppe, J.; Martin, R. Autocrine Tumor Cell Growth-inhibiting Activities from Human Malignant Melanoma. Cancer Res. 1989, 49, 5358–5363. [Google Scholar] [PubMed]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M.; et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CI-Proliferation | CI-Migration | Effect-Proliferation | Effect-Migration | |
|---|---|---|---|---|
| HTZ 349-laminin | 0.83699 | 0.38254 | synergistic | synergistic |
| BTIC-13-laminin | 0.95719 | >1 | synergistic | antagonistic |
| BTIC-18-laminin | 0.77451 | 0.68924 | synergistic | synergistic |
| U87-aminin | 0.7953 | 0.56451 | synergistic | synergistic |
| BTIC-11-laminin | 0.99912 | 0.62748 | synergistic | synergistic |
| BTIC-8-laminin | 0.74232 | 0.17014 | synergistic | synergistic |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerthofer, V.; Kreutz, M.; Renner, K.; Jachnik, B.; Dettmer, K.; Oefner, P.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Combined Modulation of Tumor Metabolism by Metformin and Diclofenac in Glioma. Int. J. Mol. Sci. 2018, 19, 2586. https://doi.org/10.3390/ijms19092586
Gerthofer V, Kreutz M, Renner K, Jachnik B, Dettmer K, Oefner P, Riemenschneider MJ, Proescholdt M, Vollmann-Zwerenz A, Hau P, et al. Combined Modulation of Tumor Metabolism by Metformin and Diclofenac in Glioma. International Journal of Molecular Sciences. 2018; 19(9):2586. https://doi.org/10.3390/ijms19092586
Chicago/Turabian StyleGerthofer, Valeria, Marina Kreutz, Kathrin Renner, Birgit Jachnik, Katja Dettmer, Peter Oefner, Markus J. Riemenschneider, Martin Proescholdt, Arabel Vollmann-Zwerenz, Peter Hau, and et al. 2018. "Combined Modulation of Tumor Metabolism by Metformin and Diclofenac in Glioma" International Journal of Molecular Sciences 19, no. 9: 2586. https://doi.org/10.3390/ijms19092586
APA StyleGerthofer, V., Kreutz, M., Renner, K., Jachnik, B., Dettmer, K., Oefner, P., Riemenschneider, M. J., Proescholdt, M., Vollmann-Zwerenz, A., Hau, P., & Seliger, C. (2018). Combined Modulation of Tumor Metabolism by Metformin and Diclofenac in Glioma. International Journal of Molecular Sciences, 19(9), 2586. https://doi.org/10.3390/ijms19092586