Macrophage Immunomodulation: The Gatekeeper for Mesenchymal Stem Cell Derived-Exosomes in Pulmonary Arterial Hypertension?

Abstract
1. Introduction
2. Macrophage-Mediated Immunity in PAH
3. Interplay of Monocytes/Macrophages with Other Immune Cell Types in PAH
4. Mesenchymal Stem/Stromal Cell (MSC) Based-Therapies in Experimental PH
5. Application of MSC-Exosome (MEx) Therapy in Experimental PH
6. MSC-Exosome (MEx) Therapy Ameliorates Core Physiological Features of Experimental PH
7. Modulation of Macrophage Function: The Gatekeeper of Exosome-Based Therapeutics
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tuder, R.M.; Archer, S.L.; Dorfmüller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Adnot, S.; Archer, S.L.; Dupuis, J.; Jones, P.L.; MacLean, M.R.; McMurtry, I.F.; Stenmark, K.R.; Thistlethwaite, P.A.; Weissmann, N.; et al. Cellular and molecular basis of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2009, 54, S20–S31. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; McGoon, M.; Torbicki, A.; Sitbon, O.; Krowka, M.J.; Olschewski, H.; Gaine, S. Diagnosis and differential assessment of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 40S–47S. [Google Scholar] [CrossRef] [PubMed]
- Farber, H.W.; Loscalzo, J. Pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Gall, H.; Felix, J.F.; Schneck, F.K.; Milger, K.; Sommer, N.; Voswinckel, R.; Franco, O.H.; Hofman, A.; Schermuly, R.T.; Weissmann, N.; et al. The Giessen pulmonary hypertension registry: Survival in pulmonary hypertension subgroups. J. Heart Lung Transplant. 2017, 36, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Brittain, E.L.; Hemnes, A.R. Vasodilator responsive pulmonary arterial hypertension: Evidence for a new disease? Ann. Intern. Med. 2015, 162, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893217752912. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Simonneau, G. Treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Lau, E.M.; Montani, D.; Jais, X.; Sitbon, O.; Simonneau, G. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation 2014, 130, 2189–2208. [Google Scholar] [CrossRef] [PubMed]
- El Chami, H.; Hassoun, P.M. Immune and inflammatory mechanisms in pulmonary arterial hypertension. Prog. Cardiovasc. Dis. 2012, 55, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Price, L.C.; Wort, S.J.; Perros, F.; Dorfmuller, P.; Huertas, A.; Montani, D.; Cohen-Kaminsky, S.; Humbert, M. Inflammation in pulmonary arterial hypertension. Chest 2012, 141, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Kuebler, W.M.; Bonnet, S.; Tabuchi, A. Inflammation and autoimmunity in pulmonary hypertension: Is there a role for endothelial adhesion molecules? (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893218757596. [Google Scholar] [CrossRef] [PubMed]
- Pullamsetti, S.S.; Savai, R.; Janssen, W.; Dahal, B.K.; Seeger, W.; Grimminger, F.; Ghofrani, H.A.; Weissmann, N.; Schermuly, R.T. Inflammation, immunological reaction and role of infection in pulmonary hypertension. Clin. Microbiol. Infect. 2011, 17, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar] [PubMed]
- Savai, R.; Pullamsetti, S.S.; Kolbe, J.; Bieniek, E.; Voswinckel, R.; Fink, L.; Scheed, A.; Ritter, C.; Dahal, B.K.; Vater, A.; et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Goel, A.; Marsboom, G.; Fang, Y.H.; Toth, P.T.; Zhang, H.J.; Kajimoto, H.; Hong, Z.; Paul, J.; Wietholt, C.; et al. A central role for CD68(+) macrophages in hepatopulmonary syndrome. Reversal by macrophage depletion. Am. J. Respir. Crit. Care Med. 2011, 183, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Hiress, M.L.; Tu, L.; Ricard, N.; Phan, C.; Thuillet, R.; Fadel, E.; Dorfmüller, P.; Montani, D.; de Man, F.; Humbert, M.; et al. Proinflammatory signature of the dysfunctional endothelium in pulmonary hypertension. Role of the macrophage migration inhibitory factor/CD74 complex. Am. J. Respir. Crit. Care Med. 2015, 192, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Yamaji-Kegan, K.; Su, Q.; Angelini, D.J.; Myers, A.C.; Cheadle, C.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMα) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4–dependent mechanism. J. Immunol. 2010, 185, 5539–5548. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Saito, T.; Nickel, N.P.; Alastalo, T.P.; Glotzbach, J.P.; Chan, R.; Haghighat, L.; Fuchs, G.; Januszyk, M.; Cao, A.; et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014, 211, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Jiang, X.; Tamosiuniene, R.; Sung, Y.K.; Qian, J.; Dhillon, G.; Gera, L.; Farkas, L.; Rabinovitch, M.; Zamanian, R.T.; et al. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci. Transl. Med. 2013, 5, 200ra117. [Google Scholar] [CrossRef] [PubMed]
- Ee, M.T.; Kantores, C.; Ivanovska, J.; Wong, M.J.; Jain, A.; Jankov, R.P. Leukotriene B4 mediates macrophage influx and pulmonary hypertension in bleomycin-induced chronic neonatal lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L292–L302. [Google Scholar] [CrossRef] [PubMed]
- Vergadi, E.; Chang, M.S.; Lee, C.; Liang, O.; Liu, X.; Fernandez-Gonzalez, A.; Mitsialis, S.A.; Kourembanas, S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 2011, 123, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Liang, O.D.; Mitsialis, S.A.; Chang, M.S.; Vergadi, E.; Lee, C.; Aslam, M.; Fernandez-Gonzalez, A.; Liu, X.; Baveja, R.; Kourembanas, S. Mesenchymal stromal cells expressing heme oxygenase-1 reverse pulmonary hypertension. Stem Cells 2011, 29, 99–107. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, K.C.; Pugliese, S.C.; Riddle, S.R.; Poth, J.M.; Anderson, A.L.; Frid, M.G.; Li, M.; Pullamsetti, S.S.; Savai, R.; Nagel, M.A.; et al. Adventitial fibroblasts induce a distinct pro-inflammatory/pro-fibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 2014, 193, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, S.C.; Kumar, S.; Janssen, W.J.; Graham, B.B.; Frid, M.G.; Riddle, S.R.; El Kasmi, K.C.; Stenmark, K.R. A time- and compartment-specific activation of lung macrophages in hypoxic pulmonary hypertension. J. Immunol. 2017, 198, 4802–4812. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Willis, G.R.; Fernandez-Gonzalez, A.; Anastas, J.; Vitali, S.H.; Liu, X.; Ericsson, M.; Kwong, A.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell exosomes ameliorate experimental bronchopulmonary dysplasia and restore lung function through macrophage immunomodulation. Am. J. Respir. Crit. Care Med. 2018, 197, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Mitsialis, S.A.; Aslam, M.; Vitali, S.H.; Vergadi, E.; Konstantinou, G.; Sdrimas, K.; Fernandez-Gonzalez, A.; Kourembanas, S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012, 126, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.; McQuattie-Pimentel, A.C.; Chen, C.I.; Reyfman, P.A.; Anekalla, K.R.; Taylor, J.; Cardona, H.J.; Williams, K.; Joshi, N.; Budinger, G.S.; et al. Long-living alveolar macrophages modulate severity of lung injury and postnatal lung development in hyperoxia-induced lung injury mouse model. FASEB J. 2017, 31, 871. [Google Scholar]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef] [PubMed]
- Frid, M.G.; Brunetti, J.A.; Burke, D.L.; Carpenter, T.C.; Davie, N.J.; Reeves, J.T.; Roedersheimer, M.T.; van Rooijen, N.; Stenmark, K.R. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 2006, 168, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Jankov, R.P.; Luo, X.; Belcastro, R.; Copland, I.; Frndova, H.; Lye, S.J.; Hoidal, J.R.; Post, M.; Tanswell, A.K. Gadolinium chloride inhibits pulmonary macrophage influx and prevents O(2)-induced pulmonary hypertension in the neonatal rat. Pediatr. Res. 2001, 50, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Zaloudikova, M.; Vytasek, R.; Vajnerova, O.; Hnilickova, O.; Vizek, M.; Hampl, V.; Herget, J. Depletion of alveolar macrophages attenuates hypoxic pulmonary hypertension but not hypoxia-induced increase in serum concentration of MCP-1. Physiol. Res. 2016, 65, 763–768. [Google Scholar] [PubMed]
- Bryant, A.J.; Shenoy, V.; Fu, C.; Marek, G.; Lorentsen, K.J.; Herzog, E.L.; Brantly, M.L.; Avram, D.; Scott, E.W. Myeloid-derived suppressor cells are necessary for development of pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2018, 58, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2014, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Florentin, J.; Coppin, E.; Vasamsetti, S.B.; Zhao, J.; Tai, Y.Y.; Tang, Y.; Zhang, Y.; Watson, A.; Sembrat, J.; Rojas, M.; et al. Inflammatory macrophage expansion in pulmonary hypertension depends upon mobilization of blood-borne monocytes. J. Immunol. 2018, 200, 3612–3625. [Google Scholar] [CrossRef] [PubMed]
- Amsellem, V.; Abid, S.; Poupel, L.; Parpaleix, A.; Rodero, M.; Gary-Bobo, G.; Latiri, M.; Dubois-Rande, J.L.; Lipskaia, L.; Combadiere, C.; et al. Roles for the CX3CL1/CX3CR1 and CCL2/CCR2 chemokine systems in hypoxic pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 2017, 56, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Amsellem, V.; Lipskaia, L.; Abid, S.; Poupel, L.; Houssaini, A.; Quarck, R.; Marcos, E.; Mouraret, N.; Parpaleix, A.; Bobe, R.; et al. CCR5 as a treatment target in pulmonary arterial hypertension. Circulation 2014, 130, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Dorfmuller, P.; Souza, R.; Durand-Gasselin, I.; Mussot, S.; Mazmanian, M.; Herve, P.; Emilie, D.; Simonneau, G.; Humbert, M. Dendritic cell recruitment in lesions of human and experimental pulmonary hypertension. Eur. Respir. J. 2007, 29, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Mindt, B.C.; Fritz, J.H.; Duerr, C.U. Group 2 Innate lymphoid cells in pulmonary immunity and tissue homeostasis. Front. Immunol. 2018, 9, 840. [Google Scholar] [CrossRef] [PubMed]
- Nicolls, M.R.; Taraseviciene-Stewart, L.; Rai, P.R.; Badesch, D.B.; Voelkel, N.F. Autoimmunity and pulmonary hypertension: A perspective. Eur. Respir. J. 2005, 26, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Zou, X.B.; Chai, Y.F.; Yao, Y.M. Macrophage polarization in inflammatory diseases. Int. J. Biol. Sci. 2014, 10, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.L.; McCarthy, K.M.; Rogers, R.A.; Schneeberger, E.E. Interstitial lung macrophages interact with dendritic cells to present antigenic peptides derived from particulate antigens to T cells. Immunology 1994, 81, 343–351. [Google Scholar] [PubMed]
- Strickland, D.H.; Thepen, T.; Kees, U.R.; Kraal, G.; Holt, P.G. Regulation of T-cell function in lung tissue by pulmonary alveolar macrophages. Immunology 1993, 80, 266–272. [Google Scholar] [PubMed]
- Kawano, H.; Kayama, H.; Nakama, T.; Hashimoto, T.; Umemoto, E.; Takeda, K. IL-10-producing lung interstitial macrophages prevent neutrophilic asthma. Int. Immunol. 2016, 28, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Bedoret, D.; Wallemacq, H.; Marichal, T.; Desmet, C.; Quesada-Calvo, F.; Henry, E.; Closset, R.; Dewals, B.; Thielen, C.; Gustin, P.; et al. Lung interstitial macrophages alter dendritic cell functions to prevent airway allergy in mice. J. Clin. Investig. 2009, 119, 3723–3738. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F. IL-10 and macrophages orchestrate gut homeostasis. Immunity 2014, 40, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Tamosiuniene, R.; Tian, W.; Dhillon, G.; Wang, L.; Sung, Y.K.; Gera, L.; Patterson, A.J.; Agrawal, R.; Rabinovitch, M.; Ambler, K.; et al. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ. Res. 2011, 109, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.C.; Lin, W.; Chen, C.M. Human mesenchymal stem cells attenuate pulmonary hypertension induced by prenatal lipopolysaccharide treatment in rats. Clin. Exp. Pharmacol. Physiol. 2016, 43, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, G.; Fernandez-Gonzalez, A.; Aslam, M.; Vitali, S.H.; Martin, T.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stem cell-mediated reversal of bronchopulmonary dysplasia and associated pulmonary hypertension. Pulm. Circ. 2012, 2, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Kanki-Horimoto, S.; Horimoto, H.; Mieno, S.; Kishida, K.; Watanabe, F.; Furuya, E.; Katsumata, T. Implantation of mesenchymal stem cells overexpressing endothelial nitric oxide synthase improves right ventricular impairments caused by pulmonary hypertension. Circulation 2006, 114, I181–I185. [Google Scholar] [CrossRef] [PubMed]
- Baber, S.R.; Deng, W.; Master, R.G.; Bunnell, B.A.; Taylor, B.K.; Murthy, S.N.; Hyman, A.L.; Kadowitz, P.J. Intratracheal mesenchymal stem cell administration attenuates monocrotaline-induced pulmonary hypertension and endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1120–H1128. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Ke, M.W.; Cheng, C.C.; Chiou, S.H.; Wann, S.R.; Shu, C.W.; Chiou, K.R.; Tseng, C.J.; Pan, H.W.; Mar, G.Y.; et al. Therapeutic benefits of induced pluripotent stem cells in monocrotaline-induced pulmonary arterial hypertension. PLoS ONE 2016, 11, e0142476. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.X.; Pan, Y.Y.; Zhao, Y.Y.; Wang, X.X. Endothelial progenitor cell-based therapy for pulmonary arterial hypertension. Cell Transplant. 2013, 22, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhu, W.; Xia, L.; Yang, Y.; Liu, H.; Shen, J.; Zhu, J.; Xu, Y.; Yang, Z.; Wang, C. Therapeutic effect of eNOS-transfected endothelial progenitor cells on hemodynamic pulmonary arterial hypertension. Hypertens. Res. 2013, 36, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Granton, J.; Langleben, D.; Kutryk, M.B.; Camack, N.; Galipeau, J.; Courtman, D.W.; Stewart, D.J. Endothelial NO-synthase gene-enhanced progenitor cell therapy for pulmonary arterial hypertension: The PHACeT Trial. Circ. Res. 2015, 117, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Tan, J.; Maleken, A.S.; Muljadi, R.; Chan, S.T.; Lau, S.N.; Elgass, K.; Leaw, B.; Mockler, J.; Chambers, D.; et al. Human amnion cells reverse acute and chronic pulmonary damage in experimental neonatal lung injury. Stem Cell Res. Ther. 2017, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Loisel, F.; Provost, B.; Haddad, F.; Guihaire, J.; Amsallem, M.; Vrtovec, B.; Fadel, E.; Uzan, G.; Mercier, O. Stem cell therapy targeting the right ventricle in pulmonary arterial hypertension: Is it a potential avenue of therapy? Pulm. Circ. 2018, 8, 2045893218755979. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; Steendijk, P.; Ypey, D.L.; Atsma, D.E.; van der Wall, E.E.; Schalij, M.J.; van der Laarse, A. Novel approaches to treat experimental pulmonary arterial hypertension: A Review. J. Biomed. Biotechnol. 2010, 2010, 702836. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Mei, S.H.J. Cell-based therapies for lung vascular diseases. Proc. Am. Thorac. Soc. 2011, 8, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Fatemeh, H. Explant culture: An advantageous method for isolation of mesenchymal stem cells from human tissues. Cell Prolif. 2017, 50. [Google Scholar] [CrossRef]
- Romanov, Y.A.; Darevskaya, A.N.; Merzlikina, N.V.; Buravkova, L.B. Mesenchymal stem cells from human bone marrow and adipose tissue: Isolation, characterization, and differentiation potentialities. Bull. Exp. Biol. Med. 2005, 140, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Goolaerts, A.; Howard, J.P.; Lee, J.W. Mesenchymal stem cells for acute lung injury: Preclinical evidence. Crit. Care Med. 2010, 38, S569–S573. [Google Scholar] [CrossRef] [PubMed]
- Glassberg, M.K.; Minkiewicz, J.; Toonkel, R.L.; Simonet, E.S.; Rubio, G.A.; DiFede, D.; Shafazand, S.; Khan, A.; Pujol, M.V.; LaRussa, V.F.; et al. Allogeneic human mesenchymal stem cells in patients with idiopathic pulmonary fibrosis via intravenous delivery (AETHER): A phase I safety clinical trial. Chest 2017, 151, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Srour, N.; Thebaud, B. Mesenchymal stromal cells in animal bleomycin pulmonary fibrosis models: A systematic review. Stem Cells Transl. Med. 2015, 4, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Shigemura, N.; Okumura, M.; Mizuno, S.; Imanishi, Y.; Nakamura, T.; Sawa, Y. Autologous transplantation of adipose tissue-derived stromal cells ameliorates pulmonary emphysema. Am. J. Transplant. 2006, 6, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.A.; Abreu, S.C.; Cruz, F.F.; Teixeira, A.C.; Lopes-Pacheco, M.; Bandeira, E.; Olsen, P.C.; Diaz, B.L.; Takyia, C.M.; Freitas, I.P.; et al. Effects of different mesenchymal stromal cell sources and delivery routes in experimental emphysema. Respir. Res. 2014, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Braza, F.; Dirou, S.; Forest, V.; Sauzeau, V.; Hassoun, D.; Chesne, J.; Cheminant-Muller, M.A.; Sagan, C.; Magnan, A.; Lemarchand, P. Mesenchymal stem cells induce suppressive macrophages through phagocytosis in a mouse model of asthma. Stem Cells 2016, 34, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Ringden, O.; Keating, A. Mesenchymal stromal cells as treatment for chronic GVHD. Bone Marrow Transplant. 2011, 46, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Bartolucci, J.; Verdugo, F.J.; Gonzalez, P.L.; Larrea, R.E.; Abarzua, E.; Goset, C.; Rojo, P.; Palma, I.; Lamich, R.; Pedreros, P.A.; et al. Safety and Efficacy of the Intravenous Infusion of Umbilical Cord Mesenchymal Stem Cells in Patients with Heart Failure: A Phase 1/2 Randomized Controlled Trial (RIMECARD Trial [Randomized Clinical Trial of Intravenous Infusion Umbilical Cord Mesenchymal Stem Cells on Cardiopathy]). Circ. Res. 2017, 121, 1192–1204. [Google Scholar] [PubMed]
- Antunes, M.A.; Laffey, J.G.; Pelosi, P.; Rocco, P.R. Mesenchymal stem cell trials for pulmonary diseases. J. Cell. Biochem. 2014, 115, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, K.M.; Karagiannis, K.; Tsitoura, E.; Bibaki, E.; Lasithiotaki, I.; Proklou, A.; Spandidos, D.A.; Tzanakis, N. Clinical applications of mesenchymal stem cells in chronic lung diseases. Biomed. Rep. 2018, 8, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; de Visser, Y.P.; Steendijk, P.; Schutte, C.I.; Laghmani el, H.; Wagenaar, G.T.; Bax, W.H.; Mantikou, E.; Pijnappels, D.A.; Atsma, D.E.; et al. Allogenic stem cell therapy improves right ventricular function by improving lung pathology in rats with pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1606–H1616. [Google Scholar] [CrossRef] [PubMed]
- Yerebakan, C.; Sandica, E.; Prietz, S.; Klopsch, C.; Ugurlucan, M.; Kaminski, A.; Abdija, S.; Lorenzen, B.; Boltze, J.; Nitzsche, B.; et al. Autologous umbilical cord blood mononuclear cell transplantation preserves right ventricular function in a novel model of chronic right ventricular volume overload. Cell Transplant. 2009, 18, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Baveja, R.; Liang, O.D.; Fernandez-Gonzalez, A.; Lee, C.; Mitsialis, S.A.; Kourembanas, S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2009, 180, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Van Haaften, T.; Byrne, R.; Bonnet, S.; Rochefort, G.Y.; Akabutu, J.; Bouchentouf, M.; Rey-Parra, G.J.; Galipeau, J.; Haromy, A.; Eaton, F.; et al. Airway delivery of mesenchymal stem cells prevents arrested alveolar growth in neonatal lung injury in rats. Am. J. Respir. Crit. Care Med. 2009, 180, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Waszak, P.; Alphonse, R.; Vadivel, A.; Ionescu, L.; Eaton, F.; Thebaud, B. Preconditioning enhances the paracrine effect of mesenchymal stem cells in preventing oxygen-induced neonatal lung injury in rats. Stem Cells Dev. 2012, 21, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Fang, X.; Krasnodembskaya, A.; Howard, J.P.; Matthay, M.A. Concise review: Mesenchymal stem cells for acute lung injury: Role of paracrine soluble factors. Stem Cells 2011, 29, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Kourembanas, S. Exosomes: Vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu. Rev. Physiol. 2015, 77, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Willis, G.R.; Mitsialis, S.A.; Kourembanas, S. ‘Good things come in small packages’: Application of exosome-based therapeutics in neonatal lung injury. Pediatr. Res. 2018, 83, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Thery, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Théry, C. Exosomes: Secreted vesicles and intercellular communications. F1000 Biol. Rep. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 15. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Willis, G.R.; Kourembanas, S.; Mitsialis, S.A. Towards exosome-based therapeutics: Isolation, heterogeneity, and fit-for-purpose potency. Front. Cardiovasc. Med. 2017, 4, 63. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, Y.; Su, X.; He, J.; Bai, W.; He, X. MSCs-derived exosomes and neuroinflammation, neurogenesis and therapy of traumatic brain injury. Front. Cell. Neurosci. 2017, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chopp, M.; Meng, Y.; Katakowski, M.; Xin, H.; Mahmood, A.; Xiong, Y. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J. Neurosurg. 2015, 122, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Shao, H.; Su, C.; Jiang, Y.; Chen, X.; Bai, L.; Zhang, Y.; Li, Q.; Zhang, X.; Li, X. Exosomes derived from MSCs ameliorate retinal laser injury partially by inhibition of MCP-1. Sci. Rep. 2016, 6, 34562. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.; Tomarev, S. Bone marrow-derived mesenchymal stem cells-derived exosomes promote survival of retinal ganglion cells through miRNA-dependent mechanisms. Stem Cells Transl. Med. 2017, 6, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Nargesi, A.A.; Lerman, L.O.; Eirin, A. Mesenchymal stem cell-derived extracellular vesicles for kidney repair: Current status and looming challenges. Stem Cell Res. Ther. 2017, 4, 273. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Pittenger, M.F. Concise Review: MSC-derived exosomes for cell-free therapy. Stem Cells 2017, 35, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Aliotta, J.M.; Pereira, M.; Wen, S.; Dooner, M.S.; Del Tatto, M.; Papa, E.; Goldberg, L.R.; Baird, G.L.; Ventetuolo, C.E.; Quesenberry, P.J.; et al. Exosomes induce and reverse monocrotaline-induced pulmonary hypertension in mice. Cardiovasc. Res. 2016, 110, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Linares, R.; Tan, S.; Gounou, C.; Arraud, N.; Brisson, A.R. High-speed centrifugation induces aggregation of extracellular vesicles. J. Extracell. Vesicles 2015, 4, 29509. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Greening, D.W.; Zhu, H.J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; An, R.; Liu, Z.-J.; Wang, J.J.; Chen, S.Z.; Hong, M.M.; Liu, J.H.; Xiao, M.Y.; Chen, Y.F. Therapeutic effects of mesenchymal stem cell-derived microvesicles on pulmonary arterial hypertension in rats. Acta Pharmacol. Sin. 2014, 35, 1121–1128. [Google Scholar] [CrossRef] [PubMed]
- Chaubey, S.; Thueson, S.; Ponnalagu, D.; Alam, M.A.; Gheorghe, C.P.; Aghai, Z.; Singh, H.; Bhandari, V. Early gestational mesenchymal stem cell secretome attenuates experimental bronchopulmonary dysplasia in part via exosome-associated factor TSG-6. Stem Cell Res. Ther. 2018, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Mitsialis, S.A.; Kourembanas, S. Stem cell–based therapies for the newborn lung and brain: Possibilities and challenges. Semin. Perinatol. 2016, 40, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Bin, Z.; Yin, Y.; Chai, L.R.; Sim, T.S.; Hwa, C.A.B.; Kiang, L.S. Mesenchymal stem cells secrete immunologically active exosomes. Stem Cells Dev. 2014, 23, 1233–1244. [Google Scholar] [CrossRef]
- Lo Sicco, C.; Reverberi, D.; Balbi, C.; Ulivi, V.; Principi, E.; Pascucci, L.; Becherini, P.; Bosco, M.C.; Varesio, L.; Franzin, C.; et al. Mesenchymal stem cell-derived extracellular vesicles as mediators of anti-inflammatory effects: Endorsement of macrophage polarization. Stem Cells Transl. Med. 2017, 6, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Hyvärinen, K.; Holopainen, M.; Skirdenko, V.; Ruhanen, H.; Lehenkari, P.; Korhonen, M.; Käkelä, R.; Laitinen, S.; Kerkelä, E. Mesenchymal stromal cells and their extracellular vesicles enhance the anti-inflammatory phenotype of regulatory macrophages by downregulating the production of interleukin (IL)-23 and IL-22. Front. Immunol. 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed]
- Ti, D.; Hao, H.; Tong, C.; Liu, J.; Dong, L.; Zheng, J.; Zhao, Y.; Liu, H.; Fu, X.; Han, W. LPS-preconditioned mesenchymal stromal cells modify macrophage polarization for resolution of chronic inflammation via exosome-shuttled let-7b. J. Transl. Med. 2015, 13, 308. [Google Scholar] [CrossRef] [PubMed]
- Lankford, K.L.; Arroyo, E.J.; Nazimek, K.; Bryniarski, K.; Askenase, P.W.; Kocsis, J.D. Intravenously delivered mesenchymal stem cell-derived exosomes target M2-type macrophages in the injured spinal cord. PLoS ONE 2018, 13, e0190358. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takahashi, Y.; Nishikawa, M.; Kato, K.; Morishita, M.; Yamashita, T.; Matsumoto, A.; Charoenviriyakul, C.; Takakura, Y. Macrophage-dependent clearance of systemically administered B16BL6-derived exosomes from the blood circulation in mice. J. Extracell. Vesicles 2015, 4, 26238. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Essandoh, K.; Li, Y.; Huo, J.; Fan, G.C. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock 2016, 46, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Self-Fordham, J.B.; Naqvi, A.R.; Uttamani, J.R.; Kulkarni, V.; Nares, S. MicroRNA: Dynamic regulators of macrophage polarization and plasticity. Front. Immunol. 2017, 8, 1062. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.J.; Bonnet, S.; Chan, S.Y. Translational advances in the field of pulmonary hypertension. translating microrna biology in pulmonary hypertension. It will take more than “miR” words. Am. J. Respir. Crit. Care Med. 2017, 195, 167–178. [Google Scholar] [CrossRef] [PubMed]



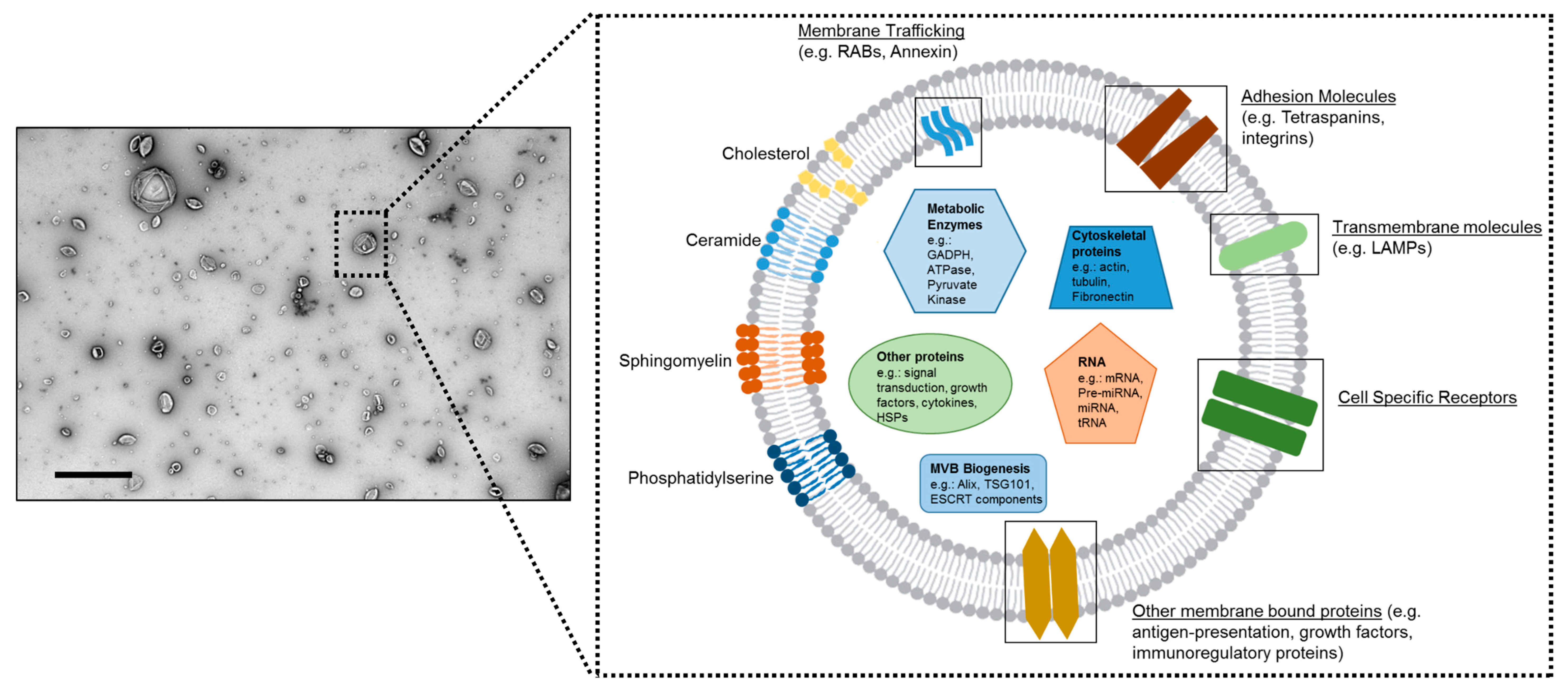{kind=link}
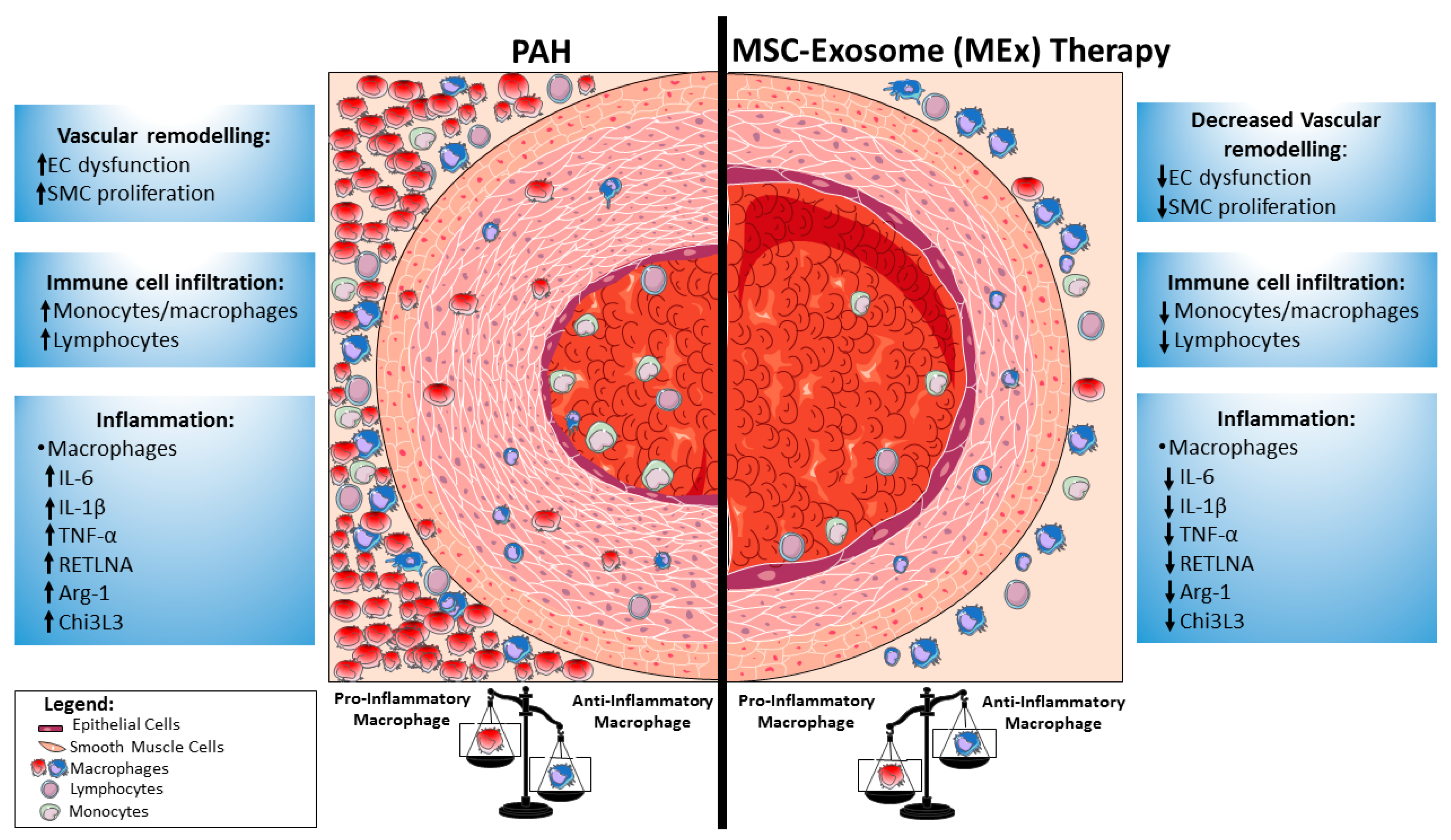{kind=link}
| Disease | Species | MSC-Product ‘Nomenclature’ | Final Isolation Step | Dose Assessment | Dose | Ref |
|---|---|---|---|---|---|---|
| Hypoxia-PH | Mouse | Exosomes | PEG-SEC | Protein | 0.1–10 μg | [29] |
| MCT-PH | Mouse | Exosomes | UC (100,000× g) | Protein | 25 μg | [104] |
| MCT-PH | Rats | Microvesicles | UC (100,000× g) | Protein | 30 μg | [108] |
| Hyperoxia-BPD | Mouse | Exosomes | Density Cushion | Cell equivalent | 0.5 × 106 | [28] |
| Hyperoxia-BPD | Mouse | Extracellular vesicles | UC (110,000× g) | Cell equivalent | 0.7 × 106 | [109] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Mitsialis, S.A.; Kourembanas, S. Macrophage Immunomodulation: The Gatekeeper for Mesenchymal Stem Cell Derived-Exosomes in Pulmonary Arterial Hypertension? Int. J. Mol. Sci. 2018, 19, 2534. https://doi.org/10.3390/ijms19092534
Willis GR, Fernandez-Gonzalez A, Reis M, Mitsialis SA, Kourembanas S. Macrophage Immunomodulation: The Gatekeeper for Mesenchymal Stem Cell Derived-Exosomes in Pulmonary Arterial Hypertension? International Journal of Molecular Sciences. 2018; 19(9):2534. https://doi.org/10.3390/ijms19092534
Chicago/Turabian StyleWillis, Gareth R., Angeles Fernandez-Gonzalez, Monica Reis, S. Alex Mitsialis, and Stella Kourembanas. 2018. "Macrophage Immunomodulation: The Gatekeeper for Mesenchymal Stem Cell Derived-Exosomes in Pulmonary Arterial Hypertension?" International Journal of Molecular Sciences 19, no. 9: 2534. https://doi.org/10.3390/ijms19092534
APA StyleWillis, G. R., Fernandez-Gonzalez, A., Reis, M., Mitsialis, S. A., & Kourembanas, S. (2018). Macrophage Immunomodulation: The Gatekeeper for Mesenchymal Stem Cell Derived-Exosomes in Pulmonary Arterial Hypertension? International Journal of Molecular Sciences, 19(9), 2534. https://doi.org/10.3390/ijms19092534