Radiation Sensitization of Basal Cell and Head and Neck Squamous Cell Carcinoma by the Hedgehog Pathway Inhibitor Vismodegib

 , ,
, , 

Abstract
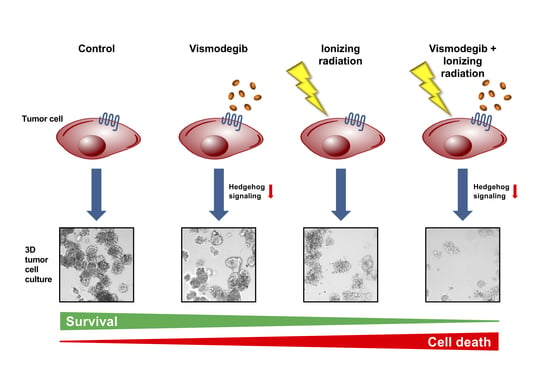
1. Introduction
2. Results
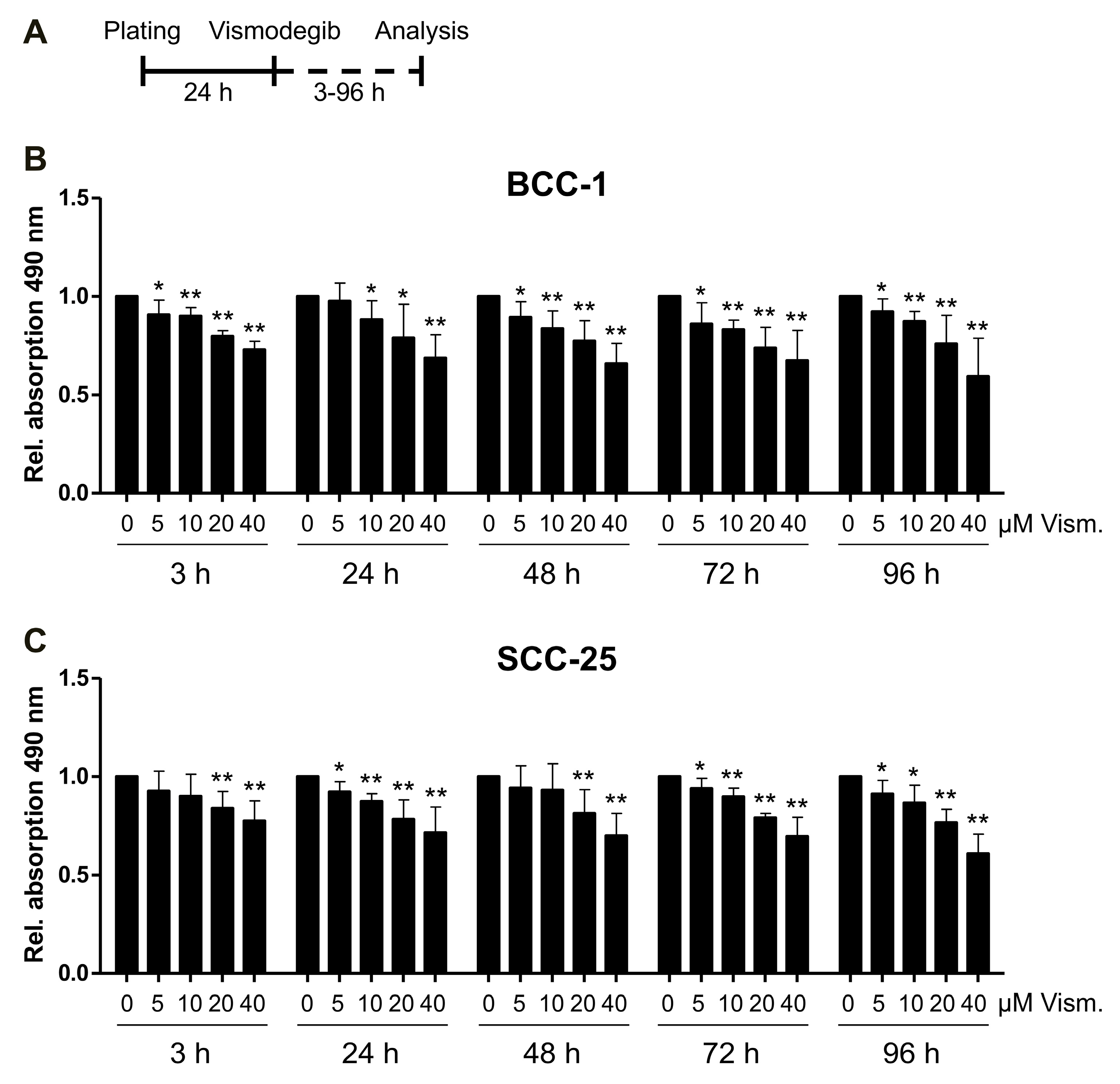
2.1. Impact of Vismodegib Concentration and Treatment Time on Cell Proliferation/Viability
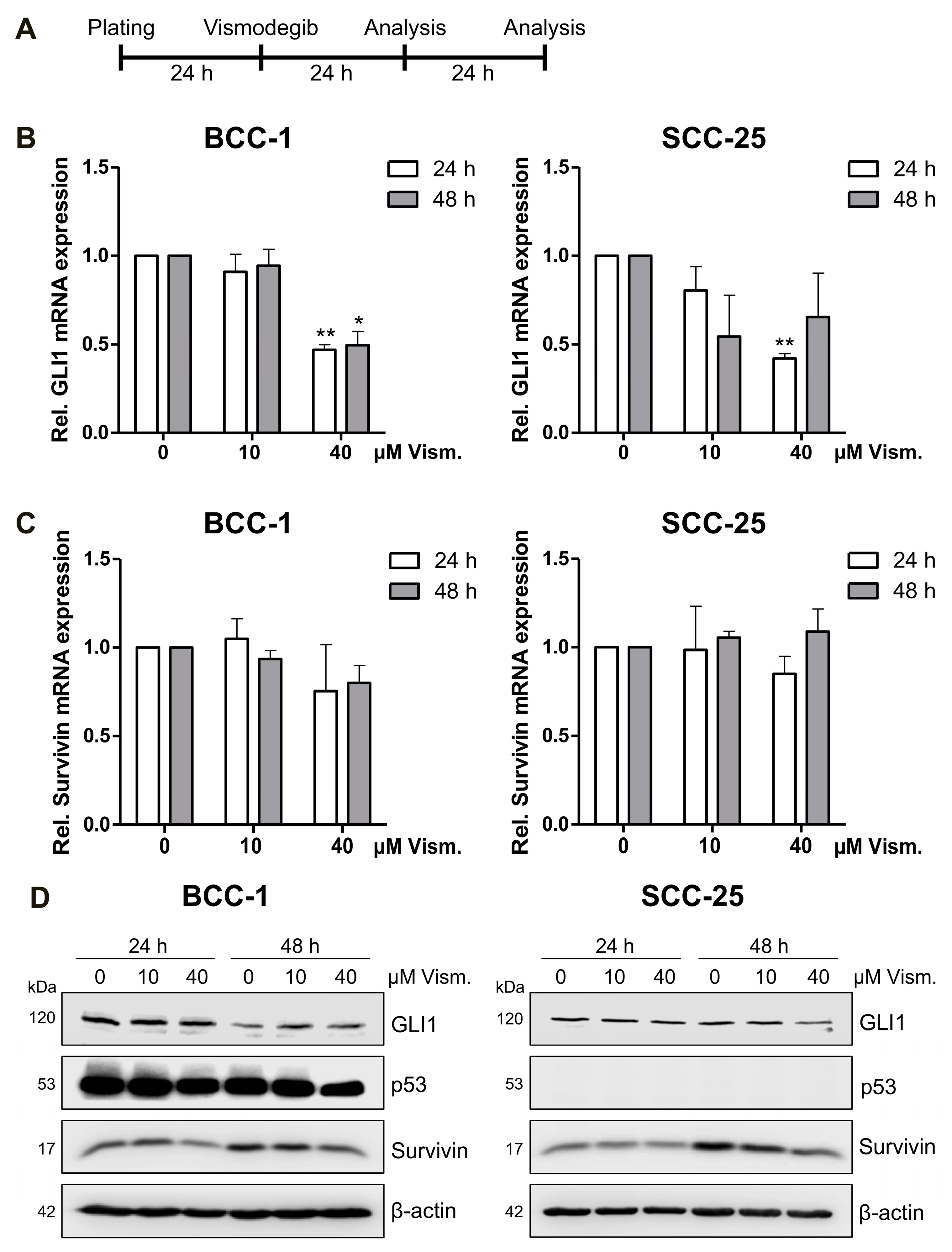
2.2. Vismodegib Decreases Hh Signaling Target Gene GLI1 and Survivin Expression in a Cell Line-Dependent Manner
2.3. Vismodegib Treatment and Irradiation Modulate Cell Viability, Cell Cycle Distribution and Cell Death
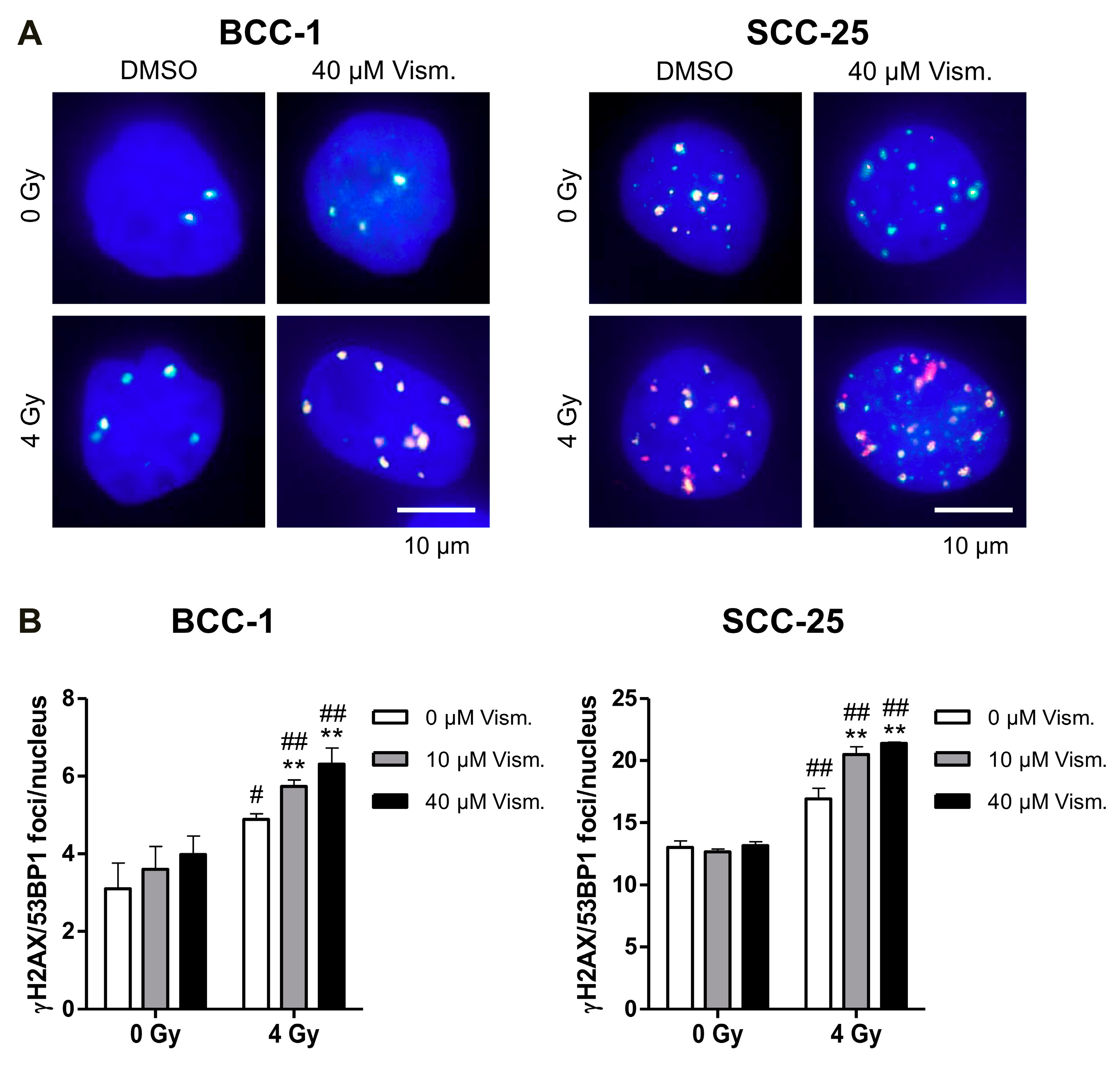
2.4. Vismodegib Increases Radiation-Induced DNA Damage of BCC-1 and SCC-25 Cells
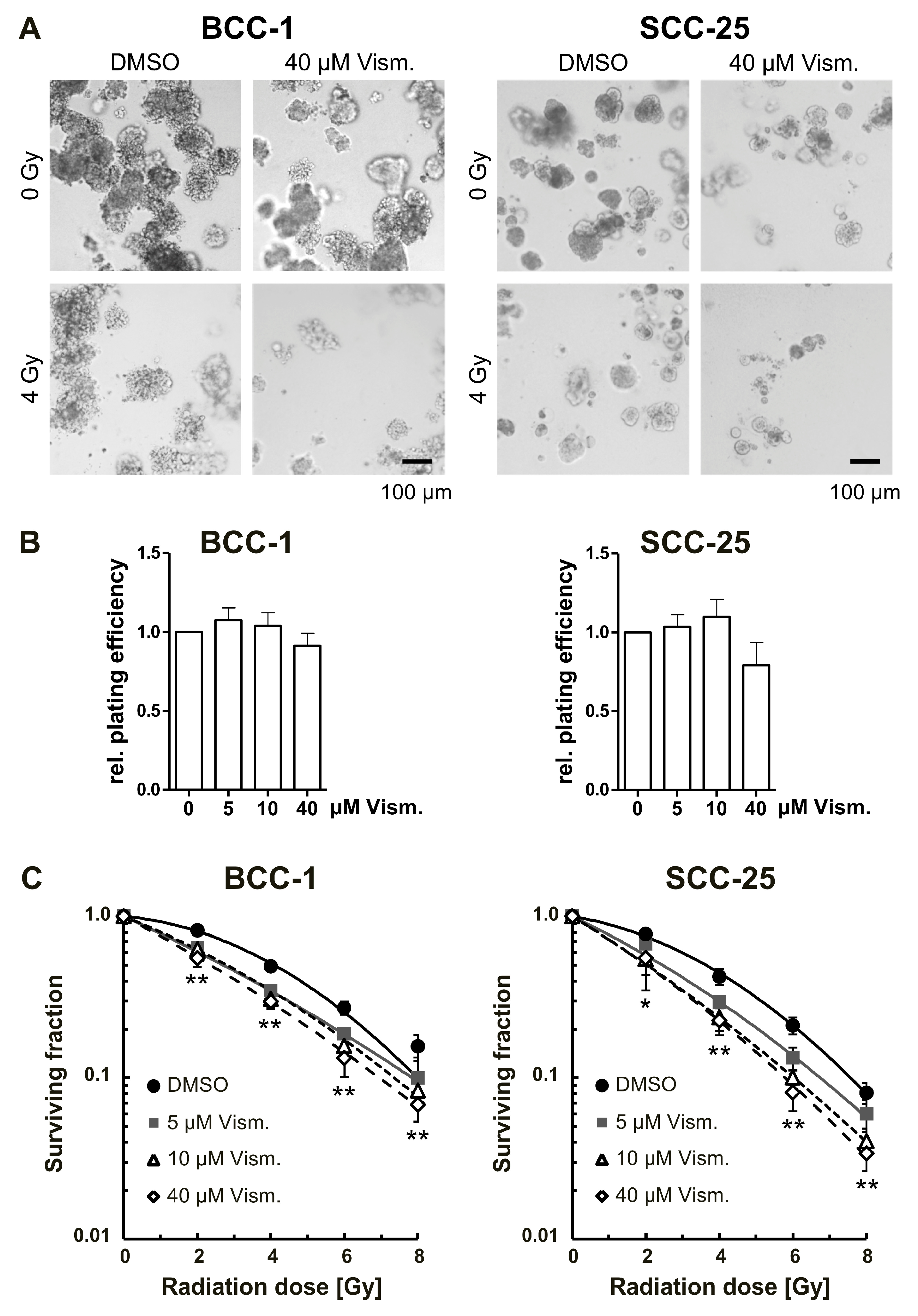
2.5. Vismodegib Radiosensitizes BCC-1 and SCC-25 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Vismodegib Treatment and Irradiation Procedure
4.3. Cell Proliferation (MTS) Assay
4.4. Western Blot Analysis
4.5. RNA Preparation and Quantitative Real-Time RT-PCR
4.6. Cell Cycle and Apoptosis Analysis
4.7. Immunofluorescence Staining and Quantification of γH2AX/53BP1 Foci Formation
4.8. 3D Colony Formation Assay
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| 53BP1 | p53-binding protein 1 |
| BCC | Basal cell carcinoma |
| DSB | Double-strand break |
| GLI | Glioma-associated oncogene homologue 1 |
| Hh | Hedgehog |
| HNSCC | Head and neck squamous cell carcinoma |
| IAP | Inhibitor of apoptosis protein |
| PARP | Poly ((adenosine diphosphate)ADP-ribose) polymerase |
| PTCH1 | Patched homologue 1 |
| SCC | Squamous cell carcinoma |
| SMO | Smoothened |
| SUFU | Suppressor of fused |
References
- Griffin, L.L.; Ali, F.R.; Lear, J.T. Non-melanoma skin cancer. Clin. Med. 2016, 16, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Lear, J.T.; Szeimies, R.M. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef]
- Bath-Hextall, F.; Leonardi-Bee, J.; Smith, C.; Meal, A.; Hubbard, R. Trends in incidence of skin basal cell carcinoma. Additional evidence from a UK primary care database study. Int. J. Cancer 2007, 121, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Kiiski, V.; de Vries, E.; Flohil, S.C.; Bijl, M.J.; Hofman, A.; Stricker, B.H.; Nijsten, T. Risk factors for single and multiple basal cell carcinomas. Arch. Dermatol. 2010, 146, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Barakat, M.T.; Humke, E.W.; Scott, M.P. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol. Med. 2010, 16, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Unden, A.B.; Zaphiropoulos, P.G.; Bruce, K.; Toftgard, R.; Stahle-Backdahl, M. Human patched (PTCH) mRNA is overexpressed consistently in tumor cells of both familial and sporadic basal cell carcinoma. Cancer Res. 1997, 57, 2336–2340. [Google Scholar] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Vlckova, K.; Ondrusova, L.; Vachtenheim, J.; Reda, J.; Dundr, P.; Zadinova, M.; Zakova, P.; Pouckova, P. Survivin, a novel target of the Hedgehog/GLI signaling pathway in human tumor cells. Cell Death Dis. 2016, 7, e2048. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Moura, U.; Opitz, I.; Soltermann, A.; Rehrauer, H.; Thies, S.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Role of hedgehog signaling in malignant pleural mesothelioma. Clin. Cancer Res. 2012, 18, 4646–4656. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; McGuirk, M.; Hockenberry, T.N.; Wu, Q.; Ashar, H.; Black, S.; Wen, S.F.; Wang, L.; Kirschmeier, P.; Bishop, W.R.; et al. Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway. Oncogene 2002, 21, 2613–2622. [Google Scholar] [CrossRef] [PubMed]
- Rodel, F.; Sprenger, T.; Kaina, B.; Liersch, T.; Rodel, C.; Fulda, S.; Hehlgans, S. Survivin as a prognostic/predictive marker and molecular target in cancer therapy. Curr. Med. Chem. 2012, 19, 3679–3688. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Ruat, M.; Hoch, L.; Faure, H.; Rognan, D. Targeting of Smoothened for therapeutic gain. Trends Pharmacol. Sci. 2014, 35, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.N.; Eagles, J.; Keysar, S.B.; Wang, G.; Glogowska, M.J.; Altunbas, C.; Anderson, R.T.; Le, P.N.; Morton, J.J.; Frederick, B.; et al. Hedgehog signaling drives radioresistance and stroma-driven tumor repopulation in head and neck squamous cancers. Cancer Res. 2014, 74, 7024–7036. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.X.; Wang, S.; Zhao, H.; Liu, N.; Chen, D.; Sun, M.; Zheng, J.H. Sonic hedgehog signaling may promote invasion and metastasis of oral squamous cell carcinoma by activating MMP-9 and E-cadherin expression. Med. Oncol. 2014, 31, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.D.; Inzunza, H.; Chang, H.; Qi, Z.; Hu, B.; Malone, D.; Cogswell, J. Mutations in the hedgehog pathway genes SMO and PTCH1 in human gastric tumors. PLoS ONE 2013, 8, e54415. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, R.; Tsujimura, T.; Tao, L.; Kamikonya, N.; Fujiwara, Y. The oncoprotein and stem cell renewal factor BMI1 associates with poor clinical outcome in oesophageal cancer patients undergoing preoperative chemoradiotherapy. BMC Cancer 2012, 12, 461. [Google Scholar] [CrossRef] [PubMed]
- Dimitrova, K.; Stoehr, M.; Dehghani, F.; Dietz, A.; Wichmann, G.; Bertolini, J.; Mozet, C. Overexpression of the Hedgehog signalling pathway in head and neck squamous cell carcinoma. Onkologie 2013, 36, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Chang, C.J.; Lin, C.P.; Chang, S.Y.; Chu, P.Y.; Tai, S.K.; Li, W.Y.; Chao, K.S.; Chen, Y.J. Expression of hedgehog signaling molecules as a prognostic indicator of oral squamous cell carcinoma. Head Neck 2012, 34, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Aziz, K.; Chettiar, S.T.; Aftab, B.T.; Armour, M.; Gajula, R.; Gandhi, N.; Salih, T.; Herman, J.M.; Wong, J.; et al. Hedgehog pathway inhibition radiosensitizes non-small cell lung cancers. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Pollom, E.L.; Bui, T.T.; Chang, A.L.; Colevas, A.D.; Hara, W.Y. Concurrent Vismodegib and Radiotherapy for Recurrent, Advanced Basal Cell Carcinoma. JAMA Dermatol. 2015, 151, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, D.R.; Algazi, A.; Arron, S.T.; Neuhaus, I.M.; Yom, S.S. Induction Hedgehog pathway inhibition followed by combined-modality radiotherapy for basal cell carcinoma. Br. J. Dermatol. 2015, 173, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Schulze, B.; Meissner, M.; Ghanaati, S.; Burck, I.; Rodel, C.; Balermpas, P. Hedgehog pathway inhibitor in combination with radiation therapy for basal cell carcinomas of the head and neck: First clinical experience with vismodegib for locally advanced disease. Strahlenther. Onkol. 2016, 192, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, G.; Dittmann, K.; Weiss, C.; Reichert, S.; Hausmann, E.; Rodel, C.; Rodel, F. Radiation-induced survivin nuclear accumulation is linked to DNA damage repair. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Hanna, A.; Samant, R.S.; Shevde, L.A. The Impact of Hedgehog Signaling Pathway on DNA Repair Mechanisms in Human Cancer. Cancers (Basel) 2015, 7, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Reichert, S.; Rodel, C.; Mirsch, J.; Harter, P.N.; Tomicic, M.T.; Mittelbronn, M.; Kaina, B.; Rodel, F. Survivin inhibition and DNA double-strand break repair: A molecular mechanism to overcome radioresistance in glioblastoma. Radiother. Oncol. 2011, 101, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Hehlgans, S.; Sandfort, V.; Cordes, N. 3D matrix-based cell cultures: Automated analysis of tumor cell survival and proliferation. Int. J. Oncol. 2016, 48, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Hehlgans, S.; Petraki, C.; Reichert, S.; Cordes, N.; Rodel, C.; Rodel, F. Double targeting of Survivin and XIAP radiosensitizes 3D grown human colorectal tumor cells and decreases migration. Radiother. Oncol. 2013, 108, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Thurnher, D.; Kloimstein, P.; Leitner, V.; Petzelbauer, P.; Pammer, J.; Brunner, M.; Erovic, B.M. Expression of the Sonic hedgehog pathway in squamous cell carcinoma of the skin and the mucosa of the head and neck. Head Neck 2011, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed]
- Steg, A.; Amm, H.M.; Novak, Z.; Frost, A.R.; Johnson, M.R. Gli3 mediates cell survival and sensitivity to cyclopamine in pancreatic cancer. Cancer Biol. Ther. 2010, 10, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hu, S.; Cheng, J.; Wang, G.; Tao, K. Smoothened antagonist GDC-0449 (Vismodegib) inhibits proliferation and triggers apoptosis in colon cancer cell lines. Exp. Ther. Med. 2017, 13, 2529–2536. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; McKee, C.M.; Dok, R.; Haustermans, K.; Muschel, R.J. The hedgehog inhibitor GANT61 sensitizes prostate cancer cells to ionizing radiation both in vitro and in vivo. Oncotarget 2016, 7, 84286–84298. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Schneider, L.; Forster, C.; Zips, D.; Kunz-Schughart, L.A.; Cordes, N. EGFR/JIP-4/JNK2 signaling attenuates cetuximab-mediated radiosensitization of squamous cell carcinoma cells. Cancer Res. 2013, 73, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell. Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Weaver, V.M.; Fischer, A.H.; Peterson, O.W.; Bissell, M.J. The importance of the microenvironment in breast cancer progression: Recapitulation of mammary tumorigenesis using a unique human mammary epithelial cell model and a three-dimensional culture assay. Biochem. Cell. Biol. 1996, 74, 833–851. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.J.; Patrick, D.R.; Bissell, M.J.; Fournier, M.V. Prognostic breast cancer signature identified from 3D culture model accurately predicts clinical outcome across independent datasets. PLoS ONE 2008, 3, e2994. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Cordes, N. Radiobiology goes 3D: How ECM and cell morphology impact on cell survival after irradiation. Radiother. Oncol. 2011, 99, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Che, J.; Sun, K.K.; Shen, X.J.; Yang, D.; Zhong, N.; Zhao, H. Cyclopamine increases the radiosensitivity of human pancreatic cancer cells by regulating the DNA repair signal pathway through an epidermal growth factor receptordependent pathway. Mol. Med. Rep. 2013, 8, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Ferreira, R.; Vitorino, R.; Ferreira-Pinto, M.J.; Ferreira, R.; Henriques-Coelho, T. Exploring the role of post-translational modifications on protein-protein interactions with survivin. Arch. Biochem. Biophys. 2013, 538, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Pasca di Magliano, M.; Hebrok, M. Hedgehog signalling in cancer formation and maintenance. Nat. Rev. Cancer 2003, 3, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Caamano, J.; Zhang, S.Y.; Rosvold, E.A.; Bauer, B.; Klein-Szanto, A.J. p53 alterations in human squamous cell carcinomas and carcinoma cell lines. Am. J. Pathol. 1993, 142, 1131–1139. [Google Scholar] [PubMed]
- Min, B.M.; Baek, J.H.; Shin, K.H.; Gujuluva, C.N.; Cherrick, H.M.; Park, N.H. Inactivation of the p53 gene by either mutation or HPV infection is extremely frequent in human oral squamous cell carcinoma cell lines. Eur. J. Cancer B Oral Oncol. 1994, 30B, 338–345. [Google Scholar] [CrossRef]
- Martinez, J.; Georgoff, I.; Levine, A.J. Cellular localization and cell cycle regulation by a temperature-sensitive p53 protein. Genes Dev. 1991, 5, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell. Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; DeVecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget 2011, 2, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Chiang, L.C.; Chiang, W.; Yu, H.S.; Sheu, H.M.; Chen, H.Y. Establishment and characterization of a continuous human basal cell carcinoma cell line from facial skin (I) cytological behavior of early passages. Gaoxiong Yi Xue Ke Xue Za Zhi 1994, 10, 170–176. [Google Scholar] [PubMed]
- Jee, S.H.; Chu, C.Y.; Chiu, H.C.; Huang, Y.L.; Tsai, W.L.; Liao, Y.H.; Kuo, M.L. Interleukin-6 induced basic fibroblast growth factor-dependent angiogenesis in basal cell carcinoma cell line via JAK/STAT3 and PI3-kinase/Akt pathways. J. Investig. Dermatol. 2004, 123, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Muller, B.M.; Kronenwett, R.; Hennig, G.; Euting, H.; Weber, K.; Bohmann, K.; Weichert, W.; Altmann, G.; Roth, C.; Winzer, K.J.; et al. Quantitative determination of estrogen receptor, progesterone receptor, and HER2 mRNA in formalin-fixed paraffin-embedded tissue—A new option for predictive biomarker assessment in breast cancer. Diagn. Mol. Pathol. 2011, 20, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Badawi, A.; Hehlgans, S.; Pfeilschifter, J.; Rodel, F.; Eberhardt, W. Silencing of the mRNA-binding protein HuR increases the sensitivity of colorectal cancer cells to ionizing radiation through upregulation of caspase-2. Cancer Lett. 2017, 393, 103–112. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line Treatment | Plating Efficiency [%] | α [Gy−1] | β [Gy−2] | Radiation Dose at 50% Cell Survival [Gy] | Sensitizer Enhancement Ratio (versus DMSO Control) | Radiation Dose at 10% Cell Survival [Gy] | Sensitizer Enhancement Ratio (versus DMSO Control) |
|---|---|---|---|---|---|---|---|
| BCC-1 | |||||||
| DMSO | 23.29 ± 3.39 | 0.0428 | 0.0307 | 4.10 | 7.99 | ||
| 5 µM Vism. | 25.00 ± 3.67 | 0.2348 | 0.0074 | 2.72 | 1.51 | 7.85 | 1.02 |
| 10 µM Vism. | 24.29 ± 4.84 | 0.2044 | 0.0147 | 2.82 | 1.45 | 7.37 | 1.08 |
| 40 µM Vism. | 21.11 ± 1.54 | 0.2724 | 0.0084 | 2.37 | 1.73 | 6.96 | 1.15 |
| SCC-25 | |||||||
| DMSO | 28.56 ± 2.52 | 0.0849 | 0.0290 | 3.64 | 7.57 | ||
| 5 µM Vism. | 29.44 ± 1.67 | 0.2479 | 0.0138 | 2.46 | 1.48 | 6.75 | 1.12 |
| 10 µM Vism. | 31.53 ± 5.59 | 0.3189 | 0.0104 | 2.04 | 1.79 | 6.03 | 1.25 |
| 40 µM Vism. | 22.39 ± 2.43 | 0.3152 | 0.0143 | 2.02 | 1.81 | 5.79 | 1.31 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hehlgans, S.; Booms, P.; Güllülü, Ö.; Sader, R.; Rödel, C.; Balermpas, P.; Rödel, F.; Ghanaati, S. Radiation Sensitization of Basal Cell and Head and Neck Squamous Cell Carcinoma by the Hedgehog Pathway Inhibitor Vismodegib. Int. J. Mol. Sci. 2018, 19, 2485. https://doi.org/10.3390/ijms19092485
Hehlgans S, Booms P, Güllülü Ö, Sader R, Rödel C, Balermpas P, Rödel F, Ghanaati S. Radiation Sensitization of Basal Cell and Head and Neck Squamous Cell Carcinoma by the Hedgehog Pathway Inhibitor Vismodegib. International Journal of Molecular Sciences. 2018; 19(9):2485. https://doi.org/10.3390/ijms19092485
Chicago/Turabian StyleHehlgans, Stephanie, Patrick Booms, Ömer Güllülü, Robert Sader, Claus Rödel, Panagiotis Balermpas, Franz Rödel, and Shahram Ghanaati. 2018. "Radiation Sensitization of Basal Cell and Head and Neck Squamous Cell Carcinoma by the Hedgehog Pathway Inhibitor Vismodegib" International Journal of Molecular Sciences 19, no. 9: 2485. https://doi.org/10.3390/ijms19092485
APA StyleHehlgans, S., Booms, P., Güllülü, Ö., Sader, R., Rödel, C., Balermpas, P., Rödel, F., & Ghanaati, S. (2018). Radiation Sensitization of Basal Cell and Head and Neck Squamous Cell Carcinoma by the Hedgehog Pathway Inhibitor Vismodegib. International Journal of Molecular Sciences, 19(9), 2485. https://doi.org/10.3390/ijms19092485