IL-6: A Potential Role in Cardiac Metabolic Homeostasis

Abstract

1. Introduction
2. The Role of IL-6 in Lipid Metabolism
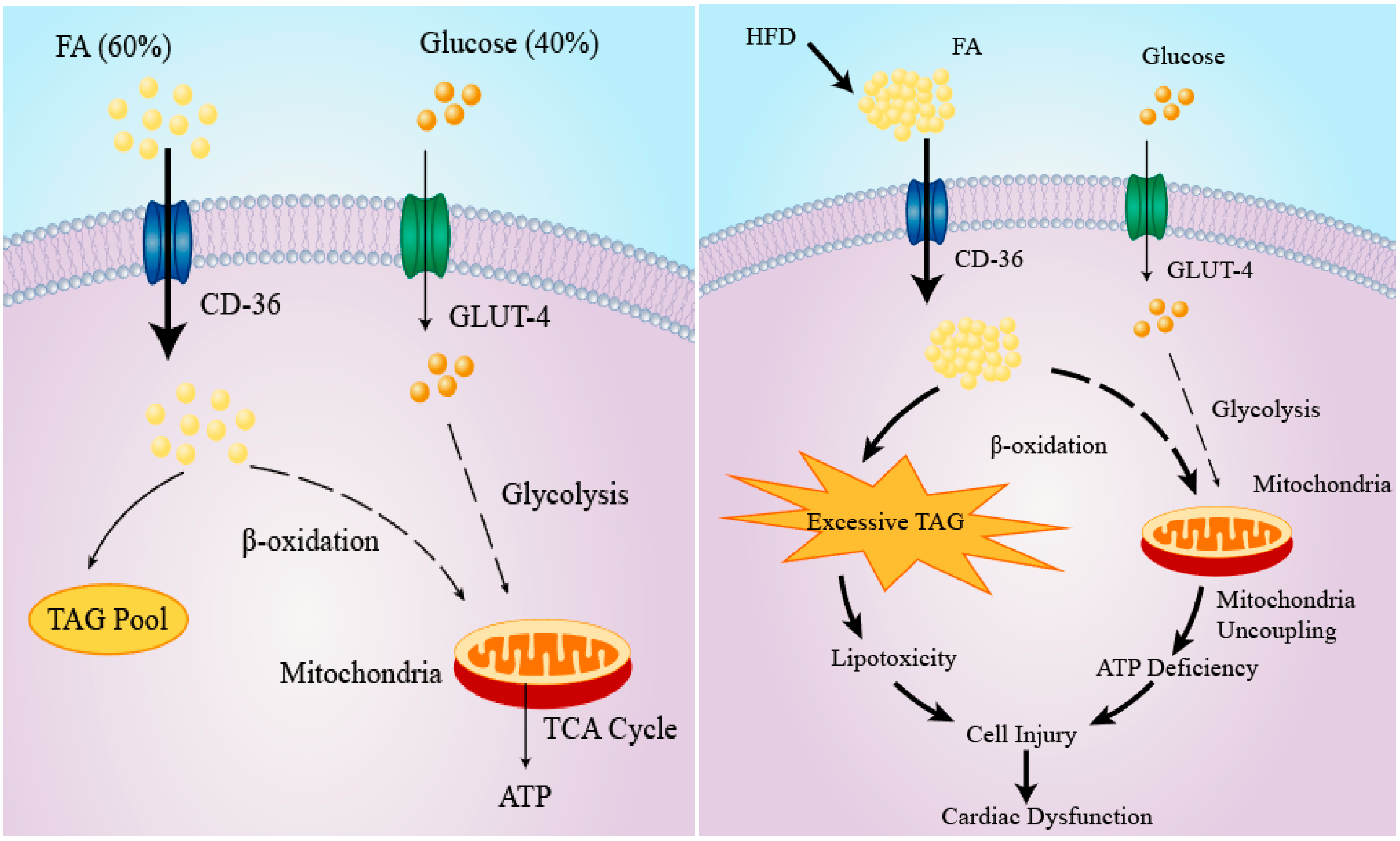
3. Cardiac Lipid Metabolism and Consequences of Lipotoxicity
4. The Role of IL-6 in Cardiac Lipotoxicity
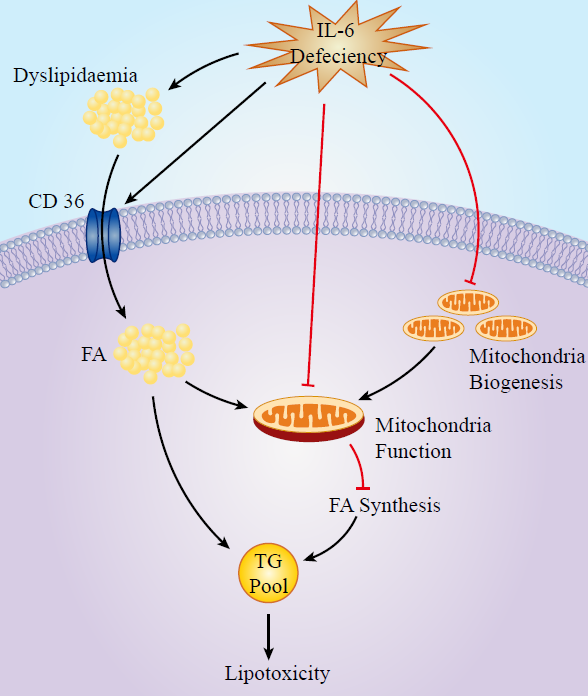
4.1. IL-6, Dyslipidaemia, and FA Transporters
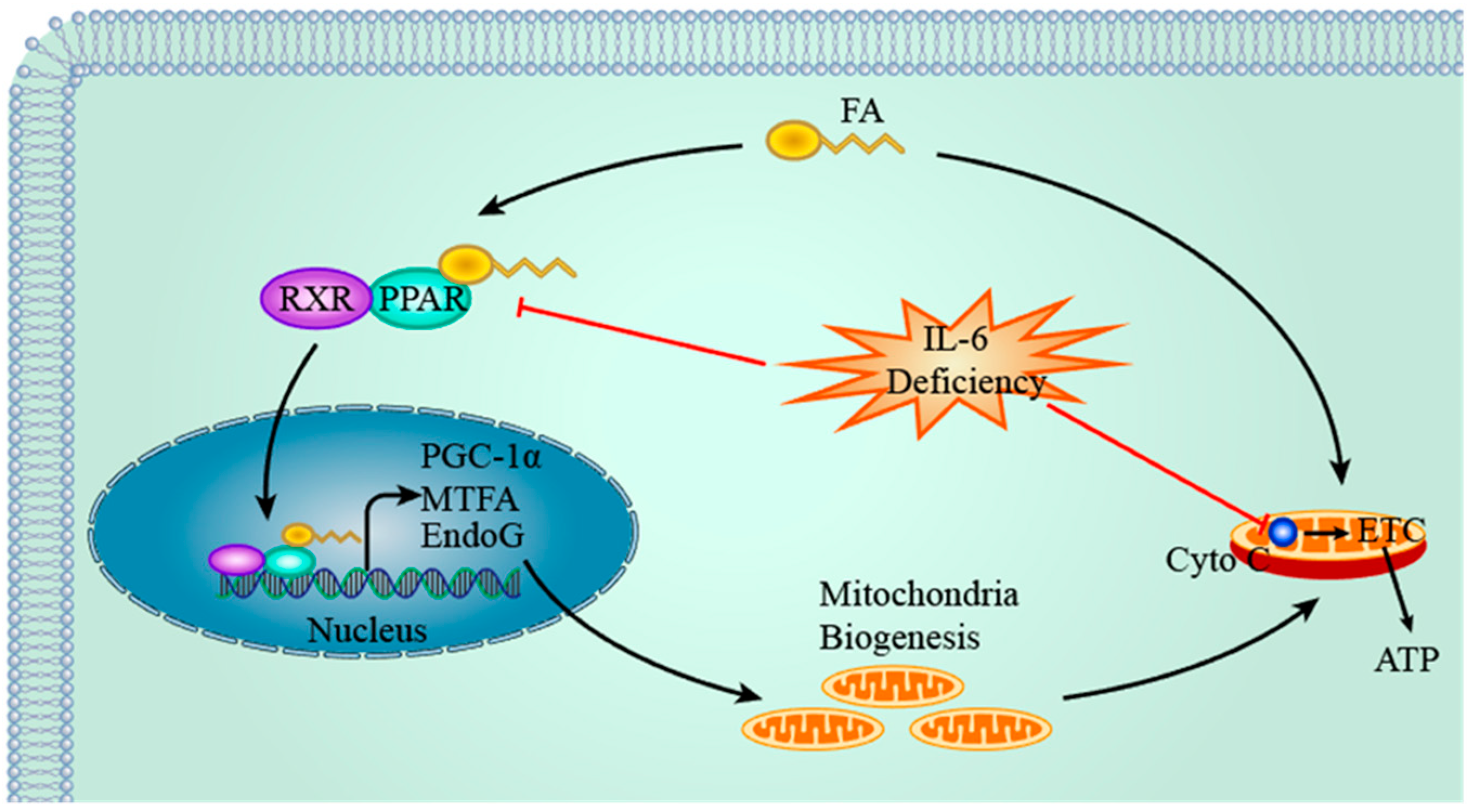
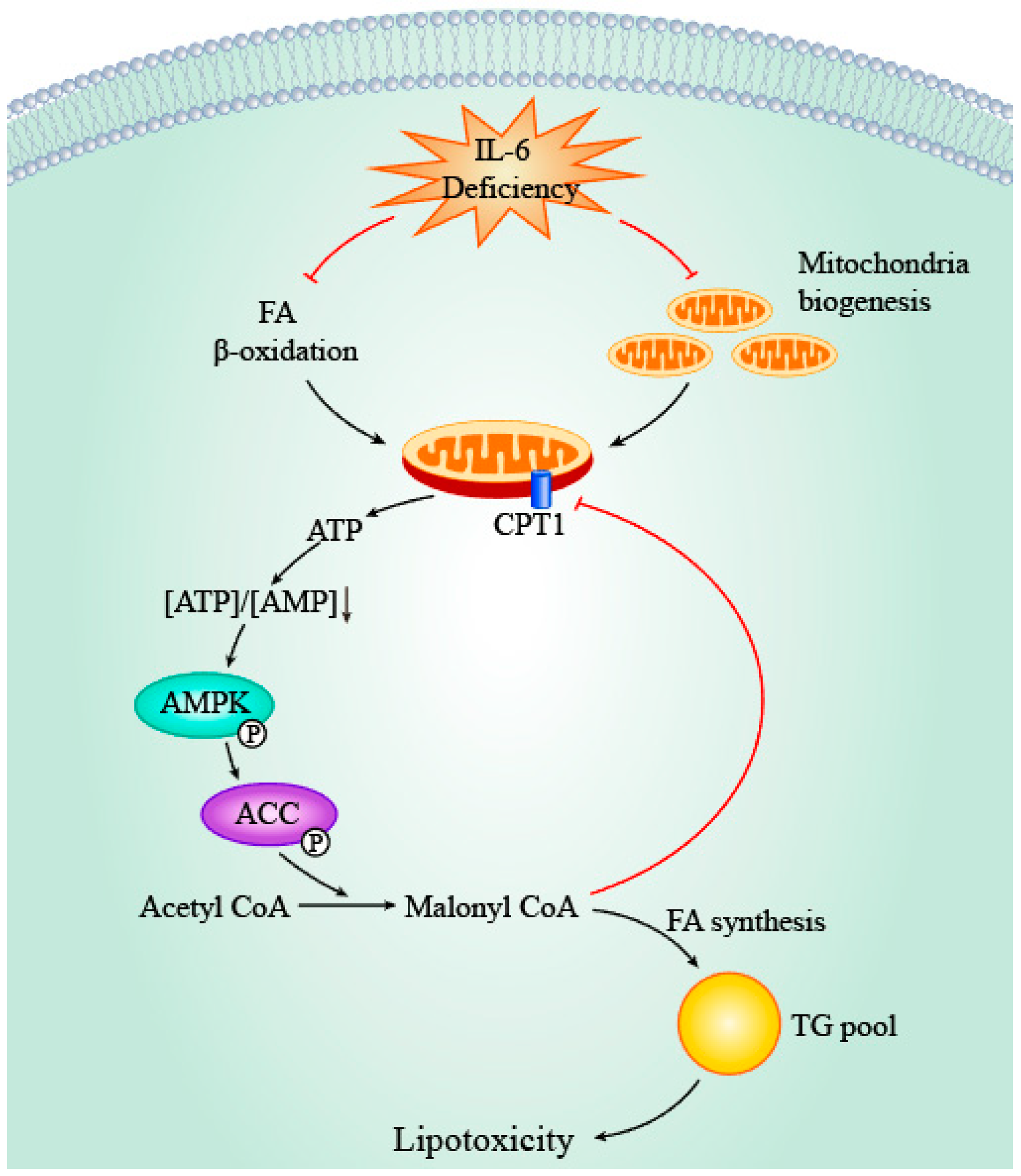
4.2. IL-6, PPAR & PGC-1α and Mitochondria
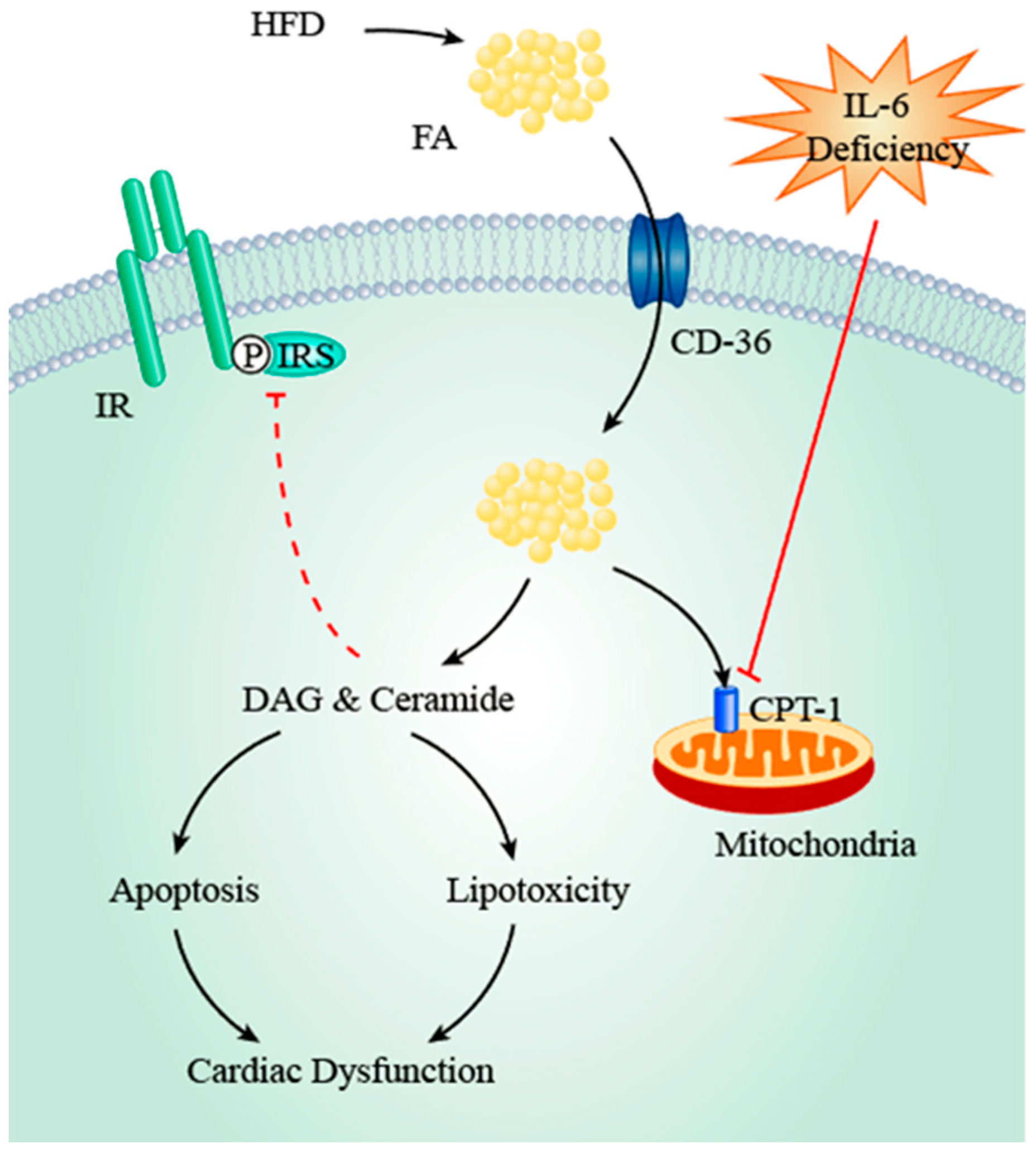
4.3. IL-6, AMPK and ACC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IL-6 | Interleukin-6 |
| FA | Fatty acid |
| rhIL-6 | Recombinant human IL-6 |
| VLDL | Very low-density lipoprotein |
| HFD | High-fat diet |
| sIL-6R | Soluble IL-6 receptor |
| TAG | Triacylglycerol |
| LPL | Lipoprotein lipase |
| mtDNA | Mitochondria DNA |
| TG | Triglycerides |
| PPAR | Peroxisome Proliferator-activated Receptor |
| PGC-1α | PPARγ coactivator 1α |
| FAT/CD36 | Fatty acid translocase |
| WT | Wild-type |
| FABPpm | Plasma membrane isoform of fatty acid binding protein |
| FATP | Fatty acid transport protein |
| PKC θ | Protein kinase C θ |
| NRF | Nuclear respiratory factor |
| AMPK | AMP-activated protein kinase |
| CPT-1 | Carnitine palmitoyltransferase-1 |
References
- Sehgal, P.B.; Helfgott, D.C.; Santhanam, U.; Tatter, S.B.; Clarick, R.H.; Ghrayeb, J.; May, L.T. Regulation of the acute phase and immune responses in viral disease. Enhanced expression of the beta 2-interferon/hepatocyte-stimulating factor/interleukin 6 gene in virus-infected human fibroblasts. J. Exp. Med. 1988, 167, 1951–1956. [Google Scholar] [CrossRef] [PubMed]
- Frei, K.; Malipiero, U.V.; Leist, T.P.; Zinkernagel, R.M.; Schwab, M.E.; Fontana, A. On the cellular source and function of interleukin 6 produced in the central nervous system in viral diseases. Eur. J. Immunol. 1989, 19, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Martinez-Maza, O.; Hirano, T.; Breen, E.; Nishanian, P.; Salazar-Gonzalez, J.; Fahey, J.; Kishimoto, T. Induction of IL-6 (B cell stimulatory factor-2/IFN-beta 2) production by HIV. J. Immunol. 1989, 142, 531–536. [Google Scholar] [PubMed]
- Nordan, R.P.; Potter, M. A macrophage-derived factor required by plasmacytomas for survival and proliferation in vitro. Science 1986, 233, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, J.; Cayphas, S.; Opdenakker, G.; Billiau, A.; Van Snick, J. Interleukin 1 and poly (rI)· poly (rC) induce production of a hybridoma growth factor by human fibroblasts. Eur. J. Immunol. 1987, 17, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, M.R.; Waage, A.; Espevik, T. Cytokine regulation of interleukin 6 production by human endothelial cells. Cell. Immunol. 1989, 121, 372–382. [Google Scholar] [CrossRef]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef]
- Jones, S.A. Directing transition from innate to acquired immunity: Defining a role for IL-6. J. Immunol. 2005, 175, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G.; Marin, V.; Montero-Julian, F.; Mantovani, A.; Farnarier, C. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentarutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; Van Hinsbergh, V.; Sozzani, S. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef]
- Hirano, T.; Taga, T.; Nakano, N.; Yasukawa, K.; Kashiwamura, S.; Shimizu, K.; Nakajima, K.; Pyun, K.H.; Kishimoto, T. Purification to homogeneity and characterization of human B-cell differentiation factor (BCDF or BSFp-2). Proc. Natl. Acad. Sci. USA 1985, 82, 5490–5494. [Google Scholar] [CrossRef] [PubMed]
- McLoughlin, R.M.; Jenkins, B.J.; Grail, D.; Williams, A.S.; Fielding, C.A.; Parker, C.R.; Ernst, M.; Topley, N.; Jones, S.A. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9589–9594. [Google Scholar] [CrossRef] [PubMed]
- Dominitzki, S.; Fantini, M.C.; Neufert, C.; Nikolaev, A.; Galle, P.R.; Scheller, J.; Monteleone, G.; Rose-John, S.; Neurath, M.F.; Becker, C. Cutting edge: Trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+ CD25− T cells. J. Immunol. 2007, 179, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Fukuda, S. Role of IL-6 in the neural stem cell differentiation. Clin. Rev. Allergy Immunol. 2005, 28, 249–256. [Google Scholar] [CrossRef]
- Streetz, K.; Luedde, T.; Manns, M.; Trautwein, C. Interleukin 6 and liver regeneration. Gut 2000, 47, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Poli, V.; Balena, R.; Fattori, E.; Markatos, A.; Yamamoto, M.; Tanaka, H.; Ciliberto, G.; Rodan, G.A.; Costantini, F. Interleukin-6 deficient mice are protected from bone loss caused by estrogen depletion. EMBO J. 1994, 13, 1189. [Google Scholar] [PubMed]
- Wallenius, V.; Wallenius, K.; Ahrén, B.; Rudling, M.; Carlsten, H.; Dickson, S.L.; Ohlsson, C.; Jansson, J.-O. Interleukin-6-deficient mice develop mature-onset obesity. Nat. Med. 2002, 8, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Gao, M.; Sun, H.; Liu, D. Interleukin-6 gene transfer reverses body weight gain and fatty liver in obese mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Wolsk, E.; Mygind, H.; Grøndahl, T.S.; Pedersen, B.K.; van Hall, G. IL-6 selectively stimulates fat metabolism in human skeletal muscle. Am. J. Physiol. Endocrinol. Metabol. 2010, 299, E832–E840. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.R.; Morgan, H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 1974, 36, 413–459. [Google Scholar] [CrossRef] [PubMed]
- Wende, A.R.; Abel, E.D. Lipotoxicity in the heart. BBA-Mol. Cell Biol. Lipids 2010, 1801, 311–319. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Nzirorera, C.; Kienesberger, P.C. Lipid metabolism and signaling in cardiac lipotoxicity. BBA-Mol. Cell Biol. Lipids 2016, 1861, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Lyngsø, D.; Simonsen, L.; Bülow, J. Metabolic effects of interleukin-6 in human splanchnic and adipose tissue. J. Physiol. 2002, 543, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.L.; Steinberg, G.R.; Macaulay, S.L.; Thomas, W.G.; Holmes, A.G.; Ramm, G.; Prelovsek, O.; Hohnen-Behrens, C.; Watt, M.J.; James, D.E. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 2006, 55, 2688–2697. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.; Carey, A.; Sacchetti, M.; Steinberg, G.; Macaulay, S.; Febbraio, M.; Pedersen, B. Acute IL-6 treatment increases fatty acid turnover in elderly humans in vivo and in tissue culture in vitro. Am. J. Physiol. Endocrinol. Metabol. 2005, 288, E155–E162. [Google Scholar] [CrossRef] [PubMed]
- Van Hall, G.; Steensberg, A.; Sacchetti, M.; Fischer, C.; Keller, C.; Schjerling, P.; Hiscock, N.; Møller, K.; Saltin, B.; Febbraio, M.A. Interleukin-6 stimulates lipolysis and fat oxidation in humans. J. Clin. Endocrinol. Metab. 2003, 88, 3005–3010. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Kanakura, Y.; Aozasa, K.; Johkoh, T.; Nakamura, M.; Nakano, S.; Nakano, N.; Ikeda, Y.; Sasaki, T.; Nishioka, K. Humanized anti–interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood 2005, 106, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Real, J.-M.; Broch, M.; Vendrell, J.; Richart, C.; Ricart, W. Interleukin-6 gene polymorphism and lipid abnormalities in healthy subjects. J. Clin. Endocrinol. Metab. 2000, 85, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.E.; Sullivan, S.; Harten, I.; Schneider, S.H.; Greenberg, A.S.; Fried, S.K. Interleukin-6 regulates human adipose tissue lipid metabolism and leptin production in vitro. J. Clin. Endocrinol. Metab. 2004, 89, 5577–5582. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalili, L.; Bouzakri, K.; Glund, S.; Lönnqvist, F.; Koistinen, H.A.; Krook, A. Signaling specificity of interleukin-6 action on glucose and lipid metabolism in skeletal muscle. Mol. Endocrinol. 2006, 20, 3364–3375. [Google Scholar] [CrossRef] [PubMed]
- Wallenius, K.; Wallenius, V.; Sunter, D.; Dickson, S.L.; Jansson, J.-O. Intracerebroventricular interleukin-6 treatment decreases body fat in rats. Biochem. Biophys. Res. Commun. 2002, 293, 560–565. [Google Scholar] [CrossRef]
- Sadagurski, M.; Norquay, L.; Farhang, J.; D’Aquino, K.; Copps, K.; White, M. Human IL6 enhances leptin action in mice. Diabetologia 2010, 53, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Klein, R.; Matheny, M.; King, M.; Meyer, E.; Scarpace, P. Induction of uncoupling protein 1 by central interleukin-6 gene delivery is dependent on sympathetic innervation of brown adipose tissue and underlies one mechanism of body weight reduction in rats. Neuroscience 2002, 115, 879–889. [Google Scholar] [CrossRef]
- Peters, M.; Schirmacher, P.; Goldschmitt, J.; Odenthal, M.; Peschel, C.; Fattori, E.; Ciliberto, G.; Dienes, H.-P.; Zum Büschenfelde, K.-H.M.; Rose-John, S. Extramedullary expansion of hematopoietic progenitor cells in interleukin (IL)-6–sIL-6R double transgenic mice. J. Exp. Med. 1997, 185, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, D.; Zhao, X.; Yang, S.; Li, Z.; Sanchis, D.; Jin, L.; Qiang, X.; Wang, K.; Xu, Y. Interleukin-6 deficiency facilitates myocardial dysfunction during high fat diet-induced obesity by promoting lipotoxicity and inflammation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 3128–3141. [Google Scholar] [CrossRef] [PubMed]
- Di Gregorio, G.B.; Hensley, L.; Lu, T.; Ranganathan, G.; Kern, P.A. Lipid and carbohydrate metabolism in mice with a targeted mutation in the IL-6 gene: Absence of development of age-related obesity. Am. J. Physiol. Endocrinol. Metabol. 2004, 287, E182–E187. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Belke, D.D.; Gamble, J.; Toshiyuki, I.; Schönekess, B.O. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim. Biophys. Acta-Lipid Lipid Met. 1994, 1213, 263–276. [Google Scholar] [CrossRef]
- Bing, R.; Siegel, A.; Ungar, I.; Gilbert, M. Metabolism of the human heart: II. Studies on fat, ketone and amino acid metabolism. Am. J. Med. 1954, 16, 504–515. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell. Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Brindley, D.N.; Kok, B.P.; Kienesberger, P.C.; Lehner, R.; Dyck, J.R. Shedding light on the enigma of myocardial lipotoxicity: The involvement of known and putative regulators of fatty acid storage and mobilization. Am. J. Physiol. Endocrinol. Metabol. 2010, 298, E897–E908. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; O’Neill, B.T.; Tathireddy, P.; Young, M.E.; Abel, E.D. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005, 112, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G. Mitochondrial energetics in the heart in obesity-related diabetes. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.; Kim, J.K. New insights into insulin resistance in the diabetic heart. Trends Endocrinol. Metab. 2011, 22, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Rinaldo, P.; Rhead, W.J.; Tian, L.; Millington, D.S.; Vockley, J.; Hamm, D.A.; Brix, A.E.; Lindsey, J.R.; Pinkert, C.A. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl. Acad. Sci. USA 1998, 95, 15592–15597. [Google Scholar] [CrossRef] [PubMed]
- Yagyu, H.; Chen, G.; Yokoyama, M.; Hirata, K.; Augustus, A.; Kako, Y.; Seo, T.; Hu, Y.; Lutz, E.P.; Merkel, M. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J. Clin. Investig. 2003, 111, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Lassers, B.W.; Kaijser, L.; Carlson, L.A. Myocardial lipid and carbohydrate metabolism in healthy, fasting men at rest: Studies during continuous infusion of 3 H-palmitate. Eur. J. Clin. Investig. 1972, 2, 348–358. [Google Scholar] [CrossRef]
- Matthews, V.; Allen, T.; Risis, S.; Chan, M.; Henstridge, D.; Watson, N.; Zaffino, L.; Babb, J.; Boon, J.; Meikle, P. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia 2010, 53, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Adeli, K.; Taghibiglou, C.; Van Iderstine, S.C.; Lewis, G.F. Mechanisms of hepatic very low-density lipoprotein overproduction in insulin resistance. Trends Cardiovasc. Med. 2001, 11, 170–176. [Google Scholar] [CrossRef]
- Kuang, M.; Febbraio, M.; Wagg, C.; Lopaschuk, G.D.; Dyck, J.R. Fatty acid translocase/CD36 deficiency does not energetically or functionally compromise hearts before or after ischemia. Circulation 2004, 109, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Luiken, J.J.; Coort, S.L.; Koonen, D.P.; Van der Horst, D.J.; Bonen, A.; Zorzano, A.; Glatz, J.F. Regulation of cardiac long-chain fatty acid and glucose uptake by translocation of substrate transporters. Pflügers Arch. 2004, 448, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chabowski, A.; Zmijewska, M.; Gorski, J.; Bonen, A.; Kaminski, K.; Winnicka, M. Effect of il-6 deficiency on myocardial expression of fatty acid transporters and intracelular lipid deposits. J. Physiol. Pharmacol. 2007, 58, 73. [Google Scholar] [PubMed]
- Luiken, J.J.; Arumugam, Y.; Dyck, D.J.; Bell, R.C.; Pelsers, M.M.; Turcotte, L.P.; Tandon, N.N.; Glatz, J.F.; Bonen, A. Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J. Biol. Chem. 2001, 276, 40567–40573. [Google Scholar] [CrossRef] [PubMed]
- Coort, S.L.; Hasselbaink, D.M.; Koonen, D.P.; Willems, J.; Coumans, W.A.; Chabowski, A.; van der Vusse, G.J.; Bonen, A.; Glatz, J.F.; Luiken, J.J. Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese zucker rats. Diabetes 2004, 53, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Angin, Y.; Steinbusch, L.K.; Simons, P.J.; Greulich, S.; Hoebers, N.T.; Douma, K.; van Zandvoort, M.A.; Coumans, W.A.; Wijnen, W.; Diamant, M. CD36 inhibition prevents lipid accumulation and contractile dysfunction in rat cardiomyocytes. Biochem. J. 2012, 448, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Ghost, S.; Baltimore, D. Activation in vitro of NF-KB by phosphorylation of its inhibitor IK-B. Nature 1990, 344, 678–682. [Google Scholar]
- Holland, W.L.; Bikman, B.T.; Wang, L.-P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid–induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, Y. Roles of PPARs on regulating myocardial energy and lipid homeostasis. J. Mol. Med. (Berl.) 2007, 85, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Madrazo, J.A.; Kelly, D.P. The PPAR trio: Regulators of myocardial energy metabolism in health and disease. J. Mol. Cell. Cardiol. 2008, 44, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Virbasius, J.V.; Scarpulla, R.C. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Cote, J.; Ruiz-Carrillo, A. Primers for mitochondrial DNA replication generated by endonuclease G. Science 1993, 261, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Bonda, T.A.; Szynaka, B.; Sokołowska, M.; Dziemidowicz, M.; Waszkiewicz, E.; Winnicka, M.M.; Bernaczyk, P.; Wawrusiewicz-Kurylonek, N.; Kamiński, K.A. Interleukin 6 modulates PPARα and PGC-1α and is involved in high-fat diet induced cardiac lipotoxicity in mouse. Int. J. Cardiol. 2016, 219, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Hennuyer, N.; Staels, B.; Fruchart, J.-C.; Fievet, C.; Gonzalez, F.J.; Auwerx, J. Alterations in lipoprotein metabolism in peroxisome proliferator-activated receptor α-deficient mice. J. Biol. Chem. 1997, 272, 27307–27312. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.-C.; Kovacs, A.; Ford, D.A.; Hsu, F.-F.; Garcia, R.; Herrero, P.; Saffitz, J.E.; Schaffer, J.E. A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 2001, 107, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Barba, I.; Chavarria, L.; Ruiz-Meana, M.; Mirabet, M.; Agulló, E.; Garcia-Dorado, D. Effect of intracellular lipid droplets on cytosolic Ca2+ and cell death during ischaemia–reperfusion injury in cardiomyocytes. J. Physiol. 2009, 587, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Diop, S.B.; Bisharat-Kernizan, J.; Birse, R.T.; Oldham, S.; Ocorr, K.; Bodmer, R. PGC-1/spargel counteracts high-fat-diet-induced obesity and cardiac lipotoxicity downstream of TOR and brummer ATGL lipase. Cell Rep. 2015, 10, 1572–1584. [Google Scholar] [CrossRef] [PubMed]
- Chambers, K.T.; Leone, T.C.; Sambandam, N.; Kovacs, A.; Wagg, C.S.; Lopaschuk, G.D.; Finck, B.N.; Kelly, D.P. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J. Biol. Chem. 2011, 286, 11155–11162. [Google Scholar] [CrossRef] [PubMed]
- Young, M.E.; Guthrie, P.H.; Razeghi, P.; Leighton, B.; Abbasi, S.; Patil, S.; Youker, K.A.; Taegtmeyer, H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes 2002, 51, 2587–2595. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Savage, D.B.; Abu-Elheiga, L.; Liu, Z.-X.; Kim, S.; Kulkarni, A.; Distefano, A.; Hwang, Y.-J.; Reznick, R.M.; Codella, R. Continuous fat oxidation in acetyl–CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc. Natl. Acad. Sci. USA 2007, 104, 16480–16485. [Google Scholar] [CrossRef] [PubMed]
- Shulman, G.I. Unraveling the cellular mechanism of insulin resistance in humans: New insights from magnetic resonance spectroscopy. Physiology 2004, 19, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.; Garland, P.; Hales, C.; Newsholme, E. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]
- Randle, P.J. Regulatory interactions between lipids and carbohydrates: The glucose fatty acid cycle after 35 years. Diabetes Metab. Res. Rev. 1998, 14, 263–283. [Google Scholar] [CrossRef]
- Su, X.; Han, X.; Mancuso, D.J.; Abendschein, D.R.; Gross, R.W. Accumulation of long-chain acylcarnitine and 3-hydroxy acylcarnitine molecular species in diabetic myocardium: Identification of alterations in mitochondrial fatty acid processing in diabetic myocardium by shotgun lipidomics. Biochemistry 2005, 44, 5234–5245. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.A.; Han, X.; Horner, C.C.; Gross, R.W. Accumulation of unsaturated acylcarnitine molecular species during acute myocardial ischemia: Metabolic compartmentalization of products of fatty acyl chain elongation in the acylcarnitine pool. Biochemistry 1996, 35, 7903–7909. [Google Scholar] [CrossRef] [PubMed]
- Haffar, T.; Bérubé-Simard, F.-A.; Bousette, N. Cardiomyocyte lipotoxicity is mediated by Il-6 and causes down-regulation of PPARs. Biochem. Biophys. Res. Commun. 2015, 459, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Itoh, H.; Doi, K.; Fukunaga, Y.; Hosoda, K.; Shintani, M.; Yamashita, J.; Chun, T.-H.; Inoue, M.; Masatsugu, K. Down regulation of peroxisome proliferator-activated receptorγ expression by inflammatory cytokines and its reversal by thiazolidinediones. Diabetologia 1999, 42, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Chew, C.-H.; Chew, G.-S.; Najimudin, N.; Tengku-Muhammad, T.S. Interleukin-6 inhibits human peroxisome proliferator activated receptor alpha gene expression via CCAAT/enhancer-binding proteins in hepatocytes. Int. J. Biochem. Cell Biol. 2007, 39, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. The AMP-activated protein kinase cascade—A unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Kelly, M.; Keller, C.; Avilucea, P.R.; Keller, P.; Luo, Z.; Xiang, X.; Giralt, M.; Hidalgo, J.; Saha, A.K.; Pedersen, B.K. AMPK activity is diminished in tissues of IL-6 knockout mice: The effect of exercise. Biochem. Biophys. Res. Commun. 2004, 320, 449–454. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, C.; Wojtaszewski, J.F.; Pedersen, B.K.; Kiens, B.; Richter, E.A. Interleukin-6 release from human skeletal muscle during exercise: Relation to AMPK activity. J. Appl. Physiol. 2003, 95, 2273–2277. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Barr, A.J.; Barr, R.L.; Desai, S.; Lopaschuk, G.D. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 1995, 270, 17513–17520. [Google Scholar] [CrossRef] [PubMed]
- Kudo, N.; Barr, A.; Barr, R.; Lopaschuk, G. 5′ AMP-activated protein kinase inhibition of acetyl CoA carboxylase can explain the high rates of fatty acid oxidation in reperfused ischemic hearts. J. Biol. Chem. 1995, 270, 17511–17520. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, G.H.; Kim, Y.M.; Lee, B.W.; Nam, H.Y.; Sim, U.-C.; Choo, S.J.; Yu, S.W.; Kim, J.J.; Kim Kwon, Y. Angiotensin II Causes Apoptosis of Adult Hippocampal Neural Stem Cells and Memory Impairment Through the Action on AMPK-PGC1α Signaling in Heart Failure. Stem Cells Transl. Med. 2017, 6, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cui, L.; Jiang, X.; Zhang, J.; Zhu, M.; Jia, J.; Zhang, Q.; Zhang, J.; Zhang, D.; Huang, Y. Extracellular pH regulates autophagy via the AMPK–ULK1 pathway in rat cardiomyocytes. FEBS Lett. 2016, 590, 3202–3212. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject/Description | Treatment | Observed Effects | Reference |
|---|---|---|---|
| Human/healthy males | rhIL-6 infusion for 4 h | Increase of lipolysis in skeletal muscle; Increase of systemic FA oxidation | [20] |
| Human/healthy males | IL-6 infusion for 2.5 h | Net increase of glycerol from subcutaneous adipose tissue; Increased uptake of FA and glycerol in splanchnic regions | [24] |
| Human/healthy males | IL-6 infusion for 4 h | Increased FA oxidation | [25] |
| Human/males with T2D vs. control | rhIL-6 infusion for 3 h | Increase of palmitate Ra and Rd in both groups | [26] |
| Human/healthy males | IL-6 infusion for 3 h | Increase of serum FA levels; Increased Ra of endogenous FA; Enhanced systemic FA oxidation | [27] |
| Human/patients with multicentric Castleman disease | Treatment of humanized anti-human IL-6 receptor monoclonal antibody | Gain of body weight; Hypertriglyceridemia; Hypercholesterinemia | [28] |
| Human/healthy females with G or C alleles at position 174 of IL-6 gene | n.d. | Trend of increased plasma IL-6 levels and elevated serum TG, VLDL-C and FFA in IL-6 G174C polymorphism | [29] |
| Species/Description | Treatment | Observed Effects | Reference |
|---|---|---|---|
| Mice/IL-6 KO | n.d. | Mature-onset obesity: Increased weight of subcutaneous fat pad | [18] |
| Mice/IL-6 KO; HFD | Intracerebroventricular IL-6 injection for 2 weeks | Decreased relative weight of mesenteric and retroperitoneal fat pads; Suppressed body weight | [32] |
| Mice/HFD; IL-6 transgenic mice (with sustained release of human IL-6) | n.d. | Decreased food intake; Increased energy expenditure; Reduced visceral fat on normal chow and free from HFD-induced obesity | [33] |
| Rat/male | Direct delivery of recombinant adeno-associated viral vector expressing murine IL-6 into hypothalamus | Suppressed weight gain and visceral adiposity | [34] |
| Mice/HFD-induced obese mice | Delivery of pLIVE-IL-6 plasmid expressing murine IL-6 | Reduction in body weight; Increased expression of enzymes involved in FA oxidation | [19] |
| Mice/Double transgenic mice co-expressing IL-6 and soluble IL-6 receptors | n.d. | Reduced body weight; Decreased body fat | [35] |
| Mice/IL-6 KO; female; HFD | n.d. | Decreased body weight gain and fat mass | [36] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Zhang, Y.; Ye, J. IL-6: A Potential Role in Cardiac Metabolic Homeostasis. Int. J. Mol. Sci. 2018, 19, 2474. https://doi.org/10.3390/ijms19092474
Xu Y, Zhang Y, Ye J. IL-6: A Potential Role in Cardiac Metabolic Homeostasis. International Journal of Molecular Sciences. 2018; 19(9):2474. https://doi.org/10.3390/ijms19092474
Chicago/Turabian StyleXu, Yitao, Yubin Zhang, and Junmei Ye. 2018. "IL-6: A Potential Role in Cardiac Metabolic Homeostasis" International Journal of Molecular Sciences 19, no. 9: 2474. https://doi.org/10.3390/ijms19092474
APA StyleXu, Y., Zhang, Y., & Ye, J. (2018). IL-6: A Potential Role in Cardiac Metabolic Homeostasis. International Journal of Molecular Sciences, 19(9), 2474. https://doi.org/10.3390/ijms19092474