Bradykinin B2 Receptor Contributes to Inflammatory Responses in Human Endothelial Cells by the Transactivation of the Fibroblast Growth Factor Receptor FGFR-1

 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
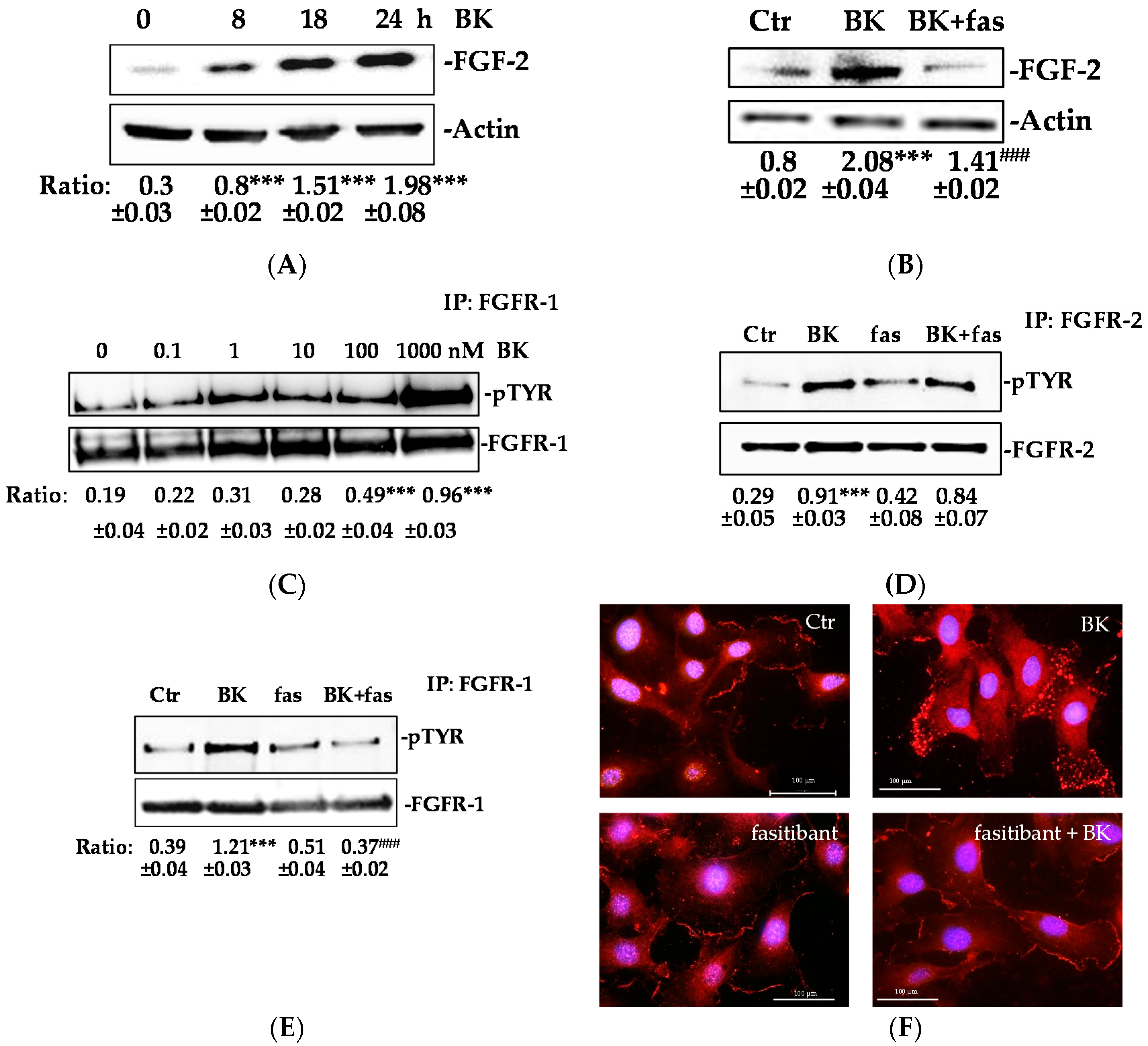
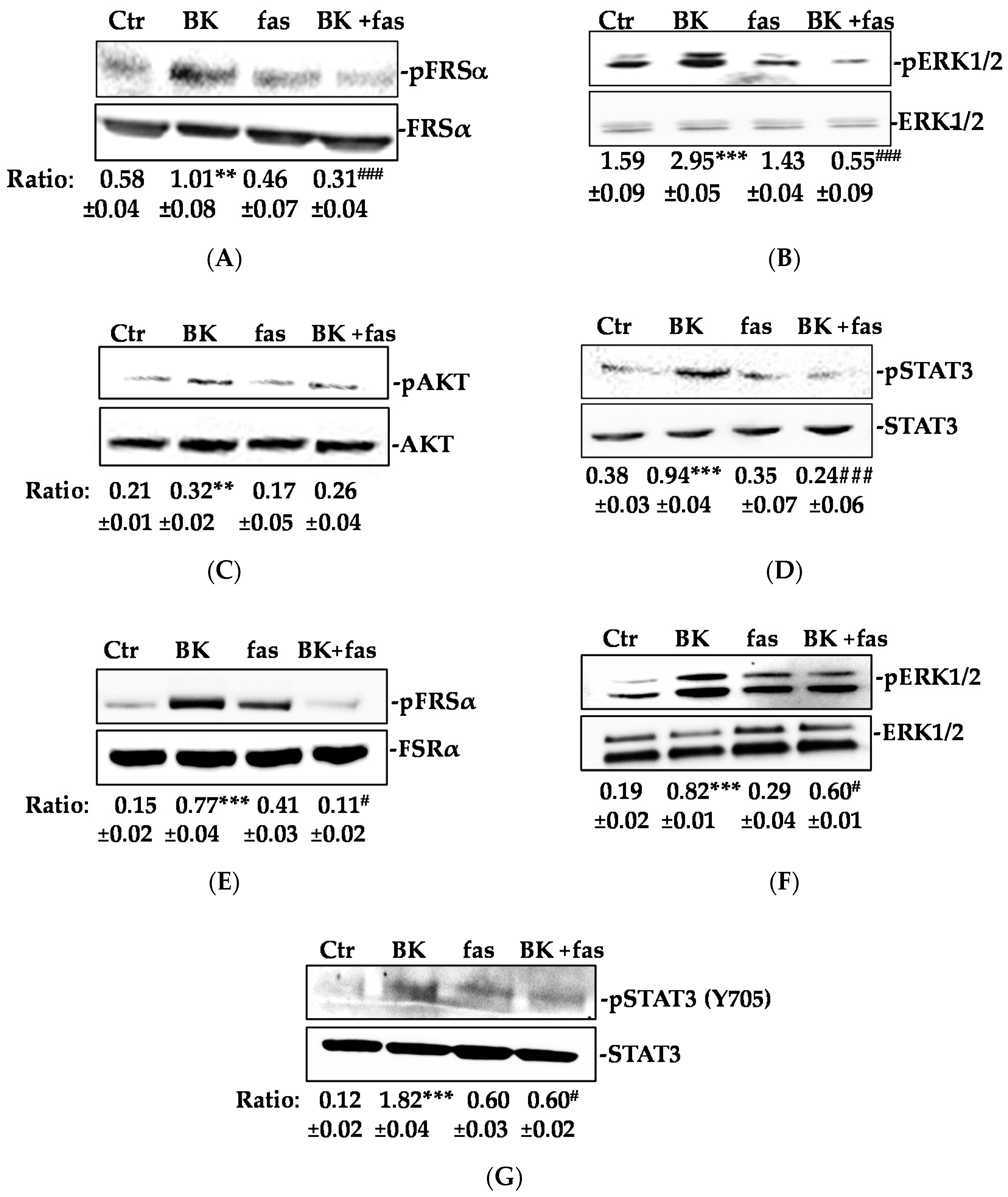
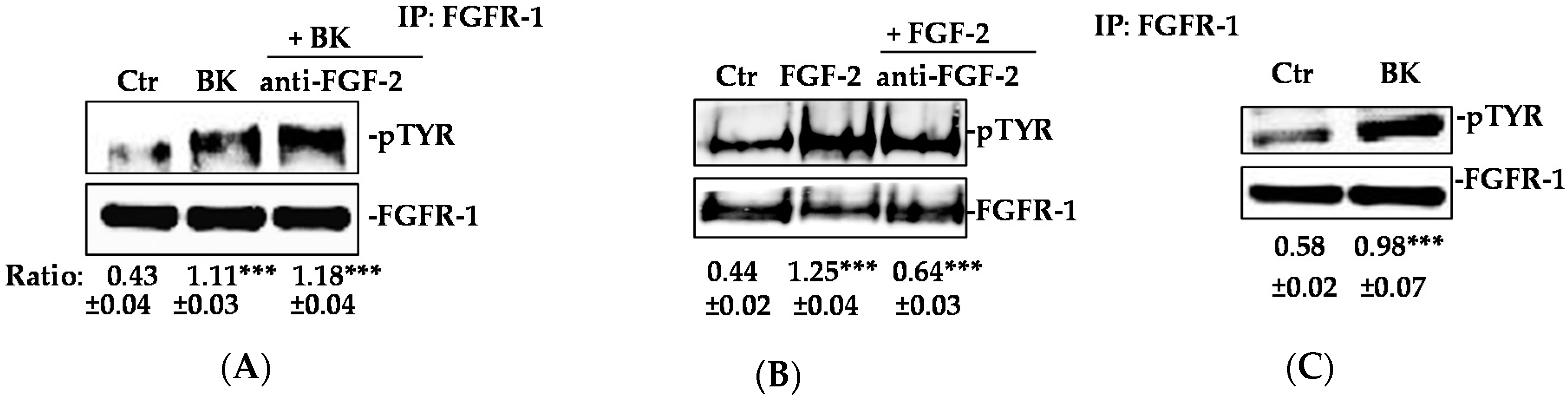
2.1. BK/B2R System Transactivates FGFR-1 and Drives Its Downstream Signaling in EC
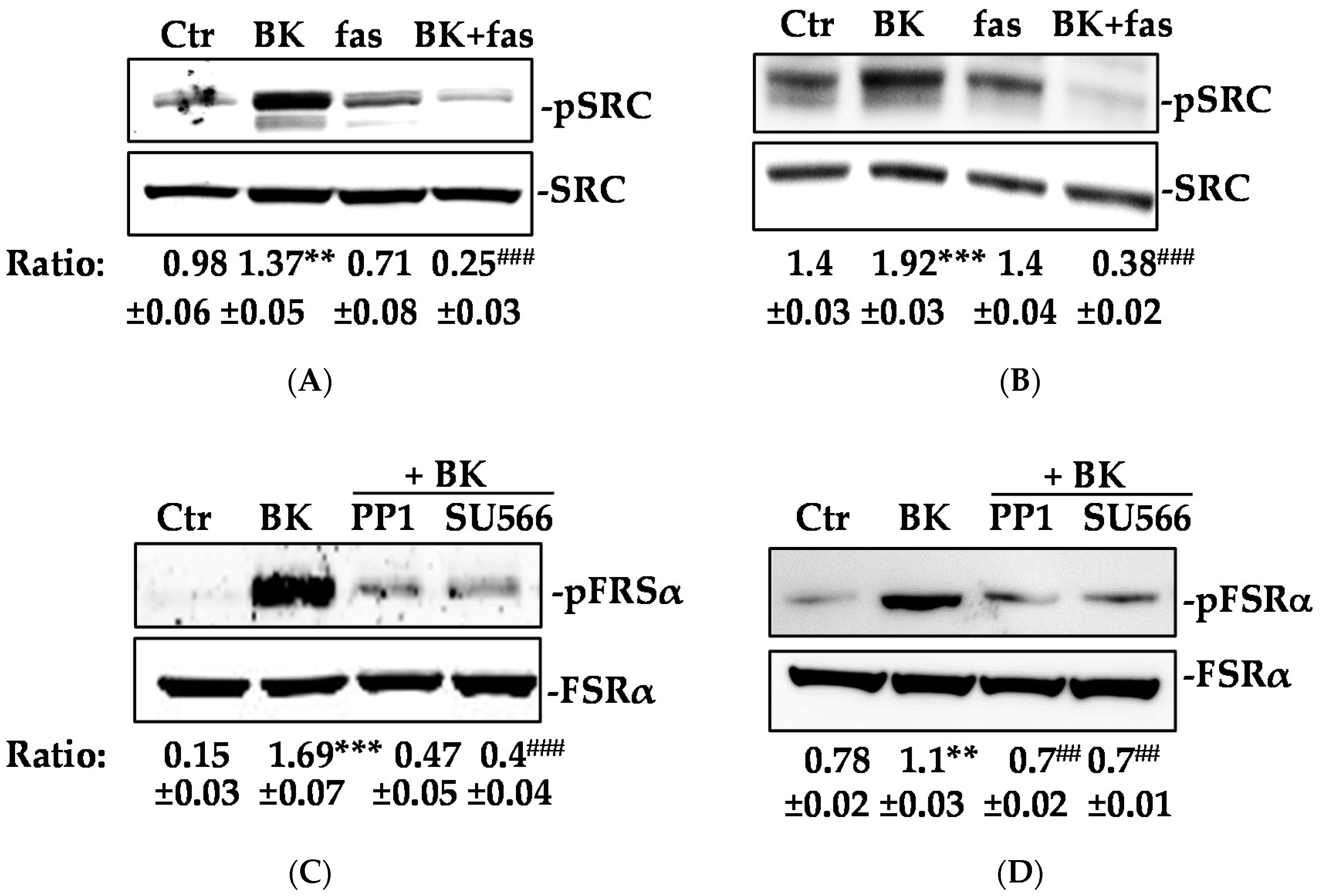
2.2. Phosphorylation of FGFR-1 by BK Requires the Activation of c-Src
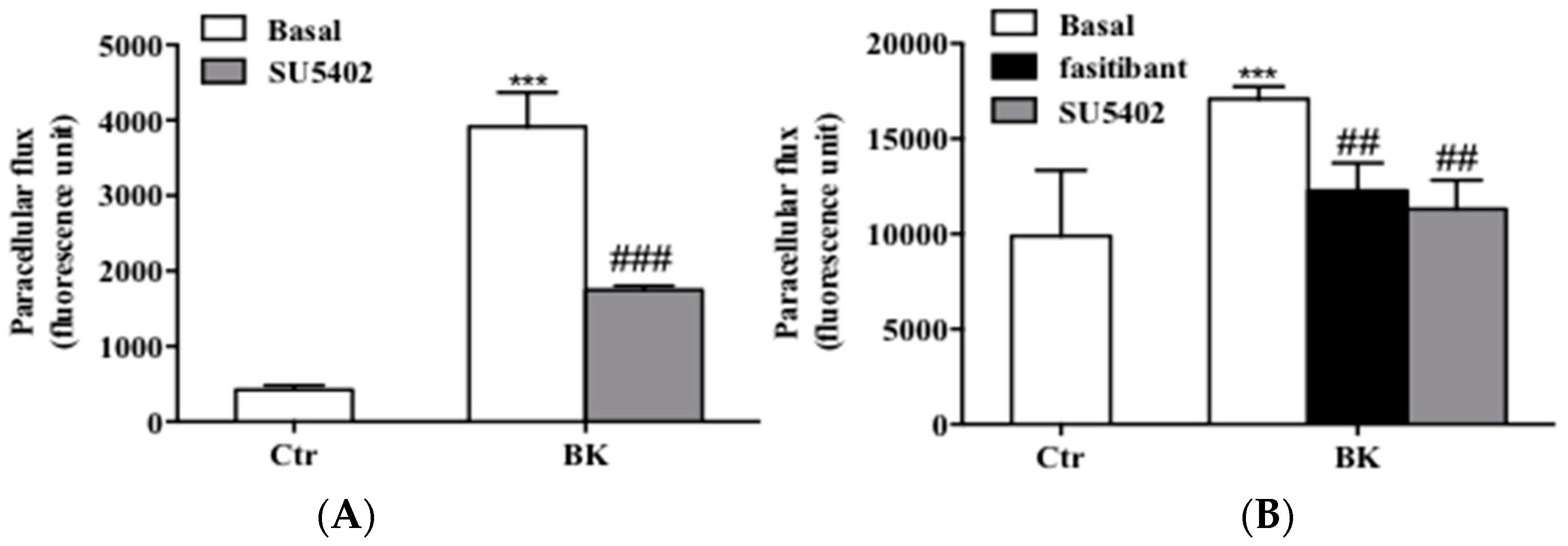
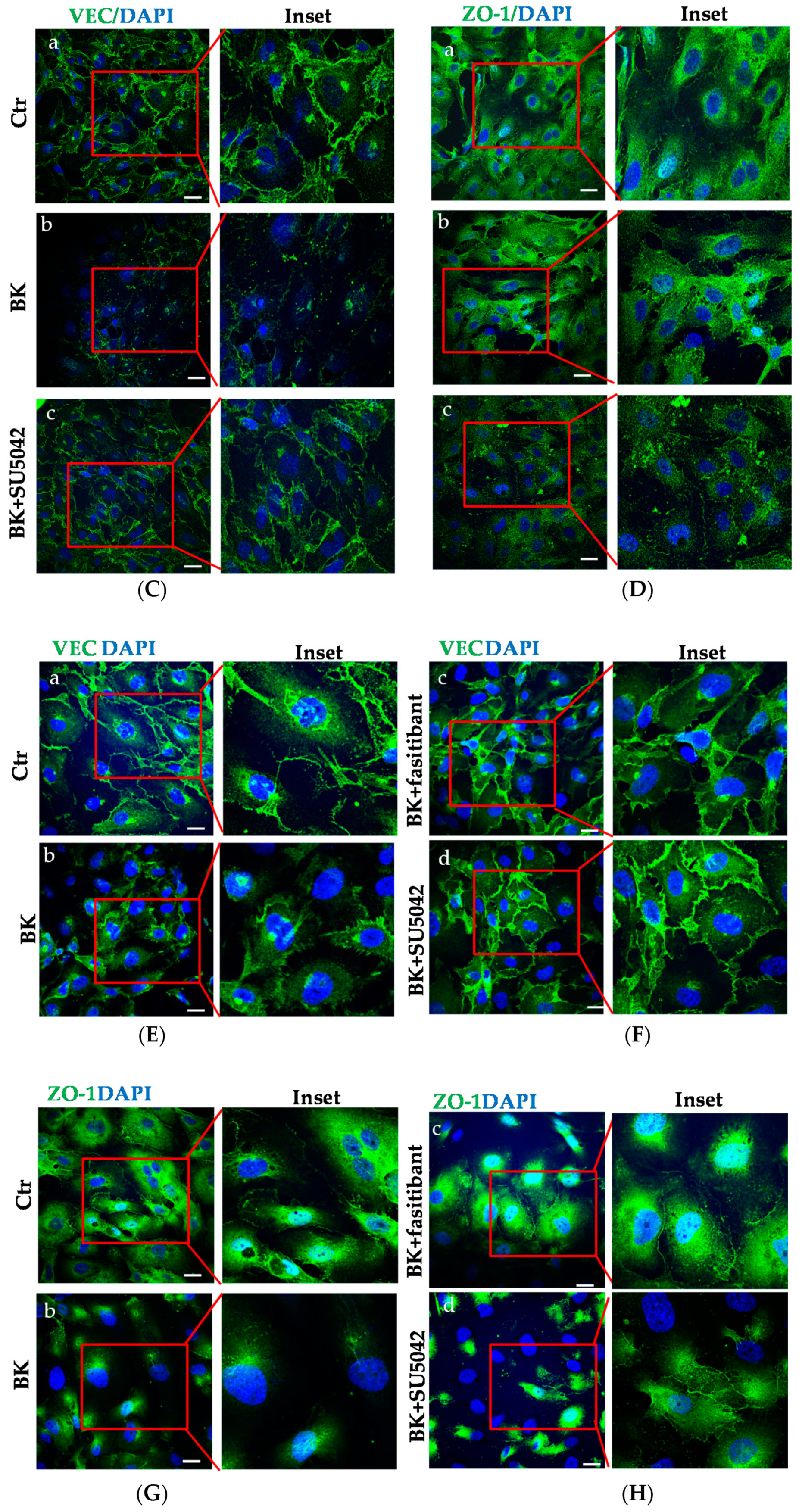
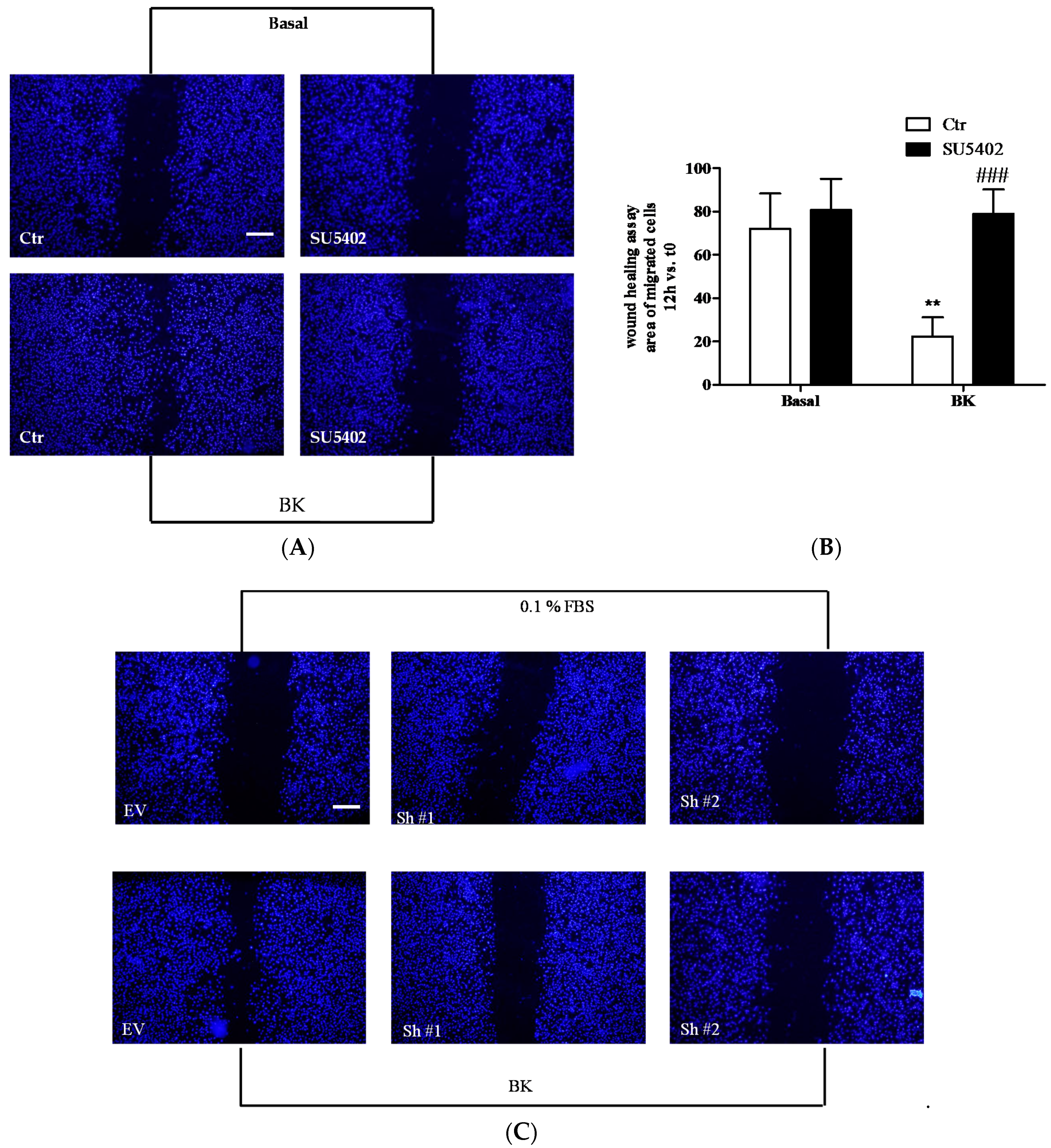
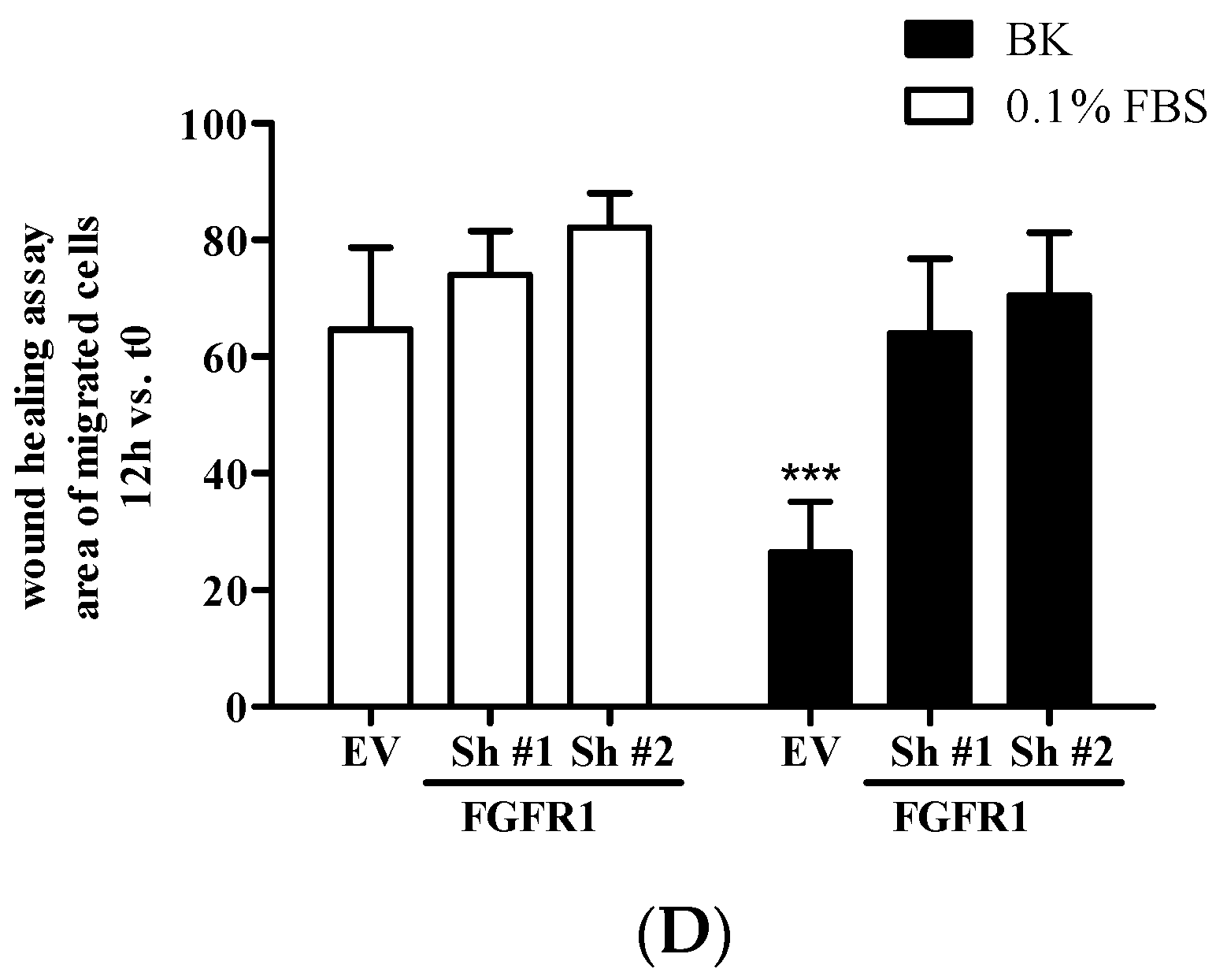
2.3. BK Induces Endothelial Permeability and Migration through FGFR-1 Activation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. FGFR-1 shRNA Transfection
4.3. Immunofluorescence Analysis
4.4. Western Blot Assay
4.5. Immunoprecipitation
4.6. Wound Assay
4.7. Permeability
4.8. Materials and Reagents
4.9. Data Analysis and Statistical Procedures
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADU | Arbitrary Density Unit |
| BK | Bradykinin |
| BSA | Bovine serum albumin |
| B2R | BK receptor 2 |
| fas | Fasitibant |
| FGF-2 | Fibroblast growth factor-2 |
| FGFR-1 | FGF-receptor-1 |
| HREC | Human retinal endothelial cells |
| NV | Neovessels |
| OIR | Oxygen induced retinopathy |
| PBS | Phosphate buffer saline |
| TYR | Tyrosine |
| VEC | Vascular endothelial cadherin |
| VEGF | Vascular endothelial growth factor |
| ZO-1 | Zona occludens 1 |
References
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Cervenak, L.; Morbidelli, L.; Donati, D.; Donnini, S.; Kambayashi, T.; Wilson, J.L.; Axelson, H.; Castaños-Velez, E.; Ljunggren, H.G.; Malefyt, R.D.; et al. Abolished angiogenicity and tumorigenicity of burkitt lymphoma by interleukin-10. Blood 2000, 96, 2568–2573. [Google Scholar] [PubMed]
- Presta, M.; Andrés, G.; Leali, D.; Dell’Era, P.; Ronca, R. Inflammatory cells and chemokines sustain fgf2-induced angiogenesis. Eur. Cytokine Netw. 2009, 20, 39–50. [Google Scholar] [PubMed]
- Murakami, M.; Elfenbein, A.; Simons, M. Non-canonical fibroblast growth factor signalling in angiogenesis. Cardiovasc. Res. 2008, 78, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Tiefenbacher, C.P.; Chilian, W.M. Basic fibroblast growth factor and heparin influence coronary arteriolar tone by causing endothelium-dependent dilation. Cardiovasc. Res. 1997, 34, 411–417. [Google Scholar] [CrossRef]
- Reuss, B.; von Bohlen und Halbach, O. Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 2003, 313, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Donnini, S.; Solito, R.; Giachetti, A.; Granger, H.J.; Ziche, M.; Morbidelli, L. Fibroblast growth factor-2 mediates angiotensin-converting enzyme inhibitor-induced angiogenesis in coronary endothelium. J. Pharmacol. Exp. Ther. 2006, 319, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Andrés, G.; Leali, D.; Mitola, S.; Coltrini, D.; Camozzi, M.; Corsini, M.; Belleri, M.; Hirsch, E.; Schwendener, R.A.; Christofori, G.; et al. A pro-inflammatory signature mediates FGF2-induced angiogenesis. J. Cell Mol. Med. 2009, 13, 2083–2108. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Heur, M. Interleukin-1β enhances cell migration through ap-1 and nf-κb pathway-dependent fgf2 expression in human corneal endothelial cells. Biol. Cell 2013, 105, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Blotnick, S.; Peoples, G.E.; Freeman, M.R.; Eberlein, T.J.; Klagsbrun, M. T lymphocytes synthesize and export heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor, mitogens for vascular cells and fibroblasts: Differential production and release by CD4+ and CD8+ T cells. Proc. Natl. Acad. Sci. USA 1994, 91, 2890–2894. [Google Scholar] [CrossRef] [PubMed]
- Peoples, G.E.; Blotnick, S.; Takahashi, K.; Freeman, M.R.; Klagsbrun, M.; Eberlein, T.J. T lymphocytes that infiltrate tumors and atherosclerotic plaques produce heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor: A potential pathologic role. Proc. Natl. Acad. Sci. USA 1995, 92, 6547–6551. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Solito, R.; Morbidelli, L.; Giachetti, A.; Ziche, M.; Donnini, S. Prostaglandin e2 regulates angiogenesis via activation of fibroblast growth factor receptor-1. J. Biol. Chem. 2008, 283, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Donnini, S.; Giachetti, A.; Morbidelli, L.; Ziche, M. Prostaglandin e(2) primes the angiogenic switch via a synergic interaction with the fibroblast growth factor-2 pathway. Circ. Res. 2009, 105, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Gualandris, A.; Rusnati, M.; Belleri, M.; Nelli, E.E.; Bastaki, M.; Molinari-Tosatti, M.P.; Bonardi, F.; Parolini, S.; Albini, A.; Morbidelli, L.; et al. Basic fibroblast growth factor overexpression in endothelial cells: An autocrine mechanism for angiogenesis and angioproliferative diseases. Cell Growth Differ. 1996, 7, 147–160. [Google Scholar] [PubMed]
- Calvani, M.; Rapisarda, A.; Uranchimeg, B.; Shoemaker, R.H.; Melillo, G. Hypoxic induction of an HIF-1alpha-dependent bFGF autocrine loop drives angiogenesis in human endothelial cells. Blood 2006, 107, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Monti, M.; Donnini, S.; Morbidelli, L.; Giachetti, A.; Mochly-Rosen, D.; Mignatti, P.; Ziche, M. Pkcε activation promotes fgf-2 exocytosis and induces endothelial cell proliferation and sprouting. J. Mol. Cell Cardiol. 2013, 63, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Meini, S.; Cucchi, P.; Catalani, C.; Cialdai, C.; Maggi, C.A.; Giachetti, A.; Ziche, M.; Donnini, S. Antagonism of bradykinin b2 receptor prevents inflammatory responses in human endothelial cells by quenching the NF-kB pathway activation. PLoS ONE 2014, 9, e84358. [Google Scholar] [CrossRef] [PubMed]
- Deacon, K.; Knox, A.J. Human airway smooth muscle cells secrete amphiregulin via bradykinin/COX-2/PGE2, inducing COX-2, CXCL8, and vegf expression in airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L237–L249. [Google Scholar] [CrossRef] [PubMed]
- Regoli, D.; Gobeil, F. Kinins and peptide receptors. Biol. Chem. 2016, 397, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Hofman, Z.L.; Relan, A.; Zeerleder, S.; Drouet, C.; Zuraw, B.; Hack, C.E. Angioedema attacks in patients with hereditary angioedema: Local manifestations of a systemic activation process. J. Allergy Clin. Immunol. 2016, 138, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Zhou, J.; Wang, B.; Dai, J.; Colman, R.W.; Song, W.; Wu, Y. A critical role for plasma kallikrein in the pathogenesis of autoantibody-induced arthritis. FASEB J. 2017, 31, 5419–5431. [Google Scholar] [CrossRef] [PubMed]
- Terzuoli, E.; Morbidelli, L.; Nannelli, G.; Giachetti, A.; Donnini, S.; Ziche, M. Involvement of bradykinin b2 receptor in pathological vascularization in oxygen-induced retinopathy in mice and rabbit cornea. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Dell'Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Breit, A.; Besik, V.; Solinski, H.J.; Muehlich, S.; Glas, E.; Yarwood, S.J.; Gudermann, T. Serine-727 phosphorylation activates hypothalamic stat-3 independently from tyrosine-705 phosphorylation. Mol. Endocrinol. 2015, 29, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.Q.; Ravindranath, L.; Mohamed, A.S.; Zohar, O.; Chen, G.H.; Lyketsos, C.G.; Etcheberrigaray, R.; Alkon, D.L. Map kinase signaling cascade dysfunction specific to alzheimer’s disease in fibroblasts. Neurobiol. Dis. 2002, 11, 166–183. [Google Scholar] [CrossRef] [PubMed]
- Bazzani, L.; Donnini, S.; Finetti, F.; Christofori, G.; Ziche, M. PGE2/EP3/SRC signaling induces egfr nuclear translocation and growth through egfr ligands release in lung adenocarcinoma cells. Oncotarget 2017, 8, 31270–31287. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Soder, R.P.; Cheng, Q.; Rovner, E.S.; Petkov, G.V. Selective inhibition of phosphodiesterase 1 relaxes urinary bladder smooth muscle: role for ryanodine receptor-mediated BK channel activation. Am. J. Physiol. Cell Physiol. 2012, 303, C1079–C1089. [Google Scholar] [CrossRef] [PubMed]
- Meini, S.; Maggi, C.A. Knee osteoarthritis: A role for bradykinin? Inflamm. Res. 2008, 57, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, S.I.; Issekutz, A.C. Basic fibroblast growth factor (bFGF, FGF-2) potentiates leukocyte recruitment to inflammation by enhancing endothelial adhesion molecule expression. Am. J. Pathol. 2006, 168, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Terasaki, H. Biological involvement of micrornas in proliferative vitreoretinopathy. Transl. Vis. Sci. Technol. 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Gudernova, I.; Vesela, I.; Balek, L.; Buchtova MDosedelova, H.; Kunova, M.; Pivnicka, J.; Jelinkova, I.; Roubalova, L.; Kozubik, A. Multikinase activity of fibroblast growth factor receptor (FGFR) inhibitors SU5402, PD173074, AZD1480, AZD4547 and BGJ398 compromises the use of small chemicals targeting FGFR catalytic activity for therapy of short-stature syndromes. Hum. Mol. Genet. 2016, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, Z.C. STAT3: A critical transcription activator in angiogenesis. Med. Res. Rev. 2008, 28, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Pintucci, G.; Yu, P.J.; Sharony, R.; Baumann, F.G.; Saponara, F.; Frasca, A.; Galloway, A.C.; Moscatelli, D.; Mignatti, P. Induction of stromelysin-1 (MMP-3) by fibroblast growth factor-2 (FGF-2) in FGF-2−/− microvascular endothelial cells requires prolonged activation of extracellular signal-regulated kinases-1 and -2 (ERK-1/2). J. Cell Biochem. 2003, 90, 1015–1025. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terzuoli, E.; Corti, F.; Nannelli, G.; Giachetti, A.; Donnini, S.; Ziche, M. Bradykinin B2 Receptor Contributes to Inflammatory Responses in Human Endothelial Cells by the Transactivation of the Fibroblast Growth Factor Receptor FGFR-1. Int. J. Mol. Sci. 2018, 19, 2638. https://doi.org/10.3390/ijms19092638
Terzuoli E, Corti F, Nannelli G, Giachetti A, Donnini S, Ziche M. Bradykinin B2 Receptor Contributes to Inflammatory Responses in Human Endothelial Cells by the Transactivation of the Fibroblast Growth Factor Receptor FGFR-1. International Journal of Molecular Sciences. 2018; 19(9):2638. https://doi.org/10.3390/ijms19092638
Chicago/Turabian StyleTerzuoli, Erika, Federico Corti, Ginevra Nannelli, Antonio Giachetti, Sandra Donnini, and Marina Ziche. 2018. "Bradykinin B2 Receptor Contributes to Inflammatory Responses in Human Endothelial Cells by the Transactivation of the Fibroblast Growth Factor Receptor FGFR-1" International Journal of Molecular Sciences 19, no. 9: 2638. https://doi.org/10.3390/ijms19092638
APA StyleTerzuoli, E., Corti, F., Nannelli, G., Giachetti, A., Donnini, S., & Ziche, M. (2018). Bradykinin B2 Receptor Contributes to Inflammatory Responses in Human Endothelial Cells by the Transactivation of the Fibroblast Growth Factor Receptor FGFR-1. International Journal of Molecular Sciences, 19(9), 2638. https://doi.org/10.3390/ijms19092638