The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma

 , , and
, , and 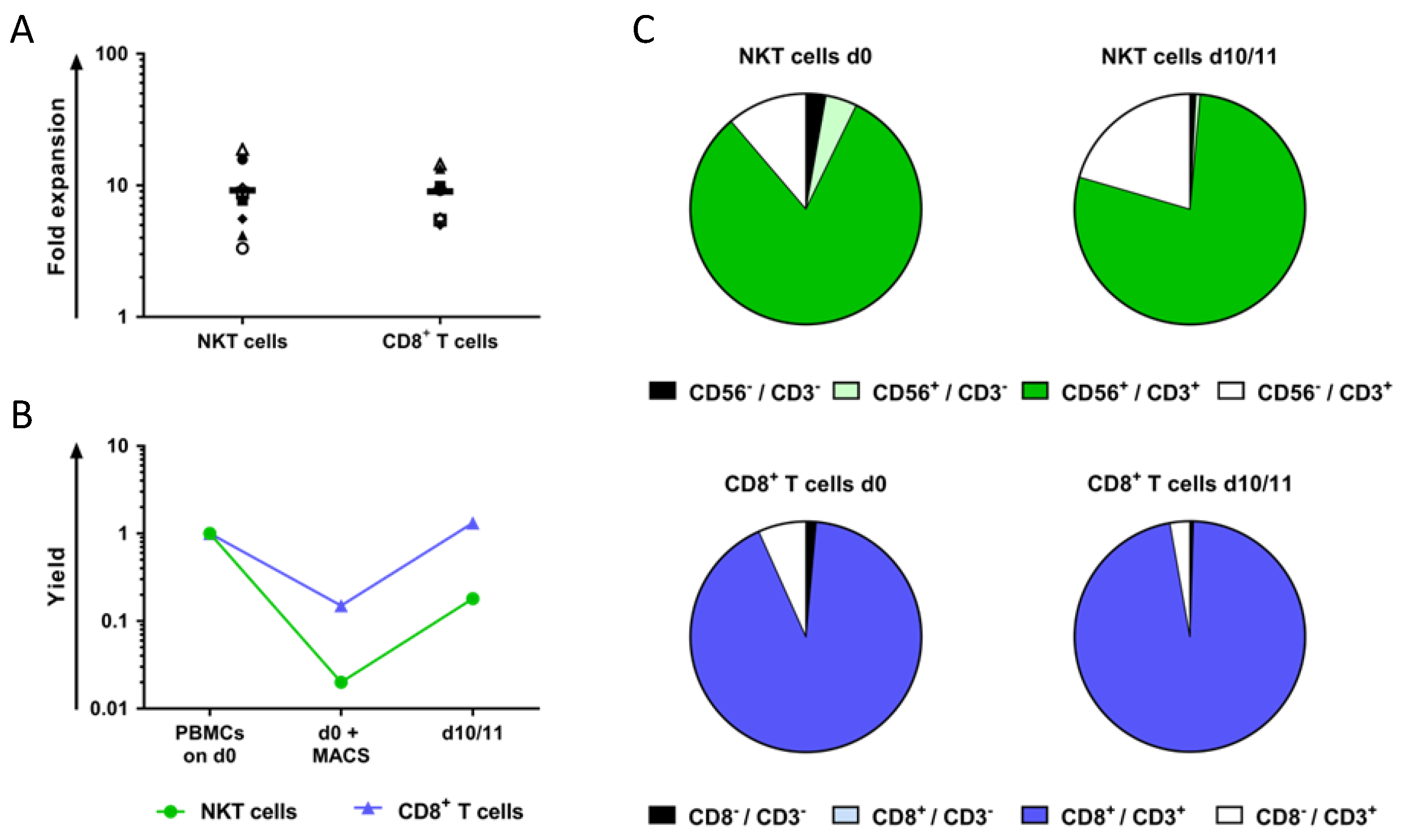{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. NKT Cells Can Be Isolated from Peripheral Blood Mononuclear Cells (PBMCs) via Magnetic Activated Cell Sorting and Subsequently Expanded Using CD3 Monoclonal Antibody (OKT-3) and Interleukin-2 (IL-2)
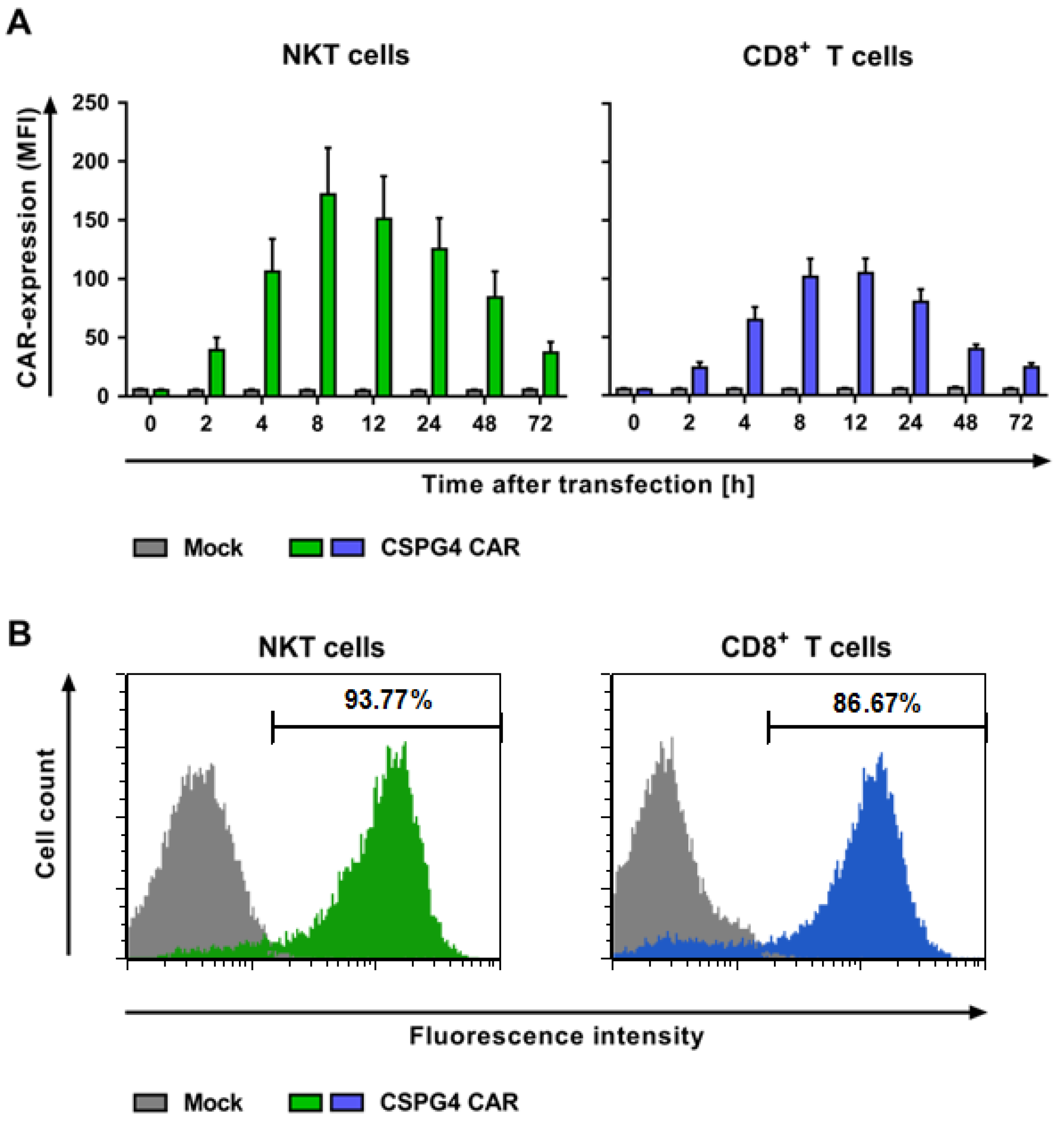
2.2. NKT Cells Can Be Efficiently Transfected with a CSPG4-Specific CAR Using mRNA Electroporation
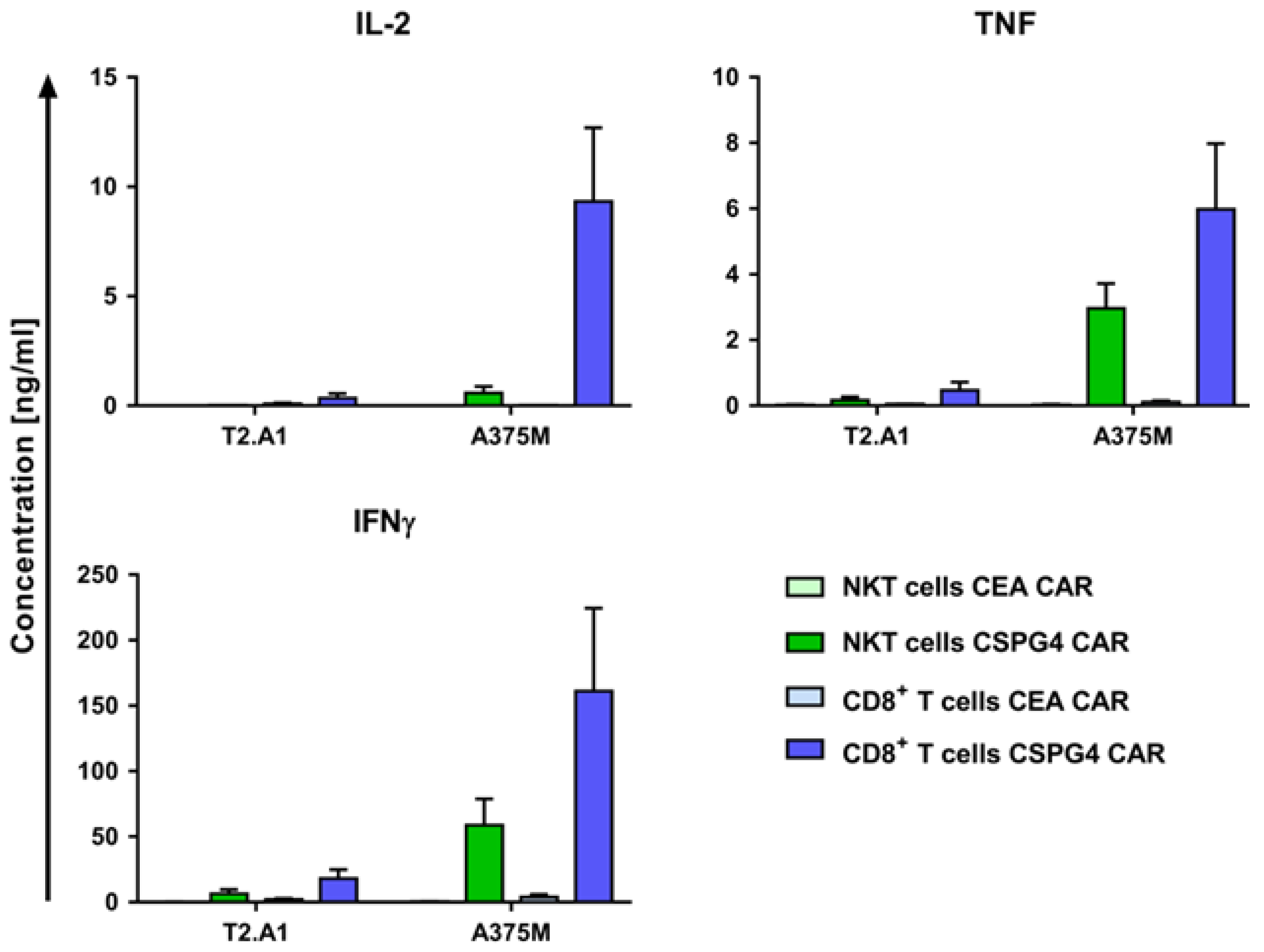
2.3. CAR-NKT Cells Specifically Produce Cytokines after Antigen Encounter
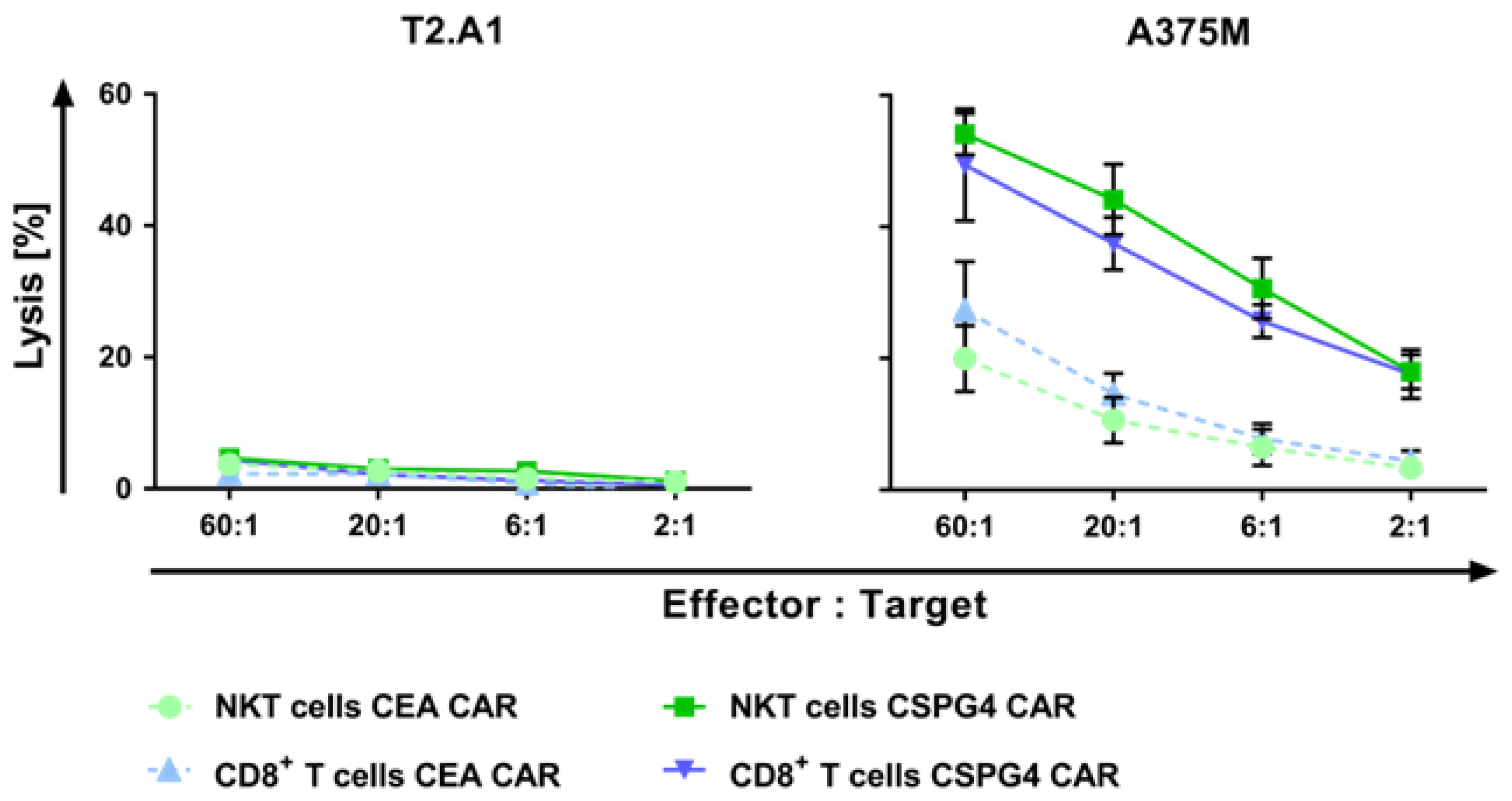
2.4. CAR-NKT Cells Lyse Human Melanoma Cells in an Antigen-Specific Manner
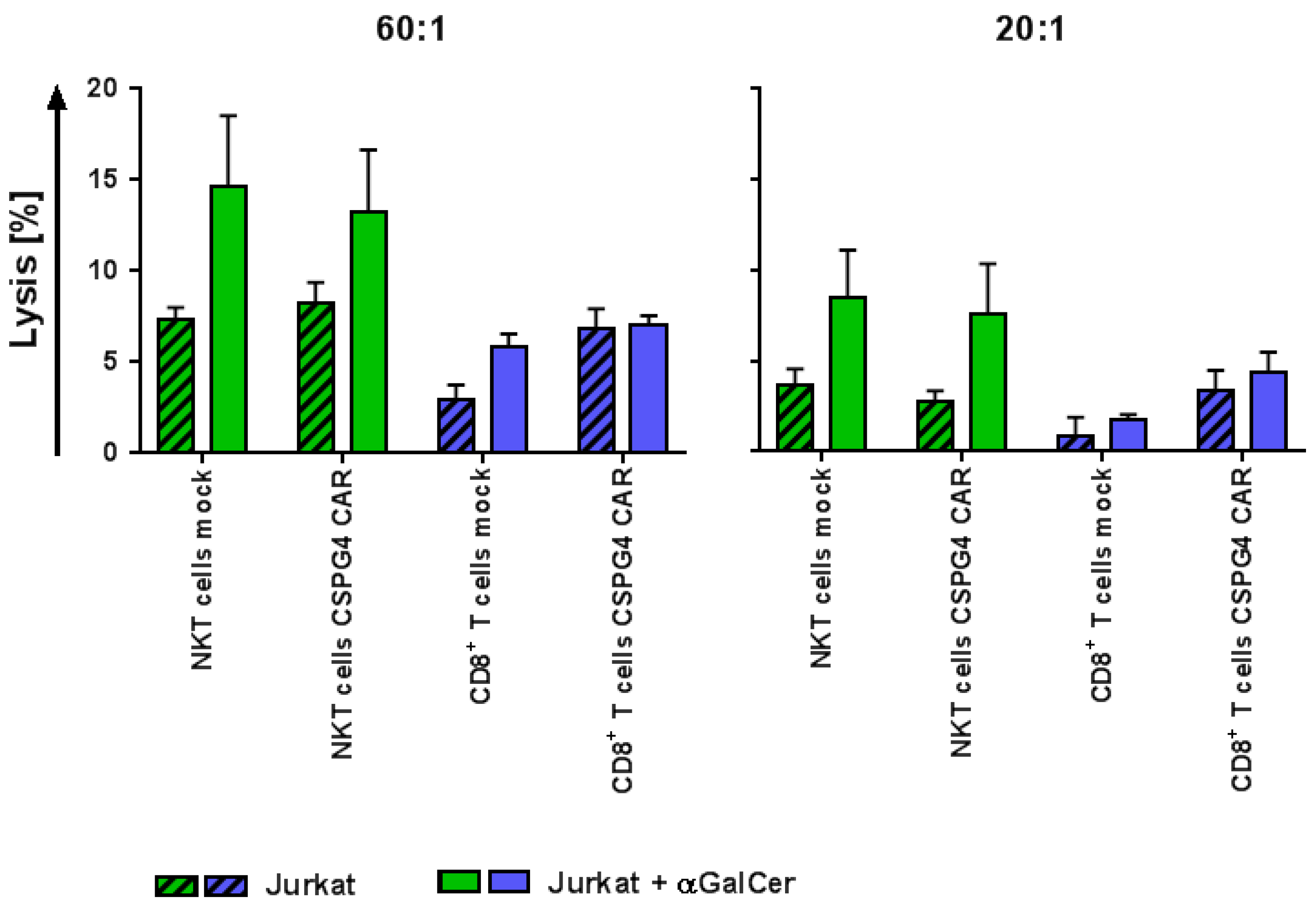
2.5. CAR-NKT Cells Retain Their Intrinsic Lytic Capacity Against α-GalCer-Loaded Jurkat Cells Expressing CD1d
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Expansion of T Cells
4.3. Flow Cytometric Analyses of Phenotypic Parameters
4.4. RNA Production and Transfection
4.5. Surface Expression of Transfected Receptors
4.6. Cytokine Production
4.7. Cytotoxicity
4.8. Figure Preparation and Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| αGalCer | alpha-galactosylceramide |
| ALL | acute lymphoblastic leukemia |
| CAR | chimeric antigen receptor |
| CEA | carcinoembyronic antigen |
| CRS | cytokine release syndrome |
| CSPG4 | chondroitin sulfate proteoglycan 4 |
| GvHD | graft versus host disease |
| HMW-MAA | high molecular weight-melanoma associated antigen |
| IFNγ | interferon-gamma |
| IL | Interleukin |
| MACS | magnetic-activated cell sorting |
| MCSP | melanoma-associated chondroitin sulfate proteoglycan |
| NK cells | natural killer cells |
| NKT cells | natural killer T cells |
| PBMCs | peripheral blood mononuclear cells |
| TCR | T-cell receptor |
| TNF | tumor necrosis factor |
References
- Park, J.H.; Riviere, I.; Gonen, M.; Wang, X.; Senechal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Xia, A.L.; Wang, X.C.; Lu, Y.J.; Lu, X.J.; Sun, B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: Challenges and opportunities. Oncotarget 2017, 8, 90521–90531. [Google Scholar] [CrossRef] [PubMed]
- Zeltsman, M.; Dozier, J.; McGee, E.; Ngai, D.; Adusumilli, P.S. CAR T-cell therapy for lung cancer and malignant pleural mesothelioma. Transl. Res. 2017, 187, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Locke, F.L.; Lin, Y.; Jain, N.; Daver, N.; Gulbis, A.M.; Adkins, S.; et al. Toxicity management after chimeric antigen receptor T cell therapy: One size does not fit ‘ALL’. Nat. Rev. Clin. Oncol. 2018, 15, 218. [Google Scholar] [CrossRef] [PubMed]
- Bedoya, F.; Frigault, M.J.; Maus, M.V. The Flipside of the Power of Engineered T Cells: Observed and Potential Toxicities of Genetically Modified T Cells as Therapy. Mol. Ther. 2017, 25, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Dorrie, J.; Schaft, N. Chimeric Antigen Receptors in Different Cell Types: New Vehicles Join the Race. Hum. Gene Ther. 2018, 29, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.L. The Influence of Invariant Natural Killer T Cells on Humoral Immunity to T-Dependent and -Independent Antigens. Front. Immunol. 2018, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Wolf, B.J.; Choi, J.E.; Exley, M.A. Novel Approaches to Exploiting Invariant NKT Cells in Cancer Immunotherapy. Front. Immunol. 2018, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; Stankovic, S.; Baxter, A.G. Raising the NKT cell family. Nat. Immunol. 2010, 11, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Cui, J.; Koezuka, Y.; Toura, I.; Kaneko, Y.; Motoki, K.; Ueno, H.; Nakagawa, R.; Sato, H.; Kondo, E.; et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science 1997, 278, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, A.; Motohashi, S.; Ishikawa, E.; Fuchida, H.; Higashino, K.; Otsuji, M.; Iizasa, T.; Nakayama, T.; Taniguchi, M.; Fujisawa, T. A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.H.; Osman, K.; Connolly, J.; Kukreja, A.; Krasovsky, J.; Pack, M.; Hutchinson, A.; Geller, M.; Liu, N.; Annable, R.; et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J. Exp. Med. 2005, 201, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Exley, M.A.; Friedlander, P.; Alatrakchi, N.; Vriend, L.; Yue, S.; Sasada, T.; Zeng, W.; Mizukami, Y.; Clark, J.; Nemer, D.; et al. Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin. Cancer Res. 2017, 23, 3510–3519. [Google Scholar] [CrossRef] [PubMed]
- Pillai, A.B.; George, T.I.; Dutt, S.; Teo, P.; Strober, S. Host NKT cells can prevent graft-versus-host disease and permit graft antitumor activity after bone marrow transplantation. J. Immunol. 2007, 178, 6242–6251. [Google Scholar] [CrossRef] [PubMed]
- Dellabona, P.; Casorati, G.; de Lalla, C.; Montagna, D.; Locatelli, F. On the use of donor-derived iNKT cells for adoptive immunotherapy to prevent leukemia recurrence in pediatric recipients of HLA haploidentical HSCT for hematological malignancies. Clin. Immunol. 2011, 140, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Slauenwhite, D.; Johnston, B. Regulation of NKT Cell Localization in Homeostasis and Infection. Front. Immunol. 2015, 6, 255. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N.; Dorrie, J.; Muller, I.; Beck, V.; Baumann, S.; Schunder, T.; Kampgen, E.; Schuler, G. A new way to generate cytolytic tumor-specific T cells: Electroporation of RNA coding for a T cell receptor into T lymphocytes. Cancer Immunol. Immunother. 2006, 55, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, Z.; Khong, H.T.; Rosenberg, S.A.; Morgan, R.A. Transduction of an HLA-DP4-restricted NY-ESO-1-specific TCR into primary human CD4+ lymphocytes. J. Immunother. 2006, 29, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, P.M.; Komarovskaya, M.E.; Ye, Z.J.; Imai, C.; Campana, D.; Bahceci, E.; Weissman, S.M. Synthetic messenger RNA as a tool for gene therapy. Hum. Gene Ther. 2006, 17, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Birkholz, K.; Paulus, A.; Schwenkert, M.; Schmidt, P.; Hoffmann, N.; Hombach, A.; Fey, G.; Abken, H.; Schuler, G.; et al. Stability and activity of MCSP-specific chimeric antigen receptors (CARs) depend on the scFv antigen-binding domain and the protein backbone. Cancer Immunol. Immunother. 2015, 64, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.M.; Zhao, Y.; Liu, X.; Jiang, S.; Carpenito, C.; Kalos, M.; Carroll, R.G.; June, C.H.; Grupp, S.A. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum. Gene Ther. 2011, 22, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific Chimeric Antigen Receptor mRNA-Engineered T cells Induce Anti-Tumor Activity in Solid Malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.R.; Chang, C.C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human high molecular weight-melanoma-associated antigen (HMW-MAA): A melanoma cell surface chondroitin sulfate proteoglycan (MSCP) with biological and clinical significance. Crit. Rev. Immunol. 2004, 24, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Godal, A.; Bruland, O.; Haug, E.; Aas, M.; Fodstad, O. Unexpected expression of the 250 kD melanoma-associated antigen in human sarcoma cells. Br. J. Cancer 1986, 53, 839–841. [Google Scholar] [CrossRef] [PubMed]
- Chekenya, M.; Rooprai, H.K.; Davies, D.; Levine, J.M.; Butt, A.M.; Pilkington, G.J. The NG2 chondroitin sulfate proteoglycan: Role in malignant progression of human brain tumours. Int. J. Dev. Neurosci. 1999, 17, 421–435. [Google Scholar] [CrossRef]
- Metelitsa, L.S.; Naidenko, O.V.; Kant, A.; Wu, H.W.; Loza, M.J.; Perussia, B.; Kronenberg, M.; Seeger, R.C. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J. Immunol. 2001, 167, 3114–3122. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.M.; McNeel, D.G. Antigen loss and tumor-mediated immunosuppression facilitate tumor recurrence. Expert. Rev. Vaccines 2012, 11, 1315–1317. [Google Scholar]
- Nieda, M.; Okai, M.; Tazbirkova, A.; Lin, H.; Yamaura, A.; Ide, K.; Abraham, R.; Juji, T.; Macfarlane, D.J.; Nicol, A.J. Therapeutic activation of Valpha24+Vbeta11+ NKT cells in human subjects results in highly coordinated secondary activation of acquired and innate immunity. Blood 2004, 103, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J. Clin. Investig. 2016, 126, 2341–2355. [Google Scholar] [CrossRef] [PubMed]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The Role of Natural Killer T Cells in Cancer- A Phenotypical and Functional Approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Haraguchi, K.; Chiba, S.; Yasukawa, M.; Shibata, Y.; Hirai, H. Valpha24+ natural killer T-cell responses against T-acute lymphoblastic leukaemia cells: Implications for immunotherapy. Br. J. Haematol. 2003, 122, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Le, R.Q.; Li, L.; Yuan, W.; Shord, S.S.; Nie, L.; Habtemariam, B.A.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Rogers, P.R.; Matsumoto, A.; Naidenko, O.; Kronenberg, M.; Mikayama, T.; Kato, S. Expansion of human Valpha24+ NKT cells by repeated stimulation with KRN7000. J. Immunol. Methods 2004, 285, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Shinga, J.; Yamasaki, S.; Kawamura, M.; Dorrie, J.; Schaft, N.; Sato, Y.; Iyoda, T.; Fujii, S. Transfer of mRNA Encoding Invariant NKT Cell Receptors Imparts Glycolipid Specific Responses to T Cells and gammadeltaT Cells. PLoS ONE 2015, 10, e0131477. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nieda, M.; Koezuka, Y.; Nicol, A.; Porcelli, S.A.; Ishikawa, Y.; Tadokoro, K.; Hirai, H.; Juji, T. Analysis of human V alpha 24+ CD4+ NKT cells activated by alpha-glycosylceramide-pulsed monocyte-derived dendritic cells. J. Immunol. 2000, 164, 4458–4464. [Google Scholar] [CrossRef] [PubMed]
- Uslu, U.; Schuler, G.; Dorrie, J.; Schaft, N. Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp. Dermatol. 2016, 25, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Harrer, D.C.; Schuler-Thurner, B.; Schaft, N.; Schuler, G.; Dorrie, J.; Uslu, U. The siRNA-mediated downregulation of PD-1 alone or simultaneously with CTLA-4 shows enhanced in-vitro CAR-T cell functionality for further clinical development towards the potential use in immunotherapy of melanoma. Exp. Dermatol. 2018, 27, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Harrer, D.C.; Simon, B.; Fujii, S.I.; Shimizu, K.; Uslu, U.; Schuler, G.; Gerer, K.F.; Hoyer, S.; Dorrie, J.; Schaft, N. RNA-transfection of gamma/delta T cells with a chimeric antigen receptor or an alpha/beta T-cell receptor: A safer alternative to genetically engineered alpha/beta T cells for the immunotherapy of melanoma. BMC Cancer 2017, 17, 551. [Google Scholar] [CrossRef] [PubMed]
- Krug, C.; Wiesinger, M.; Abken, H.; Schuler-Thurner, B.; Schuler, G.; Dorrie, J.; Schaft, N. A GMP-compliant protocol to expand and transfect cancer patient T cells with mRNA encoding a tumor-specific chimeric antigen receptor. Cancer Immunol. Immunother. 2014, 63, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [PubMed]
- Schaft, N.; Dorrie, J.; Thumann, P.; Beck, V.E.; Muller, I.; Schultz, E.S.; Kampgen, E.; Dieckmann, D.; Schuler, G. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfecting defined RNAs after rather than before maturation. J. Immunol. 2005, 174, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Hofflin, S.; Prommersberger, S.; Uslu, U.; Schuler, G.; Schmidt, C.W.; Lennerz, V.; Dorrie, J.; Schaft, N. Generation of CD8(+) T cells expressing two additional T-cell receptors (TETARs) for personalised melanoma therapy. Cancer Biol. Ther. 2015, 16, 1323–1331. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simon, B.; Wiesinger, M.; März, J.; Wistuba-Hamprecht, K.; Weide, B.; Schuler-Thurner, B.; Schuler, G.; Dörrie, J.; Uslu, U. The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. Int. J. Mol. Sci. 2018, 19, 2365. https://doi.org/10.3390/ijms19082365
Simon B, Wiesinger M, März J, Wistuba-Hamprecht K, Weide B, Schuler-Thurner B, Schuler G, Dörrie J, Uslu U. The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. International Journal of Molecular Sciences. 2018; 19(8):2365. https://doi.org/10.3390/ijms19082365
Chicago/Turabian StyleSimon, Bianca, Manuel Wiesinger, Johannes März, Kilian Wistuba-Hamprecht, Benjamin Weide, Beatrice Schuler-Thurner, Gerold Schuler, Jan Dörrie, and Ugur Uslu. 2018. "The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma" International Journal of Molecular Sciences 19, no. 8: 2365. https://doi.org/10.3390/ijms19082365
APA StyleSimon, B., Wiesinger, M., März, J., Wistuba-Hamprecht, K., Weide, B., Schuler-Thurner, B., Schuler, G., Dörrie, J., & Uslu, U. (2018). The Generation of CAR-Transfected Natural Killer T Cells for the Immunotherapy of Melanoma. International Journal of Molecular Sciences, 19(8), 2365. https://doi.org/10.3390/ijms19082365