N6-Methyladenosine Role in Acute Myeloid Leukaemia



Abstract
1. Introduction
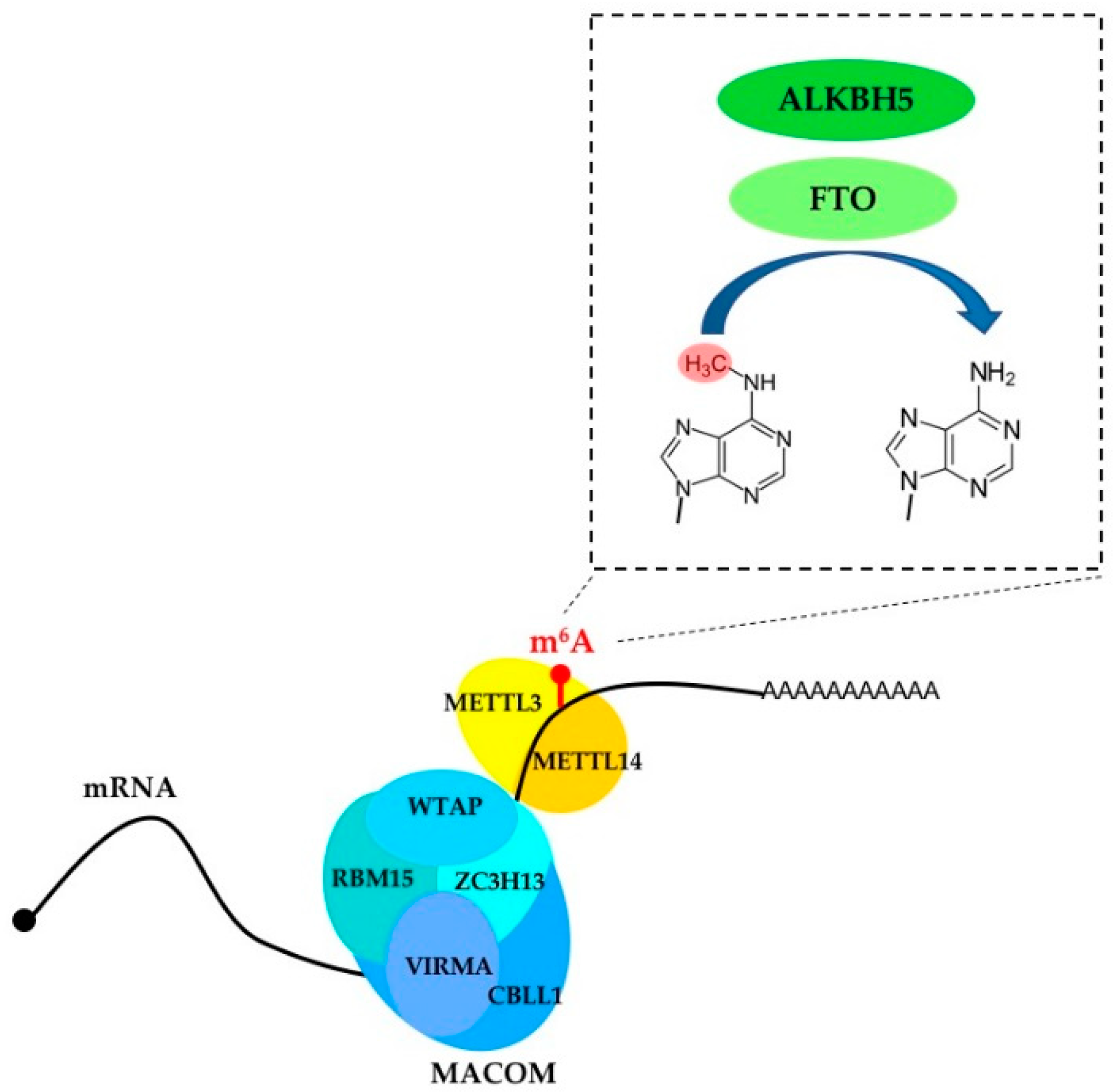
2. m6A Writers, Erasers and Readers
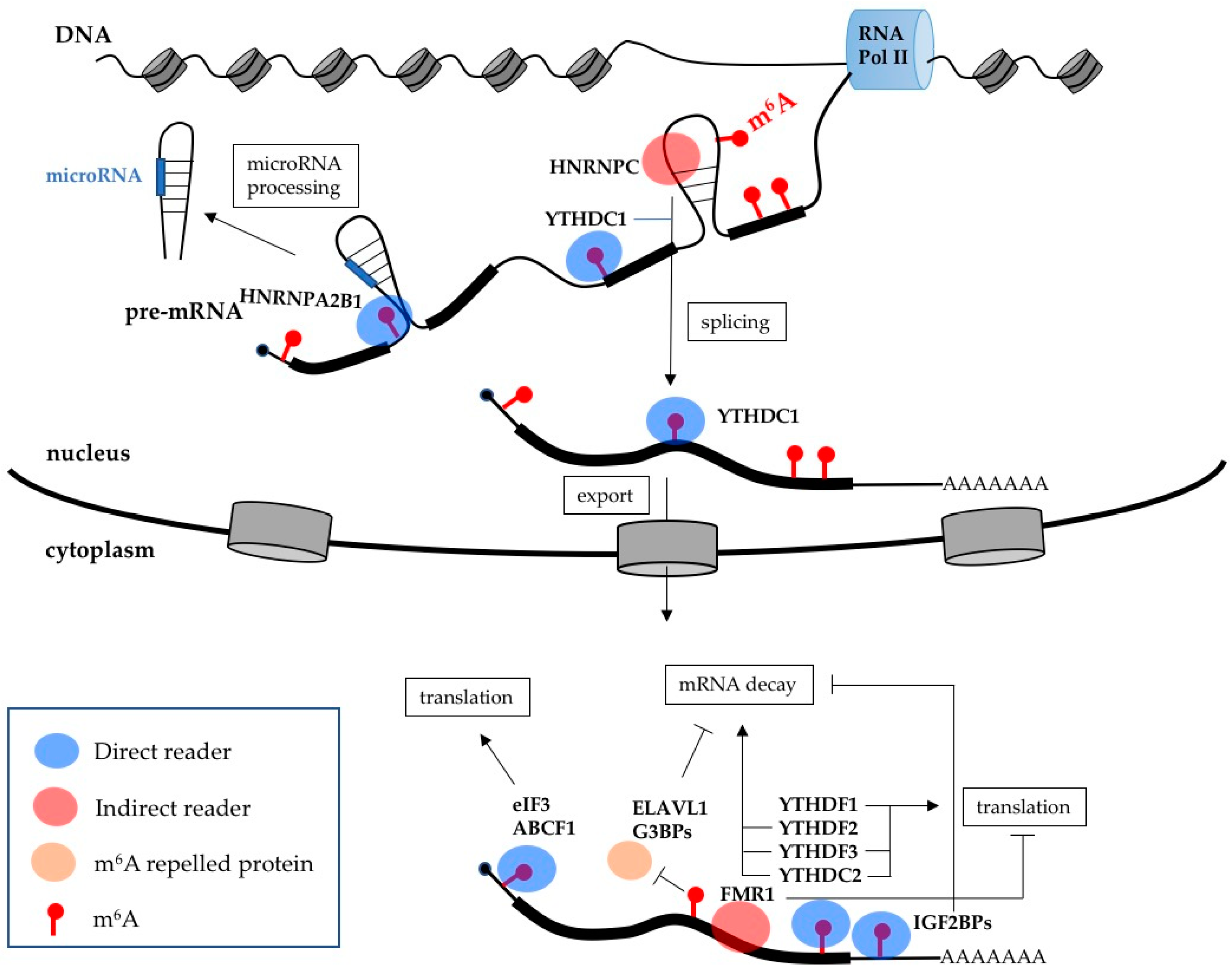
3. m6A Effects on Gene Expression
4. m6A Roles in AML (Acute Myeloid Leukaemia) and Normal Haematopoiesis
4.1. WTAP (Wilms Tumor 1-Associated Protein) in AML
4.2. Core METTL3/METTL14 (Methyltransferase-Like Protein 3/Methyltransferase-Like Protein 14) Complex in AML
4.3. RBM15 (RNA-binding motif 15) in AML
4.4. FTO (Fat Mass and Obesity-Associated) in AML
4.5. Core METTL3/METTL14 Complex in Normal Haematopoiesis
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ABCF1 | ATP binding cassette subfamily F member 1 |
| AML | Acute myeloid leukaemia |
| CEBP | CCAAT enhancer binding protein |
| CLIP | Cross-linked Immunoprecipitation |
| ELAVL1 | ELAV like RNA binding protein 1 |
| G3BP | G3BP stress granule assembly factor |
| hnRNP | Heterogeneous nuclear ribonucleoprotein |
| HSC | Haematopoietic stem cell |
| HSPC | Haematopoietic stem and progenitor cells |
| IGF2BP | Insulin-like growth factor 2 mRNA-binding protein |
| KO | Knock-out |
| LncRNA | Long non-coding RNA |
| mESC | Mouse embryonic stem cells |
| R-2HG | R-2-hydroxyglutarate |
| SAM | S-adenosylmethionine |
| UTR | Untranslated region |
References
- Tenen, D.G. Disruption of differentiation in human cancer: AML shows the way. Nat. Rev. Cancer 2003, 3, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 1, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Knuckles, P.; Bühler, M. Adenosine methylation as a molecular imprint defining the fate of RNA. FEBS Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Doxtader, K.A.; Nam, Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell 2016, 63, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C.; et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Śledź, P.; Jinek, M. Structural insights into the molecular mechanism of the m(6)A. writer complex. Elife 2016, 5, e18434. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Umetani, M.; Minami, T.; Okayama, H.; Takada, S.; Yamamoto, M.; Aburatani, H.; Reid, P.C.; Housman, D.E.; Hamakubo, T.; et al. Wilms’ tumor 1-associating protein regulates G2/M transition through stabilization of cyclin A2 mRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 17278–17283. [Google Scholar] [CrossRef] [PubMed]
- Fukusumi, Y.; Naruse, C.; Asano, M. Wtap is required for differentiation of endoderm and mesoderm in the mouse embryo. Dev. Dyn. 2008, 237, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Raffel, G.D.; Mercher, T.; Shigematsu, H.; Williams, I.R.; Cullen, D.E.; Akashi, K.; Bernard, O.A.; Gilliland, D.G. Ott1 (Rbm15) has pleiotropic roles in hematopoietic development. Proc. Natl. Acad. Sci. USA 2007, 104, 6001–6006. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 2017, 169, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Warda, A.S.; Kretschmer, J.; Hackert, P.; Lenz, C.; Urlaub, H.; Höbartner, C.; Sloan, K.E.; Bohnsack, M.T. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017, 18, 2004–2014. [Google Scholar] [CrossRef] [PubMed]
- Shima, H.; Matsumoto, M.; Ishigami, Y.; Ebina, M.; Muto, A.; Sato, Y.; Kumagai, S.; Ochiai, K.; Suzuki, T.; Igarashi, K. S-adenosylmethionine synthesis is regulated by selective N(6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell. Rep. 2017, 21, 3354–3363. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Koch, L.; Emmerling, C.; Vierkotten, J.; Peters, T.; Brüning, J.C.; Rüther, U. Inactivation of the Fto gene protects from obesity. Nature 2009, 458, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Pickering, B.F.; Jaffrey, S.R. Reading m(6)A in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. 2018, 28, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Edupuganti, R.R.; Geiger, S.; Lindeboom, R.G.H.; Shi, H.; Hsu, P.J.; Lu, Z.; Wang, S.Y.; Baltissen, M.P.A.; Jansen, P.W.T.C.; Rossa, M.; et al. N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 2017, 24, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Pandya-jones, A.; Saito, Y.; Fak, J.J.; Vagbø, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E., Jr.; Darnell, R.B. m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 2017, 31, 990–1006. [Google Scholar] [CrossRef] [PubMed]
- Louloupi, A.; Ntini, E.; Conrad, T.; Ørom, U.A.V. Transient N-6-methyladenosine transcriptome sequencing reveals a regulatory role of m6A in splicing efficiency. Cell. Rep. 2018, 23, 3429–3437. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N(6)-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Dai, Q.; Zheng, G.; He, C.; Parisien, M.; Pan, T. N(6)-methyladenosine dependent RNA structural switches regulate RNA-protein interactions. Nature 2015, 518, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Fustin, J.M.; Doi, M.; Yamaguchi, Y.; Hida, H.; Nishimura, S.; Yoshida, M.; Isagawa, T.; Morioka, M.S.; Kakeya, H.; Manabe, I.; et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 2013, 155, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA modifications in gene expression regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell. Res. 2017, 27, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Chen, Y.S.; Ping, X.L.; Yang, X.; Xiao, W.; Yang, Y.; Sun, H.Y.; Zhu, Q.; Baidya, P.; Wang, X.; et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017, 27, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, X.; Wang, W.; Shi, H.; Pan, Q.; Lu, Z.; Perez, S.P.; Suganthan, R.; He, C.; Bjørås, M.; et al. Ythdf2-mediated m(6)A mRNA clearance modulates neural development in mice. Genome Biol. 2018, 19, 69. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.L.; Wang, X.; An, R.; Cassin, J.; Vissers, C.; Liu, Y.; Liu, Y.; Xu, T.; Wang, X.; Wong, S.Z.H.; et al. Epitranscriptomic m(6)A regulation of axon regeneration in the adult mammalian nervous system. Neuron 2018, 97, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, M.N.; Pandey, R.R.; Mendel, M.; Homolka, D.; Sachidanandam, R.; Pillai, R.S. Regulation of m6A transcripts by the 3′→5′ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol. Cell 2017, 68, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.J.; Zhu, Y.; Ma, H.; Guo, Y.; Shi, X.; Liu, Y.; Qi, M.; Lu, Z.; Shi, H.; Wang, J.; et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, J.; Rao, H.; Hackert, P.; Sloan, K.E.; Höbartner, C.; Bohnsack, M.T. The m6A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5′-3′ exoribonuclease XRN1. RNA 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wan, J.; Gao, X.; Zhang, X.; Jaffrey, S.R.; Qian, S.B. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 2015, 526, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.B.; Jaffrey, S.R. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Coots, R.A.; Liu, X.M.; Mao, Y.; Dong, L.; Zhou, J.; Wan, J.; Zhang, X.; Qian, S.B. m(6)A facilitates eIF4F-independent mRNA translation. Mol. Cell 2017, 68, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N(6)-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Little, N.A.; Hastie, N.D.; Davies, R.C. Identification of WTAP, a novel Wilms’ tumour 1-associating protein. Hum. Mol. Genet. 2000, 9, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Han, Y.; Suarez Saiz, F.; Minden, M.D. A tumor suppressor and oncogene: The WT1 story. Leukemia 2007, 21, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Bansal, H.; Yihua, Q.; Iyer, S.P.; Ganapathy, S.; Proia, D.A.; Penalva, L.O.; Uren, P.J.; Suresh, U.; Carew, J.S.; Karnad, A.B.; et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 2014, 28, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N(6)-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millán-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Weng, H.; Huang, H.; Wu, H.; Qin, X.; Zhao, B.S.; Dong, L.; Shi, H.; Skibbe, J.; Shen, C.; Hu, C.; et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell 2018, 22, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Sorci, M.; Ianniello, Z.; Cruciani, S.; Larivera, S.; Ginistrelli, L.C.; Capuano, E.; Marchioni, M.; Fazi, F.; Fatica, A. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. 2018, 9, 796. [Google Scholar] [CrossRef] [PubMed]
- Herold, T.; Metzeler, K.H.; Vosberg, S.; Hartmann, L.; Röllig, C.; Stölzel, F.; Schneider, S.; Hubmann, M.; Zellmeier, E.; Ksienzyk, B.; et al. Isolated trisomy 13 defines a homogeneous AML subgroup with high frequency of mutations in spliceosome genes and poor prognosis. Blood 2014, 124, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Musialik, E.; Bujko, M.; Kober, P.; Grygorowicz, M.A.; Libura, M.; Przestrzelska, M.; Juszczyński, P.; Borg, K.; Florek, I.; Jakóbczyk, M.; et al. Comparison of promoter DNA methylation and expression levels of genes encoding CCAAT/enhancer binding proteins in AML patients. Leuk. Res. 2014, 38, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.C.H.; Rasko, J.E.J.; Wong, J.J. We skip to work: Alternative splicing in normal and malignant myelopoiesis. Leukemia 2018, 32, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m(6)A Methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Gruber, T.A.; Downing, J.R. The biology of pediatric acute megakaryoblastic leukemia. Blood 2015, 126, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tran, N.T.; Su, H.; Wang, R.; Lu, Y.; Tang, H.; Aoyagi, S.; Guo, A.; Khodadadi-Jamayran, A.; Zhou, D.; et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. Elife 2015, 4, E07938. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N(6)-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of a-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Dong, L.; Li, C.; Nachtergaele, S.; Wunderlich, M.; Qing, Y.; Deng, X.; Wang, Y.; Weng, X.; Hu, C.; et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 2018, 172, 90–105. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.R., III; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project drive: A compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 2017, 170, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Jaffrey, S.R. FTO, m(6) A.(m), and the hypothesis of reversible epitranscriptomic mRNA modifications. FEBS Lett. 2018, 592, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, Y.; Sun, B.; Wang, L.; Yang, Y.; Ma, D.; Lv, J.; Heng, J.; Ding, Y.; Xue, Y.; et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature 2017, 549, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Zhang, Y.; Gao, S.; Zhang, C.; Chen, Y.; Li, W.; Yang, Y.G.; Zhou, Q.; Liu, F. Endothelial-specific m(6)A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res. 2018, 28, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.J.; Sang, L.; Lin, M.; Yin, X.; Dong, W.; Gong, Y.; Zhou, B.O. Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ma, P.; Liu, Y.; Li, W.; Shu, Y. Multiple functions of m6A RNA methylation in cancer. J. Hematol. Oncol. 2018, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein | Classification | Function |
|---|---|---|
| METTL3 | Writer | Installs m6A in mRNA; promotes translation |
| METTL14 | Writer | Cooperates with METTL3 in m6A installation |
| METTL16 | Writer | Installs m6A in U6 snRNA and pre-mRNA |
| FTO | Eraser | Remove m6A and m6Am from mRNA |
| ALKBH5 | Eraser | Remove m6A from mRNA |
| WTAP | Component of the regulatory MACOM complex | Regulates m6A installation |
| VIRMA | Component of the regulatory MACOM complex | Regulates m6A installation |
| CBLL1 | Component of the regulatory MACOM complex | Regulates m6A installation |
| RBM15 | Component of the regulatory MACOM complex | Regulates m6A installation |
| ZC3H13 | Component of the regulatory MACOM complex | Regulates m6A installation |
| ABCF1 | Direct reader | Stimulates translation |
| eIF3 | Direct reader | Stimulates translation |
| HNRPA2B1 | Direct reader | Stimulates microRNA processing |
| IGF2BPs | Direct readers | Increase mRNA stability |
| YTHDC1 | Direct reader | Stimulates splicing and mRNA export |
| YTHDC2 | Direct reader | Stimulates mRNA decay and translation |
| YTHDF1 | Direct reader | Stimulates translation |
| YTHDF2 | Direct reader | Stimulates mRNA decay |
| YTHDF3 | Direct reader | Stimulates mRNA decay and translation |
| FMR1 | Indirect reader | Inhibits translation |
| HNRNPC | Indirect reader | Regulates splicing |
| ELAVL1 | m6A repelled protein | Increases mRNA stability |
| G3BPs | m6A repelled protein | Increase mRNA stability |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ianniello, Z.; Fatica, A. N6-Methyladenosine Role in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2018, 19, 2345. https://doi.org/10.3390/ijms19082345
Ianniello Z, Fatica A. N6-Methyladenosine Role in Acute Myeloid Leukaemia. International Journal of Molecular Sciences. 2018; 19(8):2345. https://doi.org/10.3390/ijms19082345
Chicago/Turabian StyleIanniello, Zaira, and Alessandro Fatica. 2018. "N6-Methyladenosine Role in Acute Myeloid Leukaemia" International Journal of Molecular Sciences 19, no. 8: 2345. https://doi.org/10.3390/ijms19082345
APA StyleIanniello, Z., & Fatica, A. (2018). N6-Methyladenosine Role in Acute Myeloid Leukaemia. International Journal of Molecular Sciences, 19(8), 2345. https://doi.org/10.3390/ijms19082345