Mesenchymal Stem Cell Migration during Bone Formation and Bone Diseases Therapy

 ,
,
Abstract

1. Introduction
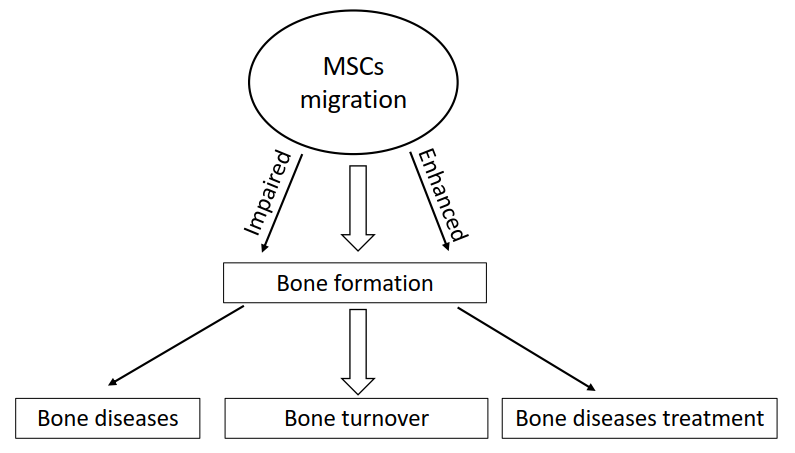
2. The Overview of MSC Migration during Bone Formation
2.1. The Skeleton System is Developed through Two Types of Bone Formation
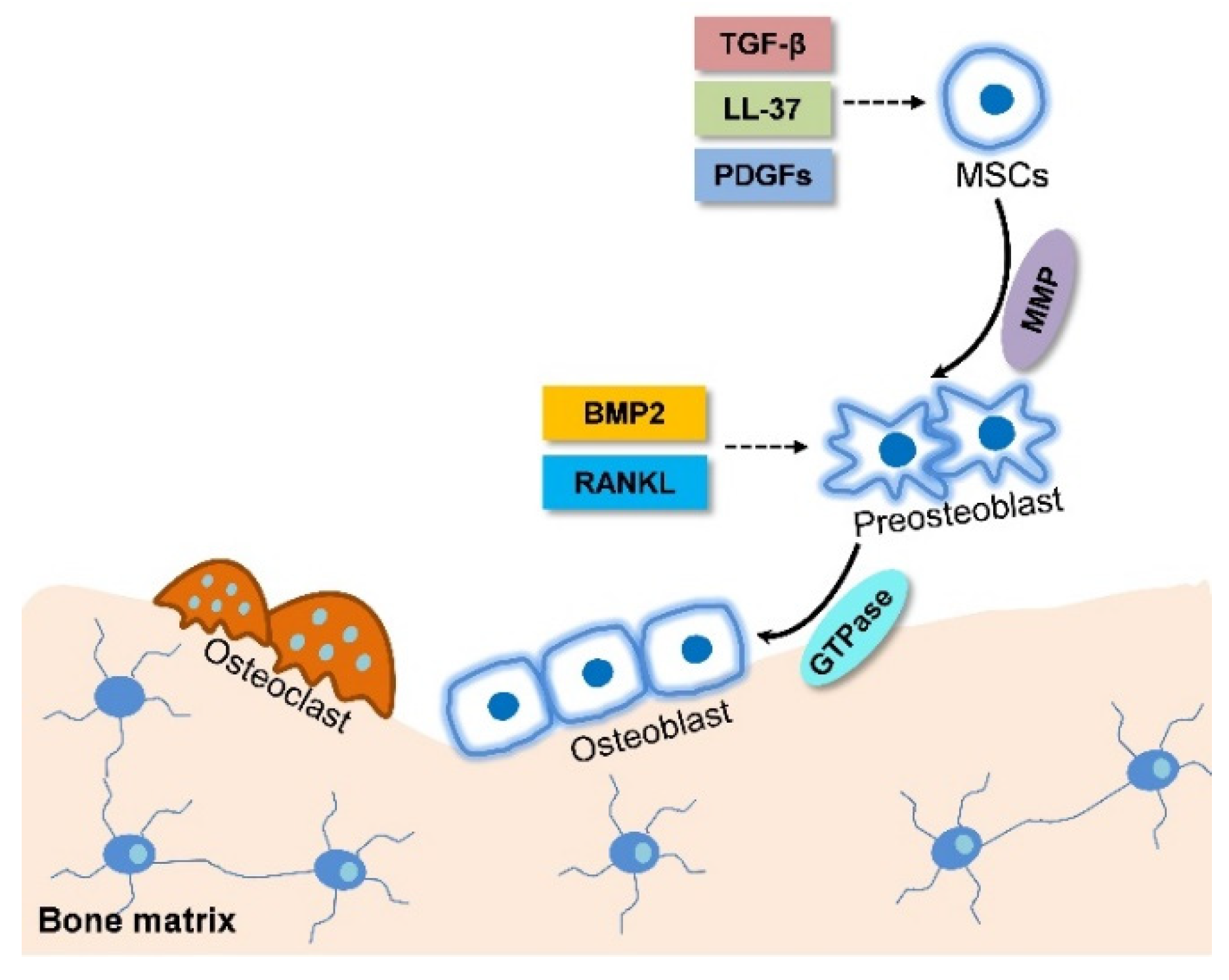
2.2. MSC Migration Initiates Endochondral Ossification
2.3. Intramembranous Ossification is Accompanied with MSC Migration
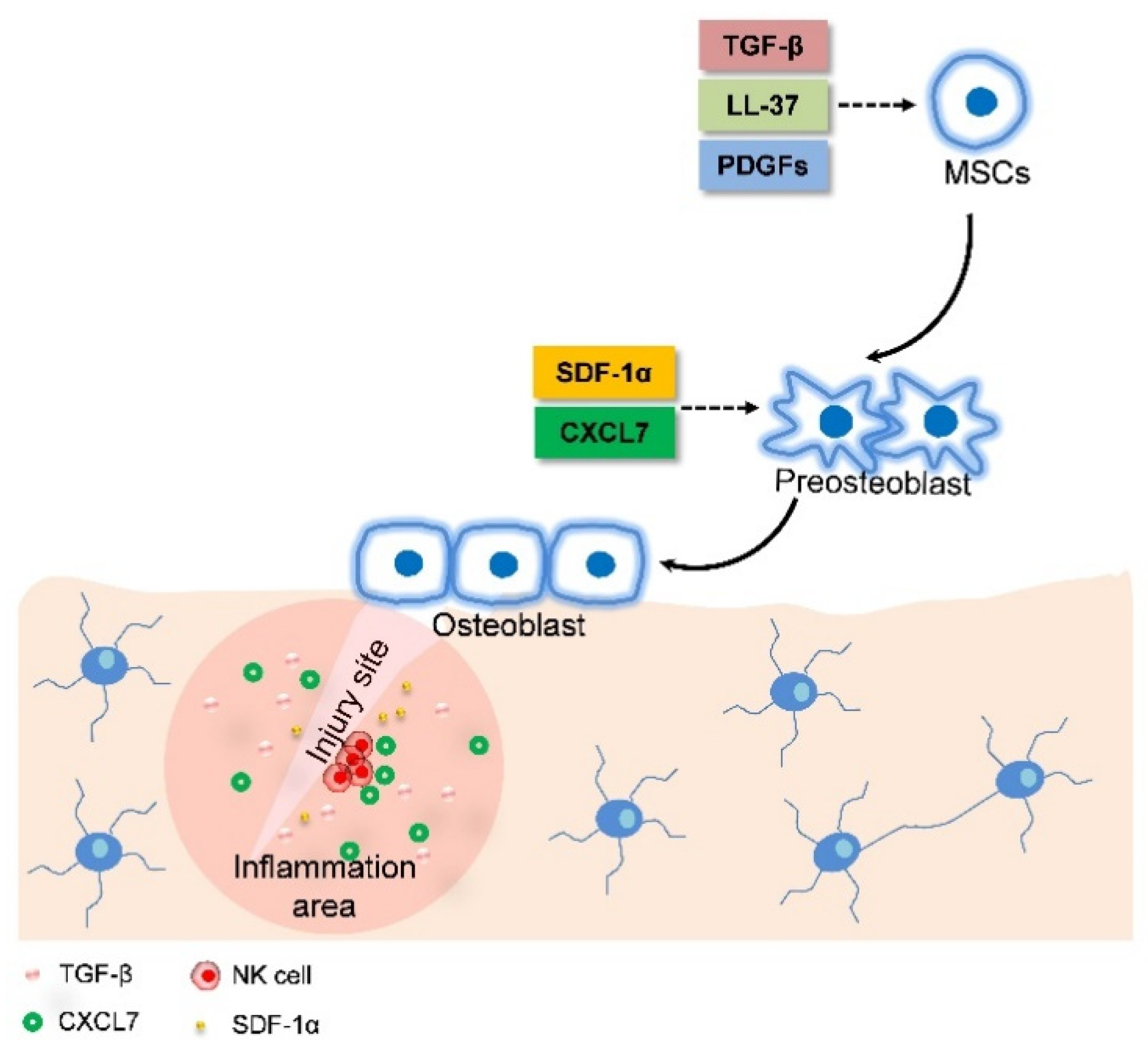
2.4. MSC Migration Induces Fracture Healing through Combining Endochondral Ossification and Intramembranous Ossification
3. The Application of MSC Migration in Bone Disease Therapy
3.1. Treatment of Osteoporosis
3.2. Bone Fracture Healing
3.3. Therapy for Other Bone Diseases
4. Enhancing MSC Migration is a New Strategy for Improving the Efficacy of Bone Disease Treatment
4.1. Pretreatment of MSCs in Culture
4.2. Genetic Modifications of MSCs or Target Tissue
4.3. Cell Surface Engineering
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALP | Alkaline phosphatase |
| ASCs | Adipose-derived stromal/stem cells |
| BMPs | Bone morphogenic proteins |
| C1q | Complement 1q |
| CCR7 | Chemokine receptor 7 |
| CXCL7 | chemokine (C-X-C motif) ligand 7 |
| DFO | Deferoxamine |
| Erk | Extracellular signal–regulated kinase |
| Flt3 | Fms-related tyrosine kinase 3 |
| GFs | Growth factors |
| GTPase | GTP hydrolase |
| HCELL | Haematopoietic cell E-selectin/L-selectin ligand |
| HGF | Hepatocyte growth factor |
| HMGB1 | High mobility group box 1 |
| IA | Intra-arterial |
| IC | Intracoronary |
| IGF | Insulin-like growth factor |
| IGFBP5 | Insulin-like growth factor binding protein 5 |
| IV | Intravenous |
| LRG1 | Leucine-rich-alpha-2-glycoprotein1 |
| MMP | Matrix metalloproteinases |
| NK | Natural killer |
| OA | Osteoarthritis |
| PDGFs | platelet-derived growth factors |
| PDLSCs | Primary period ligament stem cells |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| PTH | Parathyroid hormone |
| RAGE | Receptor for advanced glycation end |
| SATB 2 | Special AT-rich sequence binding protein 2 |
| SCF | Stem cell factor |
| SDF-1α | Stromal cell-derived factor-1 alpha; |
| SLEX | Sialyl Lewis X |
| Sox9 | Sry-related high-mobility group box 9 |
| TGF-β | Transforming growth factor-β |
| TNFα | Tumor necrosis factor alpha |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
References
- Schaff, F.; Bech, M.; Zaslansky, P.; Jud, C.; Liebi, M.; Guizar-Sicairos, M.; Pfeiffer, F. Six-dimensional real and reciprocal space small-angle X-ray scattering tomography. Nature 2015, 527, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Kaibara, K.; Ishimoto, T.; Tabata, Y.; Umakoshi, Y. Biological apatite (BAp) crystallographic orientation and texture as a new index for assessing the microstructure and function of bone regenerated by tissue engineering. Bone 2012, 51, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Gemini-Piperni, S.; Takamori, E.R.; Sartoretto, S.C.; Paiva, K.B.S.; Granjeiro, J.M.; de Oliveira, R.C.; Zambuzzi, W.F. Cellular behavior as a dynamic field for exploring bone bioengineering: A closer look at cell-biomaterial interface. Arch. Biochem. Biophys. 2014, 561, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, R.; Matsugaki, A.; Isobe, Y.; Saku, T.; Yun, H.S.; Nakano, T. Construction of human induced pluripotent stem cell-derived oriented bone matrix microstructure by using in vitro engineered anisotropic culture model. J. Biomed. Mater. Res. Part A 2018, 106, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Luu, H.H.; Song, W.X.; Luo, X.; Manning, D.; Luo, J.; Deng, Z.L.; Sharff, K.A.; Montag, A.G.; Haydon, R.C.; He, T.C. Distinct roles of bone morphogenetic proteins in osteogenic differentiation of mesenchymal stem cells. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2007, 25, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Tan, Y.Y.; Wong, R.; Wenden, A.; Zhang, L.K.; Rabie, A.B.M. The role of vascular endothelial growth factor in ossification. Int. J. Oral Sci. 2012, 4, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.M.; Lee, E.H. Transcriptional Regulatory Cascades in Runx2-Dependent Bone Development. Tissue Eng. Part B Rev. 2013, 19, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Scadden, D.T. The stem-cell niche as an entity of action. Nature 2006, 441, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lozano, F.J.; Bueno, C.; Insausti, C.L.; Meseguer, L.; Ramirez, M.C.; Blanquer, M.; Marín, N.; Martínez, S.; Moraleda, J.M. Mesenchymal stem cells derived from dental tissues. Int. Endod. J. 2011, 44, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Gou, W.L.; Lu, Q.; Peng, J.; Lu, S.B. Role of mesenchymal stem cells in bone regeneration and fracture repair: A review. Int. Orthop. 2013, 37, 2491–2498. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.W.; Dominguez-Steglich, M.A.; Guioli, S.; Kwok, C.; Weller, P.A.; Stevanovic, M.; Weissenbach, J.; Mansour, S.; Young, I.D.; Goodfellow, P.N.; et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY-related gene. Nature 1994, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2011, 13, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. CSH Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.A.; Han, J.; Kim, B.S. Stimulation of chondrogenic differentiation of mesenchymal stem cells. Int. J. Stem Cells 2012, 5, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.M.; Zhai, D.Y.; Tous, E.; Rai, R.; Mauck, R.L.; Burdick, J.A. Enhanced MSC chondrogenesis following delivery of TGF-beta 3 from alginate microspheres within hyaluronic acid hydrogels in vitro and in vivo. Biomaterials 2011, 32, 6425–6434. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, Y.; Rodriguez-Leon, J.; Izpisua Belmonte, J.C. The role of TGFβs and Sox9 during limb chondrogenesis. Curr. Opin. Cell Biol. 2006, 18, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Muneta, T.; Nagase, T.; Nimura, A.; Ju, Y.J.; Mochizuki, T.; Sekiya, I. Comparison of mesenchymal tissues-derived stem cells for in vivo chondrogenesis: Suitable conditions for cell therapy of cartilage defects in rabbit. Cell Tissue Res. 2008, 333, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Maes, C.; Kobayashi, T.; Selig, M.K.; Torrekens, S.; Roth, S.I.; Mackem, S.; Carmeliet, G.; Kronenberg, H.M. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev. Cell 2010, 19, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Fakhry, M.; Hamade, E.; Badran, B.; Buchet, R.; Magne, D. Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J. Stem Cells 2013, 5, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Stadler, H.S.; Higgins, K.M.; Capecchi, M.R. Loss of Eph-receptor expression correlates with loss of cell adhesion and chondrogenic capacity in Hoxa13 mutant limbs. Development 2001, 128, 4177–4188. [Google Scholar] [PubMed]
- Ng, J.; Wei, Y.; Zhou, B.; Burapachaisri, A.; Guo, E.; Vunjak-Novakovic, G. Extracellular matrix components and culture regimen selectively regulate cartilage formation by self-assembling human mesenchymal stem cells in vitro and in vivo. Stem Cell Res. Ther. 2016, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Choi, H.; Seon, M.; Cho, D.; Bang, S.I. LL-37 stimulates the functions of adipose-derived stromal/stem cells via early growth response 1 and the MAPK pathway. Stem Cell Res. Ther. 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Cong, Q.; Yeh, J.; Xia, X.C.; Mishina, Y.; Hao, A.J.; Li, B.J. PDGF-AA promotes osteogenic differentiation and migration of mesenchymal stem cell by down-regulating PDGFRa and derepressing BMP-Smad1 signaling. PLoS ONE 2014, 9, e113785. [Google Scholar]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-β1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Faveeuw, C.; Preece, G.; Ager, A. Transendothelial migration of lymphocytes across high endothelial venules into lymph nodes is affected by metalloproteinases. Blood 2001, 98, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.P.; Lam, A.T.L.; Newman, J.P.; Chua, K.L.M.; Kok, C.Y.L.; Chong, S.T.; Chua, M.L.K.; Lam, P.Y.P. Matrix metalloproteinase-1 facilitates MSC migration via cleavage of IGF-2/IGFBP2 complex. FEBS Open Bio 2018, 8, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Ichida, M.; Yui, Y.; Yoshioka, K.; Tanaka, T.; Wakamatsu, T.; Yoshikawa, H.; Itoh, K. Changes in cell migration of mesenchymal cells during osteogenic differentiation. FEBS Lett. 2011, 585, 4018–4024. [Google Scholar] [CrossRef] [PubMed]
- Razidlo, G.L.; Wang, Y.; Chen, J.; Krueger, E.W.; Billadeau, D.D.; McNiven, M.A. Dynamin 2 Potentiates Invasive Migration of Pancreatic Tumor Cells through Stabilization of the Rac1 GEF Vav1. Dev. Cell 2013, 24, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.J.; Lampe, M.; Merrifield, C.J. A Feedback Loop between Dynamin and Actin Recruitment during Clathrin-Mediated Endocytosis. PLoS Biol. 2012, 10, e1001302. [Google Scholar] [CrossRef] [PubMed]
- Eleniste, P.P.; Huang, S.; Wayakanon, K.; Largura, H.W.; Bruzzaniti, A. Osteoblast differentiation and migration are regulated by Dynamin GTPase activity. Int. J. Biochem. Cell Biol. 2014, 46, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Golden, D.; Saria, E.A.; Hansen, M.F. Regulation of Osteoblast Migration Involving Receptor Activator of Nuclear Factor-kappa B (RANK) Signaling. J. Cell. Physiol. 2015, 230, 2951–2960. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.; Eriksen, E.F.; Bünger, C. Bone morphogenetic protein-2 but not bone morphogenetic protein-4 and -6 stimulates chemotactic migration of human osteoblasts, human marrow osteoblasts, and U2-OS cells. Bone 1996, 18, 53–57. [Google Scholar] [CrossRef]
- Einhorn, T.A. The cell and molecular biology of fracture healing. Clin. Orthop. Relat. Res. 1998, 355, S7–S21. [Google Scholar] [CrossRef]
- Peng, H.R.; Usas, A.; Olshanski, A.; Ho, A.M.; Gearhart, B.; Cooper, G.M.; Huard, J. VEGF improves, whereas sFlt1 inhibits, BMP2-induced bone formation and bone healing through modulation of angiogenesis. J. Bone Miner. Res. 2005, 20, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.B.; Giannoudis, P.V.; Boxall, S.A.; McGonagle, D.; Jones, E. The systemic influence of platelet-derived growth factors on bone marrow mesenchymal stem cells in fracture patients. BMC Med. 2015, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.W.; Zhao, X.; Zhang, L.Y.; Zhang, J.M.; L Huillier, A.; Ling, W.F.; Roberts, A.I.; Le, A.D.; Shi, S.; Shao, C.; et al. Inflammatory Cytokine-Induced Intercellular Adhesion Molecule-1 and Vascular Cell Adhesion Molecule-1 in Mesenchymal Stem Cells Are Critical for Immunosuppression. J. Immunol. 2010, 184, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.R.; Vasconcelos, D.P.; Goncalves, R.M.; Barbosa, M.A. Enhanced mesenchymal stromal cell recruitment via natural killer cells by incorporation of inflammatory signals in biomaterials. J. R. Soc. Interface 2012, 9, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, J.; Zhang, X.; Wang, C.; Huang, Y.; Dai, K.; Zhang, X. TNF-α-induced LRG1 promotes angiogenesis and mesenchymal stem cell migration in the subchondral bone during osteoarthritis. Cell Death Dis. 2017, 8, e2715. [Google Scholar] [CrossRef] [PubMed]
- Kitaori, T.; Ito, H.; Schwarz, E.A.; Tsutsumi, R.; Yoshitomi, H.; Oishi, S.; Nakano, M.; Fujii, N.; Nagasawa, T.; Nakamura, T. Stromal Cell-Derived Factor 1/CXCR4 Signaling Is Critical for the Recruitment of Mesenchymal Stem Cells to the Fracture Site During Skeletal Repair in a Mouse Model. Arthritis Rheumatol. 2009, 60, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, Y.; Sun, X.Y.; Zhou, J.; Yang, P.S. CXCL12 overexpression promotes the angiogenesis potential of periodontal ligament stem cells. Sci. Rep.-UK 2017, 7, 10286. [Google Scholar] [CrossRef] [PubMed]
- Richter, R.; Forssmann, W.; Henschler, R. Current Developments in Mobilization of Hematopoietic Stem and Progenitor Cells and Their Interaction with Niches in Bone Marrow. Transfus. Med. Hemother. 2017, 44, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Haasters, F.; Docheva, D.; Gassner, C.; Popov, C.; Bocker, W.; Mutschler, W.; Schieker, M.; Prall, W.C. Mesenchymal stem cells from osteoporotic patients reveal reduced migration and invasion upon stimulation with BMP-2 or BMP-7. Biochem. Biophys. Res. Commun. 2014, 452, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Sanghani-Kerai, A.; Coathup, M.; Samazideh, S.; Kalia, P.; Di Silvio, L.; Idowu, B.; Blunn, G. Osteoporosis and ageing affects the migration of stem cells and this is ameliorated by transfection with CXCR4. Bone Jt. Res. 2017, 6, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, V.S.; Efstathopoulos, N.; Kontakis, G.; Kanakaris, N.K.; Giannoudis, P.V. The influence of osteoporosis in femoral fracture healing time. Injury 2009, 40, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Naderi-Meshkin, H.; Bahrami, A.R.; Bidkhori, H.R.; Mirahmadi, M.; Ahmadiankia, N. Strategies to improve homing of mesenchymal stem cells for greater efficacy in stem cell therapy. Cell Biol. Int. 2015, 39, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Lien, C.Y.; Chih-Yuan Ho, K.; Lee, O.K.; Blunn, G.W.; Su, Y. Restoration of bone mass and strength in glucocorticoid-treated mice by systemic transplantation of CXCR4 and cbfa-1 co-expressing mesenchymal stem cells. J. Bone Miner. Res. 2009, 24, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.M.; Prockop, D.J.; Fitzpatrick, L.A.; Koo, W.W.; Gordon, P.L.; Neel, M.; Sussman, M.; Orchard, P.; Marx, J.C.; Pyeritz, R.E.; et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat. Med. 1999, 5, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Yao, W.; Liu, R.; Lam, K.S.; Nolta, J.; Jia, J.; Panganiban, B.; Meng, L.; Zhou, P.; Shahnazari, M.; et al. Directing mesenchymal stem cells to bone to augment bone formation and increase bone mass. Nat. Med. 2012, 18, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Ito, H. Chemokines in mesenchymal stem cell therapy for bone repair: A novel concept of recruiting mesenchymal stem cells and the possible cell sources. Mod. Rheumatol. 2011, 21, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Obermeyer, T.S.; Yonick, D.; Lauing, K.; Stock, S.R.; Nauer, R.; Strotman, P.; Shankar, R.; Gamelli, R.; Stover, M.; Callaci, J.J. Mesenchymal stem cells facilitate fracture repair in an alcohol-induced impaired healing model. J. Orthop. Trauma 2012, 26, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Yang, L.; Zhou, Y.; Xin, Y.; Li, Q. Application of enhanced green fluorescent protein labeling technology to monitoring marrow mesenchymal stem cells migration after bone fracture. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2006, 20, 102–106. [Google Scholar] [PubMed]
- Zwingenberger, S.; Yao, Z.; Jacobi, A.; Vater, C.; Valladares, R.D.; Li, C.; Nich, C.; Rao, A.J.; Christman, J.E.; Antonios, J.K. Enhancement of BMP-2 induced bone regeneration by SDF-1α mediated stem cell recruitment. Tissue Eng. Part A 2014, 20, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Fang, T.; Qi, Y.; Yin, X.; Di, T.; Feng, G.; Lei, Z.; Zhang, Y.; Huang, Z. Combined Use of Mesenchymal Stromal Cell Sheet Transplantation and Local Injection of SDF-1 for Bone Repair in a Rat Nonunion Model. Cell Transplant. 2016, 25, 1801–1817. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Huang, S.; Hou, Y.; Liu, Y.; Ni, M.; Meng, F.; Wang, K.; Rui, Y.; Jiang, X.; Li, G. Sox11-modified mesenchymal stem cells (MSCs) accelerate bone fracture healing: Sox11 regulates differentiation and migration of MSCs. FASEB J. 2015, 29, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Lee, S.H.; Moon, S.J.; Lee, J.A.; Lee, E.J.; Kim, E.K.; Park, J.S.; Lee, J.; Min, J.K.; Kim, S.J.; et al. Overexpression of soluble RAGE in mesenchymal stem cells enhances their immunoregulatory potential for cellular therapy in autoimmune arthritis. Sci. Rep. 2016, 6, 35933. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Zhang, F.; Li, G.; Zhang, X.; Lin, X.; Yang, H.; Wang, L.; Cao, Y.; Du, J.; Fan, Z. Local application of IGFBP5 protein enhanced periodontal tissue regeneration via increasing the migration, cell proliferation and osteo/dentinogenic differentiation of mesenchymal stem cells in an inflammatory niche. Stem Cell Res. Ther. 2017, 8, 210. [Google Scholar] [CrossRef] [PubMed]
- Sheyn, D.; Shapiro, G.; Tawackoli, W.; Jun, D.S.; Koh, Y.; Kang, K.B.; Su, S.; Da, X.; Ben-David, S.; Bez, M.; et al. PTH Induces Systemically Administered Mesenchymal Stem Cells to Migrate to and Regenerate Spine Injuries. Mol. Ther. 2016, 24, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, I.; Ojima, M.; Suzuki, S.; Yamaga, M.; Horie, M.; Koga, H.; Tsuji, K.; Miyaguchi, K.; Ogishima, S.; Tanaka, H.; et al. Human mesenchymal stem cells in synovial fluid increase in the knee with degenerated cartilage and osteoarthritis. J. Orthop. Res. 2012, 30, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Okubo, Y.; Nakao, K.; Osawa, K.; Fujimura, K.; Bessho, K. Pluripotency of mesenchymal cells derived from synovial fluid in patients with temporomandibular joint disorder. Life Sci. 2011, 89, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef] [PubMed]
- De Becker, A.; Riet, I.V. Homing and migration of mesenchymal stromal cells: How to improve the efficacy of cell therapy? World J. Stem Cells 2016, 8, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, R.C. The role of chemokines in mesenchymal stem cell homing to myocardium. Stem Cell Rev. 2012, 8, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Moll, N.M.; Ransohoff, R.M. CXCL12 and CXCR4 in bone marrow physiology. Expert Rev. Hematol. 2010, 3, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Honczarenko, M.; Le, Y.; Swierkowski, M.; Ghiran, I.; Glodek, A.M.; Silberstein, L.E. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells 2006, 24, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Li, J.; Liao, L.; Chen, B.; Li, B.; Chen, L.; Jia, H.; Zhao, R.C. Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: Role in homing efficiency in NOD/SCID mice. Haematologica 2007, 92, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Priebe, W.; Glod, J.; Banerjee, D. Activation of signal transducers and activators of transcription 3 and focal adhesion kinase by stromal cell-derived factor 1 is required for migration of human mesenchymal stem cells in response to tumor cell-conditioned medium. Stem Cells 2009, 27, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Noh, M.Y.; Kim, J.Y.; Yu, H.J.; Kim, K.S.; Kim, S.H.; Koh, S.H. Direct GSK-3β inhibition enhances mesenchymal stromal cell migration by increasing expression of beta-PIX and CXCR4. Mol. Neurobiol. 2013, 47, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Lee, Y.T.; Turcotte, S.; Naud, E.; Desrosiers, R.R.; Champagne, M.; Eliopoulos, N.; Galipeau, J.; Béliveau, R. Hypoxia promotes murine bone-marrow-derived stromal cell migration and tube formation. Stem Cells 2003, 21, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xue, W.; Ge, G.; Luo, X.; Li, Y.; Xiang, H.; Ding, X.; Tian, P.; Tian, X. Hypoxic preconditioning advances CXCR4 and CXCR7 expression by activating HIF-1α in MSCs. Biochem. Biophys. Res. Commun. 2010, 401, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.K.; Wang, Z.; Munasinghe, J.; Leng, Y.; Leeds, P.; Chuang, D.M. Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke model. Stroke 2011, 42, 2932–2939. [Google Scholar] [CrossRef] [PubMed]
- Najafi, R.; Sharifi, A.M. Deferoxamine preconditioning potentiates mesenchymal stem cell homing in vitro and in streptozotocin-diabetic rats. Expert Opin. Biol. Ther. 2013, 13, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Knowles, H.J.; Tian, Y.M.; Mole, D.R.; Harris, A.L. Novel mechanism of action for hydralazine: Induction of hypoxia-inducible factor-1α, vascular endothelial growth factor, and angiogenesis by inhibition of prolyl hydroxylases. Circ. Res. 2004, 95, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M.R.; Bianchi, C.; Sellke, F.W. Hypoxia inducible factor-1α, endothelial progenitor cells, monocytes, cardiovascular risk, wound healing, cobalt and hydralazine: A unifying hypothesis. Curr. Drug Targets 2008, 9, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lu, C.; Liu, H.; Rao, S.; Cai, J.; Liu, S.; Kriegel, A.J.; Greene, A.S.; Liang, M.; Ding, X. Hypoxic preconditioning with cobalt of bone marrow mesenchymal stem cells improves cell migration and enhances therapy for treatment of ischemic acute kidney injury. PLoS ONE 2013, 8, e62703. [Google Scholar] [CrossRef] [PubMed]
- Bobis-Wozowicz, S.; Miekus, K.; Wybieralska, E.; Jarocha, D.; Zawisz, A.; Madeja, Z.; Majka, M. Genetically modified adipose tissue-derived mesenchymal stem cells overexpressing CXCR4 display increased motility, invasiveness, and homing to bone marrow of NOD/SCID mice. Exp. Hematol. 2011, 39, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ponnazhagan, S. Bone homing of mesenchymal stem cells by ectopic α4 integrin expression. FASEB J. 2007, 21, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Wiehe, J.M.; Kaya, Z.; Homann, J.M.; Wohrle, J.; Vogt, K.; Nguyen, T.; Rottbauer, W.; Torzewski, J.; Fekete, N.; Rojewski, M.; et al. GMP-adapted overexpression of CXCR4 in human mesenchymal stem cells for cardiac repair. Int. J. Cardiol. 2013, 167, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Dai, S.; Wu, W.J.; Tan, W.; Zhu, X.; Mu, J.; Guo, Y.; Bolli, R.; Rokosh, G. Stromal cell derived factor-1α confers protection against myocardial ischemia/reperfusion injury: Role of the cardiac stromal cell derived factor-1α CXCR4 axis. Circulation 2007, 116, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Penn, M.S.; Mendelsohn, F.O.; Schaer, G.L.; Sherman, W.; Farr, M.; Pastore, J.; Rouy, D.; Clemens, R.; Aras, R.; Losordo, D.W. An open-label dose escalation study to evaluate the safety of administration of nonviral stromal cell-derived factor-1 plasmid to treat symptomatic ischemic heart failure. Circ. Res. 2013, 112, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Li, S.H.; Wu, J.; Miyagi, Y.; Yau, T.M.; Rakowski, H.; Egashira, K.; Guo, J.; Weisel, R.D.; Li, R.K. Repeated and targeted transfer of angiogenic plasmids into the infarcted rat heart via ultrasound targeted microbubble destruction enhances cardiac repair. Eur. Heart J. 2011, 32, 2075–2084. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.D.; Huang, Y.; Liu, D.; Hickey, R.; Krause, D.S.; Giordano, F.J. Stromal cell-derived factor-1α plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation 2004, 110, 3300–3305. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.T.; Unzek, S.; Popovic, Z.B.; Goldman, C.K.; Forudi, F.; Kiedrowski, M.; Rovner, A.; Ellis, S.G.; Thomas, J.D.; DiCorleto, P.E.; et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet 2003, 362, 697–703. [Google Scholar] [CrossRef]
- Zhang, G.; Nakamura, Y.; Wang, X.; Hu, Q.; Suggs, L.J.; Zhang, J. Controlled release of stromal cell-derived factor-1α in situ increases c-kit+ cell homing to the infarcted heart. Tissue Eng. 2007, 13, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Liu, H.; Zhang, X.; Xu, F.; Qin, L.; Cheng, P.; Huang, H.; Guo, F.; Yang, Q.; Chen, A. Inhibition of SDF-1α /CXCR4 Signalling in Subchondral Bone Attenuates Post-Traumatic Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 943. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, D.; Millard, R.W.; Ashraf, M.; Wang, Y. Stem cell homing and angiomyogenesis in transplanted hearts are enhanced by combined intramyocardial SDF-1α delivery and endogenous cytokine signaling. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H976–H986. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, B.; Poppe, A.; Golsong, P.; Blanke, P.; Rylski, B.; Beyersdorf, F.; Schlensak, C.; Siepe, M. Functional regeneration of ischemic myocardium by transplanted cells overexpressing stromal cell-derived factor-1 (SDF-1): Intramyocardial injection versus scaffold-based application. Eur. J. Cardiothorac. Surg. 2011, 40, e135–e141. [Google Scholar] [CrossRef] [PubMed]
- Ko, I.K.; Kean, T.J.; Dennis, J.E. Targeting mesenchymal stem cells to activated endothelial cells. Biomaterials 2009, 30, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R.; Merzaban, J.S.; Cain, D.W.; Dagia, N.M.; Spencer, J.A.; Lin, C.P.; Wohlgemuth, R. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 2008, 14, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Ko, I.K.; Kim, B.G.; Awadallah, A.; Mikulan, J.; Lin, P.; Letterio, J.J.; Dennis, J.E. Targeting improves MSC treatment of inflammatory bowel disease. Mol. Ther. 2010, 18, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Zhang, W.; Xue, D.; Zhu, T.; Li, J.; Chen, E.; Yao, X.; Pan, Z. Signaling pathways involved in the effects of HMGB1 on mesenchymal stem cell migration and osteoblastic differentiation. Int. J. Mol. Med. 2016, 37, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, J.E. Dynamin and its role in membrane fission. Annu. Rev. Cell Dev. Biol. 2000, 16, 483–519. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, G.C.; Slepnev, V.I.; Neff, L.; Ringstad, N.; Takei, K.; Daniell, L.; Kim, W.; Cao, H.; McNiven, M.; Baron, R.; et al. A functional link between dynamin and the actin cytoskeleton at podosomes. J. Cell Biol. 2000, 150, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Shiga, T.; Hashimoto, J.; Yoshioka, M.; Honjo, H.; Urabe, M.; Kitajima, I.; Semba, I.; Hirasawa, Y. Osteoporosis influences the late period of fracture healing in a rat model prepared by ovariectomy and low calcium diet. J. Steroid Biochem. 1999, 68, 197–202. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, N.; Prestwich, G.D.; Wen, X. Recruitment of endogenous stem cells for tissue repair. Macromol. Biosci. 2008, 8, 836–842. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Factor | Ossification Pattern | Pathway | Efficacy | In Vitro or In Vivo | Reference |
|---|---|---|---|---|---|
| BMPs | Endochondral | Activate SMADs receptor to | MSC condensation  | In vitro | [21] |
| transduce signals | Recruitment of osteoblast | ||||
| Hoxa13 and | Endochondral | Promote cell-cell adhesion | MSC condensation | In vitro | [22] |
| Hoxd13 | Bone formation | ||||
| ECM | Endochondral | Self-assembling of MSCs | MSC condensation | In vitro | [23] |
| LL-37 | Intramembranous | MAPK pathway | Expression of EGR1 | In vitro and in vivo | [24] |
| Activation of MAPKs | |||||
| PDGFs | Intramembranous | BMP-Smad1/5/8-Twist1/Atf4 axis | Cell migration | In vitro | [25] |
| TGF-β1 | Intramembranous | SMAD signaling | Cell migration | In vitro and in vivo | [26] |
| MMP | Intramembranous | Penetrate blood vessel basement membranes | Induce MSC migration | In vitro and in vivo | [27,28] |
| NF-κB ligand | Intramembranous | Activating phosphorylation of ERK, MAPK, Akt, and NF-κB | Cell migration | In vitro | [33] |
| TNFα | Fracture healing | Induce LRG1 secretion | MSC recruitment | In vitro and in vivo | [40] |
| CXCL7 | Fracture healing | Binding CXCR2 | MSC recruitment | In vitro | [36] |
| SDF-1 | Fracture healing | Binding CXCR4 | Osteoblast migration | In vitro and in vivo | [41] |
| MSC recruitment |
: Up-regulation.| MSCs | Administration | Diseases | Efficacy | Reference |
|---|---|---|---|---|
| C3H10T1/2 | Intravenous injection | Osteoporosis | Restoration bone formation and bone mass | [48] |
| BM-MSCs | Bone marrow transplantation | Osteogenesis imperfecta | Bone mineralization density | [49] |
| Endogenous MSCs | Intravenous injection of LLP2A | Impairment of osteogenesis | Trabecular bone formation bone mass | [50] |
| Primary BM-MSCs | Intravenous transplant | Tibia fracture | Restoration both fracture callus volume and biomechanical strength | [52,53] |
| Endogenous MSCs | Implantation of fat tissue grafts expressing SDF-1α or/and BMP-2 | Bone defect | Bone formation | [49] |
| Primary MSCs sheet | Local injection | Nonunion | Bone fracture healing Inflammatory arthritis  | [55] |
| Primary GFP-MSCs | Tail vein injection | Femur fracture | Local inflammation periodontal tissue regeneration | [56] |
| sRAGE-ASCs | Tail vein injection | OA | Local inflammation | [57] |
| PDLSCs | Local injection of IGFBP5 | Periodontitis | MSC migration | [58] |
| HMSCs | Injection via ear vein | Vertebral defect | New bone formation | [59] |
: Up-regulation; : Down-regulation.| Strategy | Advantages | Main Drawbacks | Example | Reference |
|---|---|---|---|---|
| Treatment with cytokine or cytokine cocktail | Simple and fast | Unwanted genes may be expressed, expensive | Pretreat MSCs with flt3 ligand, stem cell factor (SCF) or hepatocyte growth factor (HGF). Pretreat with GSK3β inhibitor | [67,68] |
| Hypoxic condition | Simple and fast | Cells probably migrate into non-targeted organs | [72,73] | |
| Treatment with compound | Intravenous injection of LLP2A | Unwanted genes may be expressed, expensive | Treatment MSCs Valproate or lithium | [74] |
| Genetic modification of MSCs | More directed | Difficult, expensive and risk of tumorigenicity | Overexpression of CXCR4 and integrin β4 | [77,78] |
| Genetic modification of injury tissue | Targeted | Immunogenicity, Retroviral-mediated insertional mutagenesis | Transfection of SDF-1 plasmid to injury tissue | [84] |
| Injection of ectopic chemokine expressing cells | High efficiency | Safety problems, difficult and expensive | Injection of SDF-1α overexpression MSCs into tissue | [85] |
| Introduce certain protein expression | No damage for cell viability and function | Safety problems, Difficult and expensive, Risk of tumorigenicity, | Express SLEX on MSCs membrane | [89] |
| Coating of cell surface with antibodies | More targeted | Difficult and expensive | Bind VCAM-1 antibodies to MSCs bone surface | [90] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, P.; Tian, Y.; Yang, C.; Ma, X.; Wang, X.; Pei, J.; Qian, A. Mesenchymal Stem Cell Migration during Bone Formation and Bone Diseases Therapy. Int. J. Mol. Sci. 2018, 19, 2343. https://doi.org/10.3390/ijms19082343
Su P, Tian Y, Yang C, Ma X, Wang X, Pei J, Qian A. Mesenchymal Stem Cell Migration during Bone Formation and Bone Diseases Therapy. International Journal of Molecular Sciences. 2018; 19(8):2343. https://doi.org/10.3390/ijms19082343
Chicago/Turabian StyleSu, Peihong, Ye Tian, Chaofei Yang, Xiaoli Ma, Xue Wang, Jiawei Pei, and Airong Qian. 2018. "Mesenchymal Stem Cell Migration during Bone Formation and Bone Diseases Therapy" International Journal of Molecular Sciences 19, no. 8: 2343. https://doi.org/10.3390/ijms19082343
APA StyleSu, P., Tian, Y., Yang, C., Ma, X., Wang, X., Pei, J., & Qian, A. (2018). Mesenchymal Stem Cell Migration during Bone Formation and Bone Diseases Therapy. International Journal of Molecular Sciences, 19(8), 2343. https://doi.org/10.3390/ijms19082343