Interleukin-1 Beta—A Friend or Foe in Malignancies?


Abstract
1. Introduction
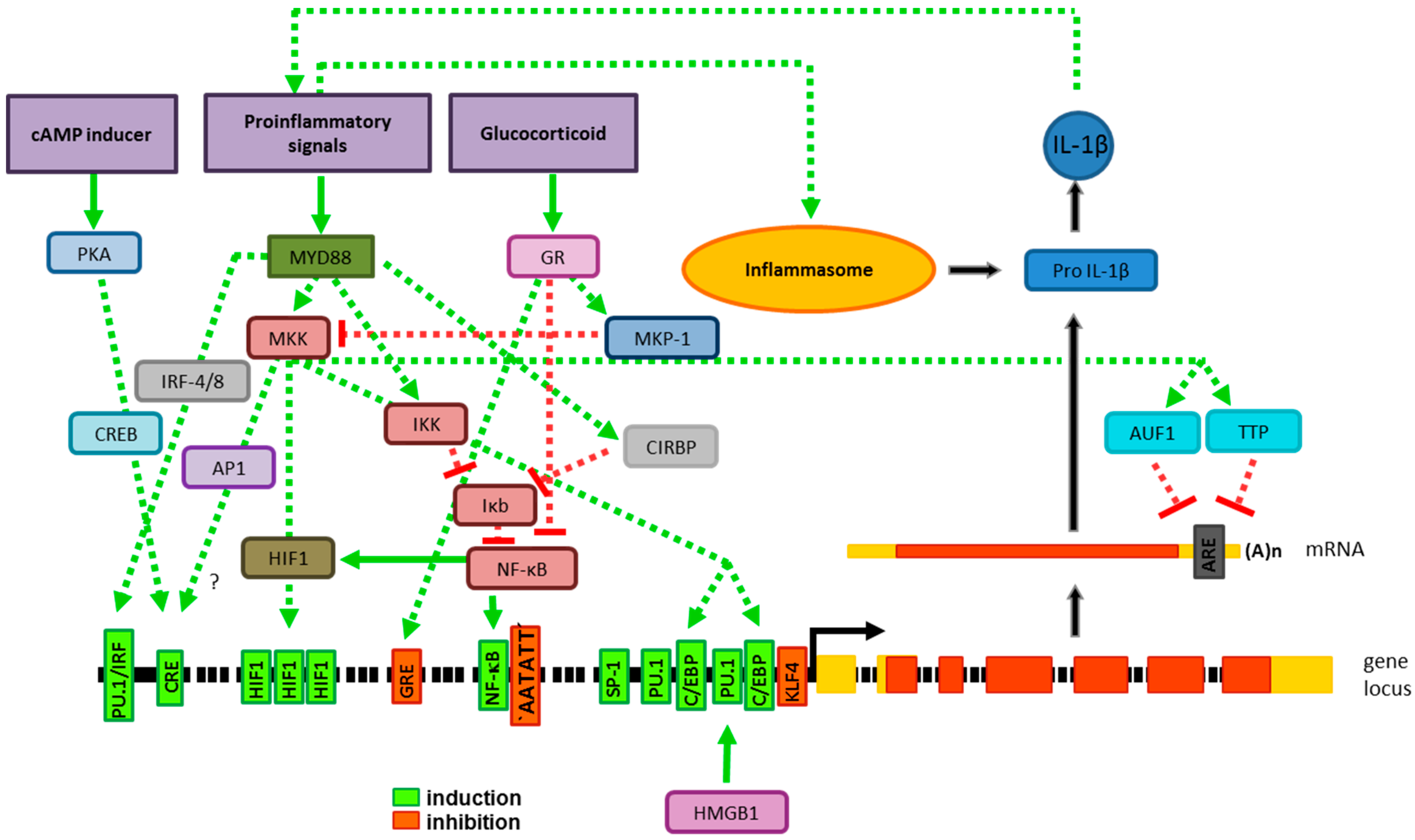
2. Production of IL-1β Requires Two Distinct Signals
2.1. Gene Expression
2.1.1. Transcriptional Gene Regulation
2.1.2. Posttranscriptional Gene Regulation
2.2. Processing of Pro-IL-1β
2.2.1. Caspases
2.2.2. Other Proteases
3. IL-1β Signaling
3.1. The IL-1 Receptor
3.2. IL-1β Signaling Cascade
3.3. IL-1Ra and IL-1R2
4. Role of IL-1 in Inflammation
5. Role of IL-1β in Tumor Development
5.1. Anti-Tumorigenic Effects of IL-1β
5.2. Tumor-Promoting Effects of IL-1β
5.2.1. Tumor Development Caused by Chronic Inflammation
5.2.2. Tumor Microenvironment
Immunosuppression
TAM
MDSC
CAF
5.2.3. Angiogenesis
5.2.4. Metastasis
6. Anti-Cancer Agents Induce IL-1β
7. Inhibition of IL-1 Signaling Inhibits Tumor Growth
7.1. Genetic Models
7.2. Pharmacological Inhibition
7.3. Immuno-Suppression
7.4. Angiogenesis
7.5. Metastasis
8. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| APC | Antigen presenting cell |
| BMDC | Bone morrow-derived macrophages |
| BRAFi | BRAF Inhibitor |
| CAF | Carcinoma-associated fibroblasts |
| DAB | Dabrafenib |
| DC | Dentritic cell |
| ENC | Encorafenib |
| HNSCC | Head and neck squamous cell carcinoma |
| IL-1α | Interleukin-1 alpha |
| IL-1β | Interleukin-1 beta |
| IL-1Ra | Interleukin-1 receptor |
| LPS | Lipopolysaccharide |
| MCA | Methylcholanthrene |
| MyD88 | Myeloid differentiation primary response 88 |
| MDSC | Myeloid-derived suppressor cells |
| NK | Natural killer cells |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B-cells |
| TAM | Tumor-associated macrophages |
| Th | T helper type T |
| TLR | Toll-like receptor |
| Treg | Regulatory T cells |
| VEGF | Vascular Endothelial Growth Factor |
References
- Dinarello, C.A. An expanding role for interleukin-1 blockade from gout to cancer. Mol. Med. 2014, 20 (Suppl. 1), S43–S58. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Awad, F.; Assrawi, E.; Louvrier, C.; Jumeau, C.; Georgin-Lavialle, S.; Grateau, G.; Amselem, S.; Giurgea, I.; Karabina, S.A. Inflammasome biology, molecular pathology and therapeutic implications. Pharmacol. Ther. 2018, 187, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Voronov, E.; Apte, R.N. Interleukin-1alpha. Semin. Immunol. 2013, 25, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Alcocer-Gomez, E.; Ryffel, B. Gain of function mutation and inflammasome driven diseases in human and mouse models. J. Autoimmun. 2018, 91, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Hajek, E.K.F.; Bent, R.; Haas, K.; Bast, A.; Steinmetz, I.; Tuettenberg, A.; Grabbe, S.; Bros, M. Braf inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Sierakowski, S.; Kucharz, E.J.; Lightfoot, R.W., Jr.; Goodwin, J.S. Interleukin-1-production by monocytes from patients with systemic lupus erythematosus. Clin. Rheumatol. 1987, 6, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Arend, W.P.; Gordon, D.F.; Wood, W.M.; Janson, R.W.; Joslin, F.G.; Jameel, S. IL-1 beta production in cultured human monocytes is regulated at multiple levels. J. Immunol. 1989, 143, 118–126. [Google Scholar] [PubMed]
- Palomo, J.; Dietrich, D.; Martin, P.; Palmer, G.; Gabay, C. The interleukin (IL)-1 cytokine family—Balance between agonists and antagonists in inflammatory diseases. Cytokine 2015, 76, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Auron, P.E.; Webb, A.C. Interleukin-1: A gene expression system regulated at multiple levels. Eur. Cytokine Netw. 1994, 5, 573–592. [Google Scholar] [PubMed]
- Kominato, Y.; Galson, D.; Waterman, W.R.; Webb, A.C.; Auron, P.E. Monocyte expression of the human prointerleukin 1 beta gene (IL1B) is dependent on promoter sequences which bind the hematopoietic transcription factor Spi-1/PU.1. Mol. Cell. Biol. 1995, 15, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wara-Aswapati, N.; Chen, C.; Tsukada, J.; Auron, P.E. NF-IL6 (C/EBPbeta) vigorously activates IL1b gene expression via a Spi-1 (PU.1) protein-protein tether. J. Biol. Chem. 2000, 275, 21272–21277. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M.; Jehnichen, P.; Jahn, B.; Schlosshan, D.; Romahn, E.; Marx, J. A novel sp-1 site in the human interleukin-1 beta promoter confers preferential transcriptional activity in keratinocytes. Eur. J. Immunol. 1996, 26, 3008–3014. [Google Scholar] [CrossRef] [PubMed]
- Toda, Y.; Tsukada, J.; Misago, M.; Kominato, Y.; Auron, P.E.; Tanaka, Y. Autocrine induction of the human pro-IL-1beta gene promoter by IL-1beta in monocytes. J. Immunol. 2002, 168, 1984–1991. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.B.; Park, H.H. Toll/interleukin-1 receptor (TIR) domain-mediated cellular signaling pathways. Apoptosis 2015, 20, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Kim, I.; Lye, E.; Shen, F.; Suzuki, N.; Suzuki, S.; Gerondakis, S.; Akira, S.; Gaffen, S.L.; Yeh, W.C.; et al. Differential role for c-rel and c/ebpbeta/delta in tlr-mediated induction of proinflammatory cytokines. J. Immunol. 2009, 182, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rom, W.N. Regulation of the interleukin-1 beta (IL-1 beta) gene by mycobacterial components and lipopolysaccharide is mediated by two nuclear factor-IL6 motifs. Mol. Cell. Biol. 1993, 13, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Godambe, S.A.; Chaplin, D.D.; Takova, T.; Bellone, C.J. Upstream nfil-6-like site located within a dnase i hypersensitivity region mediates lps-induced transcription of the murine interleukin-1 beta gene. J. Immunol. 1994, 153, 143–152. [Google Scholar] [PubMed]
- Pilzweger, C.; Holdenrieder, S. Circulating hmgb1 and rage as clinical biomarkers in malignant and autoimmune diseases. Diagnostics (Basel) 2015, 5, 219–253. [Google Scholar] [CrossRef] [PubMed]
- Mouri, F.; Tsukada, J.; Mizobe, T.; Higashi, T.; Yoshida, Y.; Minami, Y.; Izumi, H.; Kominato, Y.; Kohno, K.; Tanaka, Y. Intracellular HMGB1 transactivates the human IL1b gene promoter through association with an ets transcription factor PU.1. Eur. J. Haematol. 2008, 80, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, T.; Liu, Y.; Zhang, H.; Wang, K.; Liu, M.; Chen, G.; Xiao, X. Kruppel-like factor 4 inhibits the expression of interleukin-1 beta in lipopolysaccharide-induced raw264.7 macrophages. FEBS Lett. 2012, 586, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Cogswell, J.P.; Godlevski, M.M.; Wisely, G.B.; Clay, W.C.; Leesnitzer, L.M.; Ways, J.P.; Gray, J.G. NF-kappa B regulates IL-1 beta transcription through a consensus NF-kappa B binding site and a nonconsensus CRE-like site. J. Immunol. 1994, 153, 712–723. [Google Scholar] [PubMed]
- Roshan, M.H.; Tambo, A.; Pace, N.P. The role of TLR2, TLR4, and TLR9 in the pathogenesis of atherosclerosis. Int. J. Inflam. 2016, 2016, 1532832. [Google Scholar] [CrossRef] [PubMed]
- Brochu, C.; Cabrita, M.A.; Melanson, B.D.; Hamill, J.D.; Lau, R.; Pratt, M.A.; McKay, B.C. NF-kappab-dependent role for cold-inducible RNA binding protein in regulating interleukin 1beta. PLoS ONE 2013, 8, e57426. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, T.V.; Singh, A.K. Constitutive activity of the murine IL-1 beta promoter is regulated by a transcriptional repressor. Biochim. Biophys. Acta 1997, 1353, 32–38. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, L.; Duff, G.W. A negative regulatory region containing a glucocorticosteroid response element (nGRE) in the human interleukin-1beta gene. DNA Cell. Biol. 1997, 16, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Ratman, D.; Vanden Berghe, W.; Dejager, L.; Libert, C.; Tavernier, J.; Beck, I.M.; De Bosscher, K. How glucocorticoid receptors modulate the activity of other transcription factors: A scope beyond tethering. Mol. Cell. Endocrinol. 2013, 380, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Talwar, H.; Bauerfeld, C.; Bouhamdan, M.; Farshi, P.; Liu, Y.; Samavati, L. MKP-1 negatively regulates LPS-mediated IL-1beta production through p38 activation and HIF-1alpha expression. Cell. Signal. 2017, 34, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Petrovic, J.M.; Callaghan, D.; Jones, A.; Cui, H.; Howlett, C.; Stanimirovic, D. Evidence that hypoxia-inducible factor-1 (HIF-1) mediates transcriptional activation of interleukin-1beta (IL-1beta) in astrocyte cultures. J. Neuroimmunol. 2006, 174, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Blouin, C.C.; Page, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1alpha. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.D.; Zhang, Y.; McDevit, D.; Marecki, S.; Nikolajczyk, B.S. The interleukin-1beta gene is transcribed from a poised promoter architecture in monocytes. J. Biol. Chem. 2006, 281, 9227–9237. [Google Scholar] [CrossRef] [PubMed]
- Marecki, S.; Riendeau, C.J.; Liang, M.D.; Fenton, M.J. PU.1 and multiple IFN regulatory factor proteins synergize to mediate transcriptional activation of the human IL-1 beta gene. J. Immunol. 2001, 166, 6829–6838. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.G.; Chandra, G.; Clay, W.C.; Stinnett, S.W.; Haneline, S.A.; Lorenz, J.J.; Patel, I.R.; Wisely, G.B.; Furdon, P.J.; Taylor, J.D.; et al. A CRE/ATF-like site in the upstream regulatory sequence of the human interleukin 1 beta gene is necessary for induction in U937 and THP-1 monocytic cell lines. Mol. Cell. Biol. 1993, 13, 6678–6689. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.A.; Nisen, P.D. Inhibition of lipopolysaccharide-induced IL-1 beta transcription by cyclic adenosine monophosphate in human astrocytic cells. J. Immunol. 1995, 154, 1399–1406. [Google Scholar] [PubMed]
- Roman, J.; Ritzenthaler, J.D.; Perez, R.L.; Roser, S.L. Differential modes of regulation of interleukin-1beta expression by extracellular matrices. Immunology 1999, 98, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Roman, J.; Ritzenthaler, J.D.; Fenton, M.J.; Roser, S.; Schuyler, W. Transcriptional regulation of the human interleukin 1beta gene by fibronectin: Role of protein kinase C and activator protein 1 (AP-1). Cytokine 2000, 12, 1581–1596. [Google Scholar] [CrossRef] [PubMed]
- Loegering, D.J.; Lennartz, M.R. Protein kinase C and toll-like receptor signaling. Enzyme Res. 2011, 2011, 537821. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Tai, Y.T.; Cherng, Y.G.; Chen, T.G.; Lin, C.J.; Chen, T.L.; Chang, H.C.; Chen, R.M. GATA-2 transduces LPS-induced IL-1beta gene expression in macrophages via a toll-like receptor 4/MD88/MAPK-dependent mechanism. PLoS ONE 2013, 8, e72404. [Google Scholar]
- Wessels, I.; Fleischer, D.; Rink, L.; Uciechowski, P. Changes in chromatin structure and methylation of the human interleukin-1beta gene during monopoiesis. Immunology 2010, 130, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Yoza, B.K.; McCall, C.E. Facultative heterochromatin formation at the IL-1 beta promoter in LPS tolerance and sepsis. Cytokine 2011, 53, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wilkins, L.M.; Aziz, N.; Cannings, C.; Wyllie, D.H.; Bingle, C.; Rogus, J.; Beck, J.D.; Offenbacher, S.; Cork, M.J.; et al. Single nucleotide polymorphisms in the human interleukin-1b gene affect transcription according to haplotype context. Hum. Mol. Genet. 2006, 15, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Landvik, N.E.; Tekpli, X.; Anmarkrud, K.H.; Haugen, A.; Zienolddiny, S. Molecular characterization of a cancer-related single nucleotide polymorphism in the pro-inflammatory interleukin-1b gene. Mol. Carcinog. 2012, 51 (Suppl. 1), E168–E175. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Akopyan, G.; Garban, H.; Bonavida, B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene 2006, 25, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhou, B.; Li, S.; Yue, J.; Yang, H.; Wen, Y.; Zhan, S.; Wang, W.; Liao, M.; Zhang, M.; et al. Allele-specific induction of IL-1beta expression by c/ebpbeta and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog. 2014, 10, e1004426. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Liu, Y.; Pan, X.; Chen, R.; Li, P.; Wu, H.J.; Zhao, Z.Q.; Li, Y.P.; Huang, L.Q.; Zhuang, J.H.; et al. Interleukin-1beta promoter polymorphism enhances the risk of sleep disturbance in Alzheimer’s disease. PLoS ONE 2016, 11, e0149945. [Google Scholar]
- Cubino, N.; Montilla, C.; Usategui-Martin, R.; Cieza-Borrela, C.; Carranco, T.; Calero-Paniagua, I.; Quesada, A.; Canete, J.D.; Queiro, R.; Sanchez, M.D.; et al. Association of IL1beta (-511 A/C) and il6 (-174 G > C) polymorphisms with higher disease activity and clinical pattern of psoriatic arthritis. Clin. Rheumatol. 2016, 35, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Tayel, S.I.; Fouda, E.A.M.; Elshayeb, E.I.; Eldakamawy, A.R.A.; El-Kousy, S.M. Biochemical and molecular study on interleukin-1beta gene expression and relation of single nucleotide polymorphism in promoter region with type 2 diabetes mellitus. J. Cell. Biochem. 2018, 119, 5343–5349. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ding, Q.; Jiang, H.W. Genetic polymorphism of interleukin-1a (IL-1a), IL-1b, and IL-1 receptor antagonist (IL-1rn) and prostate cancer risk. Asian Pac. J. Cancer Prev. 2014, 15, 8741–8747. [Google Scholar] [CrossRef] [PubMed]
- Marucha, P.T.; Zeff, R.A.; Kreutzer, D.L. Cytokine-induced IL-1 beta gene expression in the human polymorphonuclear leukocyte: Transcriptional and post-transcriptional regulation by tumor necrosis factor and IL-1. J. Immunol. 1991, 147, 2603–2608. [Google Scholar] [PubMed]
- Donnelly, R.P.; Fenton, M.J.; Kaufman, J.D.; Gerrard, T.L. IL-1 expression in human monocytes is transcriptionally and posttranscriptionally regulated by IL-4. J. Immunol. 1991, 146, 3431–3436. [Google Scholar] [PubMed]
- Singh, A.K.; Aryal, B.; Zhang, X.; Fan, Y.; Price, N.L.; Suarez, Y.; Fernandez-Hernando, C. Posttranscriptional regulation of lipid metabolism by non-coding RNAs and RNA binding proteins. Semin Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Sirenko, O.I.; Lofquist, A.K.; DeMaria, C.T.; Morris, J.S.; Brewer, G.; Haskill, J.S. Adhesion-dependent regulation of an a+u-rich element-binding activity associated with AUF1. Mol. Cell. Biol. 1997, 17, 3898–3906. [Google Scholar] [CrossRef] [PubMed]
- Bros, M.; Wiechmann, N.; Besche, V.; Art, J.; Pautz, A.; Grabbe, S.; Kleinert, H.; Reske-Kunz, A.B. The rna binding protein tristetraprolin influences the activation state of murine dendritic cells. Mol. Immunol. 2010, 47, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Kwak, A.; Lee, Y.; Kim, H.; Kim, S. Intracellular interleukin (IL)-1 family cytokine processing enzyme. Arch. Pharm. Res. 2016, 39, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Muller, C.; Martin, S.J.; Beyaert, R. Proteolytic processing of interleukin-1 family cytokines: Variations on a common theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Awad, F.; Assrawi, E.; Jumeau, C.; Georgin-Lavialle, S.; Cobret, L.; Duquesnoy, P.; Piterboth, W.; Thomas, L.; Stankovic-Stojanovic, K.; Louvrier, C.; et al. Impact of human monocyte and macrophage polarization on nlr expression and NLRP3 inflammasome activation. PLoS ONE 2017, 12, e0175336. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.; Masters, S.L. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, C.; Peng, H.; Zhang, J.; Feng, Q. IL1 receptor antagonist gene IL1-RN variable number of tandem repeats polymorphism and cancer risk: A literature review and meta-analysis. PLoS ONE 2012, 7, e46017. [Google Scholar] [CrossRef] [PubMed]
- Aziz, F. The emerging role of miR-223 as novel potential diagnostic and therapeutic target for inflammatory disorders. Cell. Immunol. 2016, 303, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate nlrp3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of nlrp3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, T.; Imamura, R.; Kushiyama, H.; Suda, T. NLRP3 mediates nf-kappab activation and cytokine induction in microbially induced and sterile inflammation. PLoS ONE 2015, 10, e0119179. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting edge: Nitric oxide inhibits the NLRP3 inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef] [PubMed]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, M.; Chen, L.Y.; Liu, Y.; Martinez-Anton, A.; Qi, H.Y.; Logun, C.; Alsaaty, S.; Park, Y.H.; Kastner, D.L.; Chae, J.J.; et al. Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic amp in human macrophages. J. Immunol. 2015, 194, 5472–5487. [Google Scholar] [CrossRef] [PubMed]
- Hua, K.F.; Chou, J.C.; Ka, S.M.; Tasi, Y.L.; Chen, A.; Wu, S.H.; Chiu, H.W.; Wong, W.T.; Wang, Y.F.; Tsai, C.L.; et al. Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1beta production. J. Cell. Physiol. 2015, 230, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Lorey, M.B.; Rossi, K.; Eklund, K.K.; Nyman, T.A.; Matikainen, S. Global characterization of protein secretion from human macrophages following non-canonical caspase-4/5 inflammasome activation. Mol. Cell. Proteom. 2017, 16, S187–S199. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.L.; Zhai, J.H.; Chai, Y.F. Recent advances in the molecular mechanisms underlying pyroptosis in sepsis. Mediat. Inflamm. 2018, 2018, 5823823. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human monocytes engage an alternative inflammasome pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, C.; Russo, H.M.; El Sanadi, C.; Martin, B.N.; Li, X.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Caspase-8 as an effector and regulator of NLRP3 inflammasome signaling. J. Biol. Chem. 2015, 290, 20167–20184. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.D. Regulation of inflammasome activation. Immunol. Rev. 2015, 265, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Bossaller, L.; Chiang, P.I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.; Mocarski, E.S.; Subramanian, D.; Green, D.R.; Silverman, N.; et al. Cutting edge: FAS (CD95) mediates noncanonical IL-1beta and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J. Immunol. 2015, 194, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; MacDonald, J.A.; Chadee, K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease caps mutations. Nat. Immunol. 2016, 17, 1176–1186. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Sasaki, Y.; Abe, T.; Kano, H.; Yasutomo, K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 2014, 211, 2385–2396. [Google Scholar] [CrossRef] [PubMed]
- Baroja-Mazo, A.; Martin-Sanchez, F.; Gomez, A.I.; Martinez, C.M.; Amores-Iniesta, J.; Compan, V.; Barbera-Cremades, M.; Yague, J.; Ruiz-Ortiz, E.; Anton, J.; et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.; Li, H.; Fang, Y.; Liu, Y.; Li, M.; Zhong, B.; Yang, F.; Zou, Q.; Wu, Y. Nlrp1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han Chinese. Arthritis Rheum 2012, 64, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, M.; Laddha, N.C.; Mansuri, M.S.; Marfatia, Y.S.; Begum, R. Association of NLRP1 genetic variants and mrna overexpression with generalized vitiligo and disease activity in a gujarat population. Br. J. Dermatol. 2013, 169, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Alfakry, H.; Malle, E.; Koyani, C.N.; Pussinen, P.J.; Sorsa, T. Neutrophil proteolytic activation cascades: A possible mechanistic link between chronic periodontitis and coronary heart disease. Innate Immun. 2016, 22, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Bin, N.R.; Sugita, S. Diverse exocytic pathways for mast cell mediators. Biochem. Soc. Trans. 2018, 46, 235–247. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, A.; Wilson, H.L.; Kiss-Toth, E.; Dower, S.K.; North, R.A.; Surprenant, A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 2001, 15, 825–835. [Google Scholar] [CrossRef]
- Pizzirani, C.; Ferrari, D.; Chiozzi, P.; Adinolfi, E.; Sandona, D.; Savaglio, E.; Di Virgilio, F. Stimulation of P2 receptors causes release of IL-1beta-loaded microvesicles from human dendritic cells. Blood 2007, 109, 3856–3864. [Google Scholar] [CrossRef] [PubMed]
- Le Feuvre, R.; Brough, D.; Rothwell, N. Extracellular ATP and P2X7 receptors in neurodegeneration. Eur. J. Pharmacol. 2002, 447, 261–269. [Google Scholar] [CrossRef]
- Stoeckle, C.; Sommandas, V.; Adamopoulou, E.; Belisle, K.; Schiekofer, S.; Melms, A.; Weber, E.; Driessen, C.; Boehm, B.O.; Tolosa, E.; et al. Cathepsin g is differentially expressed in primary human antigen-presenting cells. Cell. Immunol. 2009, 255, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Trzybulska, D.; Olewicz-Gawlik, A.; Graniczna, K.; Kisiel, K.; Moskal, M.; Cieslak, D.; Sikora, J.; Hrycaj, P. Quantitative analysis of elastase and cathepsin G mRNA levels in peripheral blood CD14(+) cells from patients with rheumatoid arthritis. Cell. Immunol. 2014, 292, 40–44. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. The interleukin-1 receptor/toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef] [PubMed]
- Mcmahan, C.J.; Slack, J.L.; Mosley, B.; Cosman, D.; Lupton, S.D.; Brunton, L.L.; Grubin, C.E.; Wignall, J.M.; Jenkins, N.A.; Brannan, C.I.; et al. A novel IL-1 receptor, cloned from B-cells by mammalian expression, is expressed in many cell-types. EMBO J. 1991, 10, 2821–2832. [Google Scholar] [PubMed]
- Colotta, F.; Dower, S.K.; Sims, J.E.; Mantovani, A. The type ii ‘decoy’ receptor: A novel regulatory pathway for interleukin 1. Immunol. Today 1994, 15, 562–566. [Google Scholar] [CrossRef]
- Colotta, F.; Re, F.; Muzio, M.; Bertini, R.; Polentarutti, N.; Sironi, M.; Giri, J.G.; Dower, S.K.; Sims, J.E.; Mantovani, A. Interleukin-1 type-II receptor—A decoy target for IL-1 that is regulated by IL-4. Science 1993, 261, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Palmer, G.; Vigne, S.; Lamacchia, C.; Rodriguez, E.; Talabot-Ayer, D.; Rose-John, S.; Chalaris, A.; Gabay, C. Mouse neutrophils express the decoy type 2 interleukin-1 receptor (IL-1R2) constitutively and in acute inflammatory conditions. J. Leukoc. Biol. 2013, 94, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Nakajima, A.; Sudo, K.; Liu, Y.; Mizoroki, A.; Ikarashi, T.; Horai, R.; Kakuta, S.; Watanabe, T.; Iwakura, Y. IL-1 receptor type 2 suppresses collagen-induced arthritis by inhibiting IL-1 signal on macrophages. J. Immunol. 2015, 194, 3156–3168. [Google Scholar] [CrossRef] [PubMed]
- Lingel, A.; Weiss, T.M.; Niebuhr, M.; Pan, B.; Appleton, B.A.; Wiesmann, C.; Bazan, J.F.; Fairbrother, W.J. Structure of il-33 and its interaction with the ST2 and IL-1RACP receptors-insight into heterotrimeric IL-1 signaling complexes. Structure 2009, 17, 1398–1410. [Google Scholar] [CrossRef] [PubMed]
- Vigers, G.P.; Anderson, L.J.; Caffes, P.; Brandhuber, B.J. Crystal structure of the type-i interleukin-1 receptor complexed with interleukin-1beta. Nature 1997, 386, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Schreuder, H.; Tardif, C.; Trump-Kallmeyer, S.; Soffientini, A.; Sarubbi, E.; Akeson, A.; Bowlin, T.; Yanofsky, S.; Barrett, R.W. A new cytokine-receptor binding mode revealed by the crystal structure of the IL-1 receptor with an antagonist. Nature 1997, 386, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Korherr, C.; Hofmeister, R.; Wesche, H.; Falk, W. A critical role for interleukin-1 receptor accessory protein in interleukin-1 signaling. Eur. J. Immunol. 1997, 27, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Knop, J.; Wesche, H.; Raffetseder, U.; Kurrle, R.; Boraschi, D.; Martin, M.U. The type ii IL-1 receptor interacts with the IL-1 receptor accessory protein: A novel mechanism of regulation of IL-1 responsiveness. J. Immunol. 1998, 161, 6871–6877. [Google Scholar] [PubMed]
- Greenfeder, S.A.; Nunes, P.; Kwee, L.; Labow, M.; Chizzonite, R.A.; Ju, G. Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. J. Biol. Chem. 1995, 270, 13757–13765. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Ni, J.; Feng, P.; Dixit, V.M. IRAK (pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 1997, 278, 1612–1615. [Google Scholar] [CrossRef] [PubMed]
- Wesche, H.; Henzel, W.J.; Shillinglaw, W.; Li, S.; Cao, Z.D. MyD88: An adapter that recruits IRAK to the IL-1 receptor complex. Immunity 1997, 7, 837–847. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.Q.; Ghosh, S.; Janeway, C.A. MyD88 is an adaptor protein in the htoll/IL-1 receptor family signaling pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Burns, K.; Martinon, F.; Esslinger, C.; Pahl, H.; Schneider, P.; Bodmer, J.L.; Di Marco, F.; French, L.; Tschopp, J. MyD88, an adapter protein involved in interleukin-1 signaling. J. Biol. Chem. 1998, 273, 12203–12209. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, R.; Wu, H. Helical assembly in the death domain (DD) superfamily. Curr. Opin. Struct. Biol. 2012, 22, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An oligomeric signaling platform formed by the toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [PubMed]
- Dossang, A.C.G.; Motshwene, P.G.; Yang, Y.; Symmons, M.F.; Bryant, C.E.; Borman, S.; George, J.; Weber, A.N.R.; Gay, N.J. The N-terminal loop of IRAK-4 death domain regulates ordered assembly of the myddosome signalling scaffold. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.; Clatworthy, J.; Martin, L.; Martinon, F.; Plumpton, C.; Maschera, B.; Lewis, A.; Ray, K.; Tschopp, J.; Volpe, F. Tollip, a new component of the IL-1RI pathway, links irak to the IL-1 receptor. Nat. Cell Biol. 2000, 2, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Moynagh, P.N. The roles of pellino e3 ubiquitin ligases in immunity. Nat. Rev. Immunol. 2014, 14, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Strelow, A.; Fontana, E.J.; Wesche, H. Irak-4: A novel member of the irak family with the properties of an irak-kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 5567–5572. [Google Scholar] [CrossRef] [PubMed]
- Kollewe, C.; Mackensen, A.C.; Neumann, D.; Knop, J.; Cao, P.; Li, S.U.; Wesche, H.; Martin, M.U. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J. Biol. Chem. 2004, 279, 5227–5236. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Liu, Z.G.; Bennett, B.; Suzuki, N.; Xia, Y.; Karin, M. Signaling by proinflammatory cytokines: Oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999, 13, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Smith, H.; Windheim, M.; Peggie, M.; Carrick, E.; Morrice, N.; Cohen, P. The IRAK-catalysed activation of the E3 ligase function of pellino isoforms induces the Lys63-linked polyubiquitination of IRAK1. Biochem. J. 2008, 409, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Peggie, M.; Campbell, D.G.; Vandermoere, F.; Carrick, E.; Cohen, P. Identification of the phosphorylation sites on the E3 ubiquitin ligase pellino that are critical for activation by IRAK1 and IRAK4. Proc. Natl. Acad. Sci. USA 2009, 106, 4584–4590. [Google Scholar] [CrossRef] [PubMed]
- Strickson, S.; Emmerich, C.H.; Goh, E.T.H.; Zhang, J.; Kelsall, I.R.; Macartney, T.; Hastie, C.J.; Knebel, A.; Peggie, M.; Marchesi, F.; et al. Roles of the TRAF6 and pellino E3 ligases in MyD88 and RANKl signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E3481–E3489. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Xiao, C.C.; Paschal, A.E.; Bailey, S.T.; Rao, P.; Hayden, M.S.; Lee, K.Y.; Bussey, C.; Steckel, M.; Tanaka, N.; et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 2005, 19, 2668–2681. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-translational modifications of the TAK1-TAB complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef] [PubMed]
- Pauls, E.; Nanda, S.K.; Smith, H.; Toth, R.; Arthur, J.S.C.; Cohen, P. Two phases of inflammatory mediator production defined by the study of IRAK2 and IRAK1 knock-in mice. J. Immunol. 2013, 191, 2717–2730. [Google Scholar] [CrossRef] [PubMed]
- Vigers, G.P.; Caffes, P.; Evans, R.J.; Thompson, R.C.; Eisenberg, S.P.; Brandhuber, B.J. X-ray structure of interleukin-1 receptor antagonist at 2.0-A resolution. J. Biol. Chem. 1994, 269, 12874–12879. [Google Scholar] [PubMed]
- Arend, W.P.; Guthridge, C.J. Biological role of interleukin 1 receptor antagonist isoforms. Ann. Rheum. Dis. 2000, 59 (Suppl. 1), i60–i64. [Google Scholar] [CrossRef] [PubMed]
- Bellehumeur, C.; Blanchet, J.; Fontaine, J.Y.; Bourcier, N.; Akoum, A. Interleukin 1 regulates its own receptors in human endometrial cells via distinct mechanisms. Hum. Reprod. 2009, 24, 2193–2204. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.J.; Ferguson, J.F.; Feng, R.; Wang, F.; Patel, P.N.; Li, M.; Xue, C.; Qu, L.; Liu, Y.; Boyd, J.H.; et al. A functional synonymous coding variant in the IL1rn gene is associated with survival in septic shock. Am. J. Respir. Crit. Care Med. 2014, 190, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Kollewe, C.; Neumann, D.; Martin, M.U. The first two N-terminal immunoglobulin-like domains of soluble human IL-1 receptor type II are sufficient to bind and neutralize IL-1beta. FEBS Lett. 2000, 487, 189–193. [Google Scholar] [CrossRef]
- Neumann, D.; Kollewe, C.; Martin, M.U.; Boraschi, D. The membrane form of the type ii IL-1 receptor accounts for inhibitory function. J. Immunol. 2000, 165, 3350–3357. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Slack, J.L.; Davis, R.; Cerretti, D.P.; Kozlosky, C.J.; Blanton, R.A.; Shows, D.; Peschon, J.J.; Black, R.A. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 2000, 275, 14608–14614. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Lokau, J.; Dusterhoft, S.; Trad, A.; Garbers, C.; Scheller, J.; Rose-John, S.; Grotzinger, J. The membrane-proximal domain of A Disintegrin and Metalloprotease 17 (ADAM17) is responsible for recognition of the interleukin-6 receptor and interleukin-1 receptor II. FEBS Lett. 2012, 586, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.L.; Rouhani, F.N.; Hawari, F.; Levine, S.J. Shedding of the type II IL-1 decoy receptor requires a multifunctional aminopeptidase, aminopeptidase regulator of tnf receptor type 1 shedding. J. Immunol. 2003, 171, 6814–6819. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Marjaux, E.; Imhof, A.; De Strooper, B.; Haass, C.; Lichtenthaler, S.F. Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J. Biol. Chem. 2007, 282, 11982–11995. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Hart, R.P.; Liu, X.J.; Clevenger, W.; Maki, R.A.; DeSouza, E.B. Cloning and characterization of an alternatively processed human type ii interleukin-1 receptor mrna. J. Biol. Chem. 1996, 271, 20965–20972. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.E.; Muzio, M.; Mantovani, A.; Whitehead, A.S. IL-1 signaling cascade in liver cells and the involvement of a soluble form of the IL-1 receptor accessory protein. J. Immunol. 2000, 164, 5277–5286. [Google Scholar] [CrossRef] [PubMed]
- Peters, V.A.; Joesting, J.J.; Freund, G.G. IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav. Immun. 2013, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vambutas, A.; DeVoti, J.; Goldofsky, E.; Gordon, M.; Lesser, M.; Bonagura, V. Alternate splicing of interleukin-1 receptor type ii (IL1R2) in vitro correlates with clinical glucocorticoid responsiveness in patients with aied. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Mora-Buch, R.; Dotti, I.; Planell, N.; Calderon-Gomez, E.; Jung, P.; Masamunt, M.C.; Llach, J.; Ricart, E.; Batlle, E.; Panes, J.; et al. Epithelial IL-1R2 acts as a homeostatic regulator during remission of ulcerative colitis. Mucosal Immunol. 2016, 9, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, D.Y.; Eisenman, J.R.; Conlon, P.J.; Larsen, A.D.; Tushinski, R.J. Interleukin 1 regulates hematopoietic activity, a role previously ascribed to hemopoietin 1. Proc. Natl. Acad. Sci. USA 1987, 84, 5267–5271. [Google Scholar] [CrossRef] [PubMed]
- Neta, R.; Douches, S.; Oppenheim, J.J. Interleukin 1 is a radioprotector. J. Immunol. 1986, 136, 2483–2485. [Google Scholar] [PubMed]
- Yoshimura, A.; Mori, H.; Ohishi, M.; Aki, D.; Hanada, T. Negative regulation of cytokine signaling influences inflammation. Curr. Opin. Immunol. 2003, 15, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Kim, G.K.; Maurizio, P.L.; Molnar, E.E.; Choi, Y. TRAF6 autoubiquitination-independent activation of the NFkappab and MAPK pathways in response to IL-1 and RANKL. PLoS ONE 2008, 3, e4064. [Google Scholar] [CrossRef] [PubMed]
- Brinson, C.W.; Lu, Z.; Li, Y.; Lopes-Virella, M.F.; Huang, Y. Lipopolysaccharide and IL-1beta coordinate a synergy on cytokine production by upregulating MyD88 expression in human gingival fibroblasts. Mol. Immunol. 2016, 79, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Koide, S.L.; Inaba, K.; Steinman, R.M. Interleukin 1 enhances T-dependent immune responses by amplifying the function of dendritic cells. J. Exp. Med. 1987, 165, 515–530. [Google Scholar] [CrossRef] [PubMed]
- Heufler, C.; Koch, F.; Schuler, G. Granulocyte/macrophage colony-stimulating factor and interleukin 1 mediate the maturation of murine epidermal langerhans cells into potent immunostimulatory dendritic cells. J. Exp. Med. 1988, 167, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.; Fabri, M.; Krutzik, S.R.; Lee, D.J.; Vu, D.M.; Sieling, P.A.; Montoya, D.; Liu, P.T.; Modlin, R.L. Interleukin-1beta triggers the differentiation of macrophages with enhanced capacity to present mycobacterial antigen to t cells. Immunology 2014, 141, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Lipsky, P.E.; Thompson, P.A.; Rosenwasser, L.J.; Dinarello, C.A. The role of interleukin 1 in human b cell activation: Inhibition of B cell proliferation and the generation of immunoglobulin-secreting cells by an antibody against human leukocytic pyrogen. J. Immunol. 1983, 130, 2708–2714. [Google Scholar] [PubMed]
- Ben Aribia, M.H.; Leroy, E.; Lantz, O.; Metivier, D.; Autran, B.; Charpentier, B.; Hercend, T.; Senik, A. rIL 2-induced proliferation of human circulating nk cells and t lymphocytes: Synergistic effects of IL 1 and IL 2. J. Immunol. 1987, 139, 443–451. [Google Scholar] [PubMed]
- Ben-Sasson, S.Z.; Wang, K.; Cohen, J.; Paul, W.E. IL-1beta strikingly enhances antigen-driven CD4 and CD8 T-cell responses. Cold Spring Harb. Symp. Quant. Biol. 2013, 78, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 2016, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.G.; Raulet, D.H. CD28-induced costimulation of T helper type 2 cells mediated by induction of responsiveness to interleukin 4. J. Exp. Med. 1993, 178, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Napolitani, G.; Lanzavecchia, A.; Sallusto, F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human t helper cells. Nat. Immunol. 2007, 8, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing t cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Saijo, S.; Murayama, M.A.; Shimizu, K.; Akitsu, A.; Iwakura, Y. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-beta-induced Foxp3 expression. J. Immunol. 2014, 192, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Mailer, R.K.; Joly, A.L.; Liu, S.; Elias, S.; Tegner, J.; Andersson, J. IL-1beta promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci. Rep. 2015, 5, 14674. [Google Scholar] [CrossRef] [PubMed]
- Ilarregui, J.M.; van Beelen, A.J.; Fehres, C.M.; Bruijns, S.C.; Garcia-Vallejo, J.J.; van Kooyk, Y. New roles for CD14 and IL-beta linking inflammatory dendritic cells to IL-17 production in memory CD4(+) t cells. Immunol. Cell. Biol. 2016, 94, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human Th17 cells produce ifn-gamma or IL-10 and are regulated by IL-1beta. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta t cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kuldo, J.M.; Ogawara, K.I.; Werner, N.; Asgeirsdottir, S.A.; Kamps, J.A.; Kok, R.J.; Molema, G. Molecular pathways of endothelial cell activation for (targeted) pharmacological intervention of chronic inflammatory diseases. Curr. Vasc. Pharmacol. 2005, 3, 11–39. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; O’Connell, R.M.; Gutierrez, M.A.; Pietras, E.M.; Shahangian, A.; Gross, C.E.; Thirumala, A.; Cheung, A.L.; Cheng, G.; Modlin, R.L. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against staphylococcus aureus. Immunity 2006, 24, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Canada, J.M.; Van Tassell, B.W.; Wise, C.M.; Dinarello, C.A. Interleukin-1 blockade in rheumatoid arthritis and heart failure: A missed opportunity? Int. J. Cardiol. 2014, 171, e125–e126. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.N.; Parry-Jones, A.R.; Allan, S.M. Interleukin-1 and acute brain injury. Front. Cell. Neurosci. 2015, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.C.; Zhang, J.G.; Tan, M.S.; Chen, H.; Meng, D.W.; Jiang, T.; Meng, X.F.; Li, Y.; Sun, Z.; Li, M.M.; et al. NLRP1 inflammasome is activated in patients with medial temporal lobe epilepsy and contributes to neuronal pyroptosis in amygdala kindling-induced rat model. J. Neuroinflamm. 2015, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.; Lim, Y.A.; Cheng, Y.L.; Lok, K.Z.; Chunduri, P.; Baik, S.H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.G.; et al. Evidence that NF-kappab and MAPK signaling promotes nlrp inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The nlrp3 inflammasome in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Mamik, M.K.; Power, C. Inflammasomes in neurological diseases: Emerging pathogenic and therapeutic concepts. Brain J. Neurol. 2017, 140, 2273–2285. [Google Scholar] [CrossRef] [PubMed]
- Dumusc, A.; So, A. Interleukin-1 as a therapeutic target in gout. Curr. Opin. Rheumatol. 2015, 27, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Pilli, D.; Zou, A.; Tea, F.; Dale, R.C.; Brilot, F. Expanding role of t cells in human autoimmune diseases of the central nervous system. Front. Immunol. 2017, 8, 652. [Google Scholar] [CrossRef] [PubMed]
- Striz, I. Cytokines of the IL-1 family: Recognized targets in chronic inflammation underrated in organ transplantations. Clin. Sci. (Lond.) 2017, 131, 2241–2256. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Sergeev, P.; Ris, F.; Oberholzer, J.; Joller-Jemelka, H.I.; Spinas, G.A.; Kaiser, N.; Halban, P.A.; Donath, M.Y. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Investig. 2002, 110, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Westwell-Roper, C.Y.; Ehses, J.A.; Verchere, C.B. Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1beta production and beta-cell dysfunction. Diabetes 2014, 63, 1698–1711. [Google Scholar] [CrossRef] [PubMed]
- Boni-Schnetzler, M.; Donath, M.Y. How biologics targeting the IL-1 system are being considered for the treatment of type 2 diabetes. Br. J. Clin. Pharmacol. 2013, 76, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chen, R.; Zhou, J. E2f1 and nf-kappab: Key mediators of inflammation-associated cancers and potential therapeutic targets. Curr. Cancer Drug Targets 2016, 16, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Compromised mapk signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Tang, Y.; Hua, S. Immunological approaches towards cancer and inflammation: A cross talk. Front. Immunol. 2018, 9, 563. [Google Scholar] [CrossRef] [PubMed]
- Liegel, J.; Avigan, D.; Rosenblatt, J. Cellular immunotherapy as a therapeutic approach in multiple myeloma. Expert Rev. Hematol. 2018, 11, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Remstein, E.D.; Therneau, T.M.; Dispenzieri, A.; Kurtin, P.J.; Hodnefield, J.M.; Larson, D.R.; Plevak, M.F.; Jelinek, D.F.; Fonseca, R.; et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N. Engl. J. Med. 2007, 356, 2582–2590. [Google Scholar] [CrossRef] [PubMed]
- Costes, V.; Portier, M.; Lu, Z.Y.; Rossi, J.F.; Bataille, R.; Klein, B. Interleukin-1 in multiple myeloma: Producer cells and their role in the control of IL-6 production. Br. J. Haematol. 1998, 103, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Witzig, T.E.; Moon-Tasson, L.L.; Dinarello, C.A.; Donovan, K.A. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am. J. Hematol. 2016, 91, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Nakata, K.; Kashimoto, S.; Yoshida, H.; Yamada, M. Antitumor effect of recombinant human interleukin 1 alpha against murine syngeneic tumors. Jpn. J. Cancer Res. 1986, 77, 767–773. [Google Scholar] [PubMed]
- North, R.J.; Neubauer, R.H.; Huang, J.J.; Newton, R.C.; Loveless, S.E. Interleukin 1-induced, T cell-mediated regression of immunogenic murine tumors. Requirement for an adequate level of already acquired host concomitant immunity. J. Exp. Med. 1988, 168, 2031–2043. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.M.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Haabeth, O.A.; Lorvik, K.B.; Hammarstrom, C.; Donaldson, I.M.; Haraldsen, G.; Bogen, B.; Corthay, A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat. Commun 2011, 2, 240. [Google Scholar] [CrossRef] [PubMed]
- Haabeth, O.A.; Lorvik, K.B.; Yagita, H.; Bogen, B.; Corthay, A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology 2016, 5, e1039763. [Google Scholar] [CrossRef] [PubMed]
- Veltri, S.; Smith, J.W., 2nd. Interleukin 1 trials in cancer patients: A review of the toxicity, antitumor and hematopoietic effects. Stem Cells 1996, 14, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Mullerad, J.; Cohen, S.; Benharroch, D.; Apte, R.N. Local delivery of IL-1 alpha polymeric microspheres for the immunotherapy of an experimental fibrosarcoma. Cancer Investig. 2003, 21, 720–728. [Google Scholar] [CrossRef]
- Mullerad, J.; Cohen, S.; Voronov, E.; Apte, R.N. Macrophage activation for the production of immunostimulatory cytokines by delivering interleukin 1 via biodegradable microspheres. Cytokine 2000, 12, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, K.E.; Dai, M.; Hellstrom, I. Curing tumor-bearing mice by shifting a Th2 to a Th1 anti-tumor response. Hum. Antibodies 2017, 25, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Geis, A.L.; Thiele Orberg, E.; Housseau, F. Interleukin-17 and type 17 helper T cells in cancer management and research. Immunotargets Ther. 2014, 3, 39–54. [Google Scholar] [PubMed]
- Mansilla, S.; Llovera, L.; Portugal, J. Chemotherapeutic targeting of cell death pathways. Anticancer Agents Med. Chem. 2012, 12, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Pulskens, W.P.; Sadler, J.J.; Butter, L.M.; Teske, G.J.; Ulland, T.K.; Eisenbarth, S.C.; Florquin, S.; Flavell, R.A.; Leemans, J.C.; et al. Necrotic cells trigger a sterile inflammatory response through the nlrp3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 20388–20393. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the nlrp3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Mattarollo, S.R.; Loi, S.; Duret, H.; Ma, Y.; Zitvogel, L.; Smyth, M.J. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res. 2011, 71, 4809–4820. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.L.; Terhorst, L.; Collins, K.P.; Geller, D.A.; Vodovotz, Y.; Kim, J.; Krane, A.; Antoni, M.; Marsh, J.W.; Burke, L.E.; et al. Prospective analyses of cytokine mediation of sleep and survival in the context of advanced cancer. Psychosom. Med. 2018, 80, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Krelin, Y.; Song, X.; Dotan, S.; Recih, E.; Elkabets, M.; Carmi, Y.; Dvorkin, T.; White, R.M.; Gayvoronsky, L.; et al. Effects of micro-environment- and malignant cell-derived interleukin-1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur. J. Cancer 2006, 42, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Maker, A.V.; Katabi, N.; Qin, L.X.; Klimstra, D.S.; Schattner, M.; Brennan, M.F.; Jarnagin, W.R.; Allen, P.J. Cyst fluid interleukin-1beta (IL1beta) levels predict the risk of carcinoma in intraductal papillary mucinous neoplasms of the pancreas. Clin. Cancer Res. 2011, 17, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.N.; Voronov, E. Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol. Rev. 2008, 222, 222–241. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Grivennikov, S.I.; Karin, M. The unholy trinity: Inflammation, cytokines, and stat3 shape the cancer microenvironment. Cancer Cell 2011, 19, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J.; Ben-Neriah, Y.; Pikarsky, E. Inflammation and cancer: Is the link as simple as we think? J. Investig. Dermatol. 2005, 124, x–xiv. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M. Risk of hepatocellular carcinoma in hepatitis b and prevention through treatment. Clevel. Clin. J. Med. 2009, 76 (Suppl. 3), S6–S9. [Google Scholar] [CrossRef] [PubMed]
- Yi, G.; Liang, M.; Li, M.; Fang, X.; Liu, J.; Lai, Y.; Chen, J.; Yao, W.; Feng, X.; Hu, L.; et al. A large lung gene expression study identifying IL1b as a novel player in airway inflammation in copd airway epithelial cells. Inflamm. Res. 2018, 67, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.B.; Zuo, W.; Wang, A.J.; Lu, N.H. Helicobacter pylori infection synergistic with IL-1beta gene polymorphisms potentially contributes to the carcinogenesis of gastric cancer. Int. J. Med. Sci. 2016, 13, 298–303. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C.; Rothman, N.; et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, Y.; Niwa, T.; Rehnberg, E.; Toyoda, T.; Yoshida, S.; Mori, A.; Wakabayashi, M.; Iwakura, Y.; Ichinose, M.; Kim, Y.J.; et al. Interleukin-1beta induced by helicobacter pylori infection enhances mouse gastric carcinogenesis. Cancer Lett. 2013, 340, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.Y.; Chan, A.O.; Rashid, A.; Wong, D.K.; Seto, W.K.; Cho, C.H.; Lai, C.L.; Yuen, M.F. Interleukin-1beta increases the risk of gastric cancer through induction of aberrant DNA methylation in a mouse model. Oncol. Lett. 2016, 11, 2919–2924. [Google Scholar] [CrossRef] [PubMed]
- Sandri, B.J.; Kaplan, A.; Hodgson, S.W.; Peterson, M.; Avdulov, S.; Higgins, L.; Markowski, T.; Yang, P.; Limper, A.H.; Griffin, T.J.; et al. Multi-omic molecular profiling of lung cancer in chronic obstructive pulmonary disease. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef] [PubMed]
- Krelin, Y.; Voronov, E.; Dotan, S.; Elkabets, M.; Reich, E.; Fogel, M.; Huszar, M.; Iwakura, Y.; Segal, S.; Dinarello, C.A.; et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007, 67, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Nakamoto, Y. Emergence of immunotherapy as a novel way to treat hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 1839–1858. [Google Scholar] [CrossRef] [PubMed]
- Kanterman, J.; Sade-Feldman, M.; Baniyash, M. New insights into chronic inflammation-induced immunosuppression. Semin. Cancer Biol. 2012, 22, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef] [PubMed]
- Mishalian, I.; Granot, Z.; Fridlender, Z.G. The diversity of circulating neutrophils in cancer. Immunobiology 2017, 222, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Belgiovine, C.; D’Incalci, M.; Allavena, P.; Frapolli, R. Tumor-associated macrophages and anti-tumor therapies: Complex links. Cell. Mol. Life Sci. 2016, 73, 2411–2424. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-associated macrophages: Functional diversity, clinical significance, and open questions. Semin. Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: Critical cells driving immune suppression in the tumor microenvironment. Adv. Cancer Res. 2015, 128, 95–139. [Google Scholar] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Baniyash, M. Myeloid-derived suppressor cells as intruders and targets: Clinical implications in cancer therapy. Cancer Immunol. Immunother. 2016, 65, 857–867. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, F.; Solito, S.; Ugel, S.; Molon, B.; Bronte, V.; Marigo, I. Mdscs in cancer: Conceiving new prognostic and therapeutic targets. Biochim. Biophys. Acta 2016, 1865, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Keskinov, A.A.; Shurin, M.R. Myeloid regulatory cells in tumor spreading and metastasis. Immunobiology 2015, 220, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Apte, R.N. Targeting the tumor microenvironment by intervention in interleukin-1 biology. Curr. Pharm. Des. 2017, 23, 4893–4905. [Google Scholar] [PubMed]
- Nishikawa, H. [Regulatory T cells in cancer immunotherapy]. Rinsho Ketsueki 2014, 55, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Plitas, G.; Rudensky, A.Y. Regulatory t cells: Differentiation and function. Cancer Immunol. Res. 2016, 4, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, Y.; Jia, T.; Sun, Y. Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumour. Biol. 2015, 36, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, C.F.; Li, Y.C.; Deng, W.W.; Mao, L.; Wu, L.; Zhang, W.F.; Zhang, L.; Sun, Z.J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Tanchot, C.; Terme, M.; Pere, H.; Tran, T.; Benhamouda, N.; Strioga, M.; Banissi, C.; Galluzzi, L.; Kroemer, G.; Tartour, E. Tumor-infiltrating regulatory t cells: Phenotype, role, mechanism of expansion in situ and clinical significance. Cancer Microenviron. 2013, 6, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.F.; Nierkens, S.; Figdor, C.G.; de Vries, I.J.; Adema, G.J. Regulatory t cells in melanoma: The final hurdle towards effective immunotherapy? Lancet Oncol. 2012, 13, e32–e42. [Google Scholar] [CrossRef]
- Minnema-Luiting, J.; Vroman, H.; Aerts, J.; Cornelissen, R. Heterogeneity in immune cell content in malignant pleural mesothelioma. Int. J. Mol. Sci. 2018, 19, 1041. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized m2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wei, G.; Cheng, W.A.; Dong, Z.; Sun, H.; Lee, V.Y.; Cha, S.C.; Smith, D.L.; Kwak, L.W.; Qin, H. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol. Immunother. 2018, 1036, 105–128. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef] [PubMed]
- Peranzoni, E.; Zilio, S.; Marigo, I.; Dolcetti, L.; Zanovello, P.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 2010, 22, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Poschke, I.; Mougiakakos, D.; Hansson, J.; Masucci, G.V.; Kiessling, R. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 2010, 70, 4335–4345. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Sevko, A.; Ramacher, M.; Bazhin, A.V.; Falk, C.S.; Osen, W.; Borrello, I.; Kato, M.; Schadendorf, D.; Baniyash, M.; et al. Chronic inflammation promotes myeloid-derived suppressor cell activation blocking antitumor immunity in transgenic mouse melanoma model. Proc. Natl. Acad. Sci. USA 2011, 108, 17111–17116. [Google Scholar] [CrossRef] [PubMed]
- Filipazzi, P.; Huber, V.; Rivoltini, L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol. Immunother. 2012, 61, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 2006, 176, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhang, J.; Han, X.; Li, H.; Xie, M.; Sun, Y.; Liu, W.; Ba, X.; Zeng, X. Recruited monocytic myeloid-derived suppressor cells promote the arrest of tumor cells in the premetastatic niche through an IL-1beta-mediated increase in E-selectin expression. Int. J. Cancer 2017, 140, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gebhardt, C.; Umansky, L.; Beckhove, P.; Schulze, T.J.; Utikal, J.; Umansky, V. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int. J. Cancer 2015, 136, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin e2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The role of IL-1beta in the early tumor cell-induced angiogenic response. J. Immunol. 2013, 190, 3500–3509. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Vanaclocha, F.; Fantuzzi, G.; Mendoza, L.; Fuentes, A.M.; Anasagasti, M.J.; Martin, J.; Carrascal, T.; Walsh, P.; Reznikov, L.L.; Kim, S.H.; et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc. Natl. Acad. Sci. USA 2000, 97, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Chirivi, R.G.; Garofalo, A.; Padura, I.M.; Mantovani, A.; Giavazzi, R. Interleukin 1 receptor antagonist inhibits the augmentation of metastasis induced by interleukin 1 or lipopolysaccharide in a human melanoma/nude mouse system. Cancer Res. 1993, 53, 5051–5054. [Google Scholar] [PubMed]
- Erez, N.; Glanz, S.; Raz, Y.; Avivi, C.; Barshack, I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 2013, 437, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer associated fibroblasts: The dark side of the coin. Am. J. Cancer Res. 2011, 1, 482–497. [Google Scholar] [PubMed]
- Kalluri, R.; Zeisberg, E. Controlling angiogenesis in heart valves. Nat. Med. 2006, 12, 1118–1119. [Google Scholar] [CrossRef] [PubMed]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer associated fibroblasts: The architects of stroma remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Oren, M. New plays in the p53 theater. Curr. Opin. Genet. Dev. 2011, 21, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Schauer, I.G.; Zhang, J.; Xing, Z.; Guo, X.; Mercado-Uribe, I.; Sood, A.K.; Huang, P.; Liu, J. Interleukin-1beta promotes ovarian tumorigenesis through a p53/NF-kappab-mediated inflammatory response in stromal fibroblasts. Neoplasia 2013, 15, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Xiao, N.; Wang, J.; Wang, Z.; Zheng, S.; Shan, S.; Wang, J.; Du, J.; Wang, J. Smc1a recruits tumor-associated-fibroblasts (TAFs) and promotes colorectal cancer metastasis. Cancer Lett. 2017, 385, 39–45. [Google Scholar] [CrossRef] [PubMed]
- De Marco, P.; Lappano, R.; De Francesco, E.M.; Cirillo, F.; Pupo, M.; Avino, S.; Vivacqua, A.; Abonante, S.; Picard, D.; Maggiolini, M. Gper signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1beta/IL1R1 response. Sci. Rep. 2016, 6, 24354. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour-host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Tanaka, M.; Miki, M.; Usui, K.; Suzuki, T.; Maemondo, M.; Hong, X.; Tazawa, R.; Kikuchi, T.; Matsushima, K.; et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of lewis lung carcinoma by induction of angiogenic factors: In vivo analysis of tumor-stromal interaction. J. Immunol. 2002, 169, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Nakao, S.; Kuwano, T.; Tsutsumi-Miyahara, C.; Ueda, S.; Kimura, Y.N.; Hamano, S.; Sonoda, K.H.; Saijo, Y.; Nukiwa, T.; Strieter, R.M.; et al. Infiltration of COX-2-expressing macrophages is a prerequisite for IL-1 beta-induced neovascularization and tumor growth. J. Clin. Investig. 2005, 115, 2979–2991. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, E.C.; Chuaqui, R.F.; Liotta, L.A. General mechanisms of metastasis. Cancer 1997, 80, 1529–1537. [Google Scholar] [CrossRef]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Sceneay, J.; Smyth, M.J.; Moller, A. The pre-metastatic niche: Finding common ground. Cancer Metastasis Rev. 2013, 32, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Giavazzi, R.; Garofalo, A.; Bani, M.R.; Abbate, M.; Ghezzi, P.; Boraschi, D.; Mantovani, A.; Dejana, E. Interleukin 1-induced augmentation of experimental metastases from a human melanoma in nude mice. Cancer Res. 1990, 50, 4771–4775. [Google Scholar] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. Vegfr1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, Q.; Feng, X.; Zhang, R.; Li, J.; Chen, F. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer 2018, 18, 500. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Vanaclocha, F.; Amezaga, C.; Asumendi, A.; Kaplanski, G.; Dinarello, C.A. Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res. 1994, 54, 2667–2672. [Google Scholar] [PubMed]
- Vidal-Vanaclocha, F.; Alvarez, A.; Asumendi, A.; Urcelay, B.; Tonino, P.; Dinarello, C.A. Interleukin 1 (IL-1)-dependent melanoma hepatic metastasis in vivo; increased endothelial adherence by IL-1-induced mannose receptors and growth factor production in vitro. J. Natl. Cancer Inst. 1996, 88, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Carrascal, M.T.; Mendoza, L.; Valcarcel, M.; Salado, C.; Egilegor, E.; Telleria, N.; Vidal-Vanaclocha, F.; Dinarello, C.A. Interleukin-18 binding protein reduces B16 melanoma hepatic metastasis by neutralizing adhesiveness and growth factors of sinusoidal endothelium. Cancer Res. 2003, 63, 491–497. [Google Scholar] [PubMed]
- Smith, L.B.; Leo, M.C.; Anderson, C.; Wright, T.J.; Weymann, K.B.; Wood, L.J. The role of IL-1beta and TNF-alpha signaling in the genesis of cancer treatment related symptoms (CTRS): A study using cytokine receptor-deficient mice. Brain Behav. Immun. 2014, 38, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Tran, L.T.; Magun, E.A.; Magun, B.E.; Wood, L.J. Production of IL-1beta by bone marrow-derived macrophages in response to chemotherapeutic drugs: Synergistic effects of doxorubicin and vincristine. Cancer Biol. Ther. 2014, 15, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with braf-mutant melanoma (columbus): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Kulkarni, D.; Song, K.; Briley, L.; King, K.; Dabrowski, C.; Mookerjee, B.; Legos, J.; Spraggs, C. Pyrexia in dabrafenib-treated melanoma patients is not associated with common genetic variation or HLA polymorphisms. Pharmacogenomics 2016, 17, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Westbom, C.; Thompson, J.K.; Leggett, A.; MacPherson, M.; Beuschel, S.; Pass, H.; Vacek, P.; Shukla, A. Inflammasome modulation by chemotherapeutics in malignant mesothelioma. PLoS ONE 2015, 10, e0145404. [Google Scholar] [CrossRef] [PubMed]
- Young, H.L.; Rowling, E.J.; Bugatti, M.; Giurisato, E.; Luheshi, N.; Arozarena, I.; Acosta, J.C.; Kamarashev, J.; Frederick, D.T.; Cooper, Z.A.; et al. An adaptive signaling network in melanoma inflammatory niches confers tolerance to mapk signaling inhibition. J. Exp. Med. 2017, 214, 1691–1710. [Google Scholar] [CrossRef] [PubMed]
- Voloshin, T.; Alishekevitz, D.; Kaneti, L.; Miller, V.; Isakov, E.; Kaplanov, I.; Voronov, E.; Fremder, E.; Benhar, M.; Machluf, M.; et al. Blocking il1beta pathway following paclitaxel chemotherapy slightly inhibits primary tumor growth but promotes spontaneous metastasis. Mol. Cancer Ther. 2015, 14, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.Y.; Liu, F.; Huang, G.L.; Liu, Y.; Shen, J.X.; Zhou, P.; Liu, W.M.; Shen, D.Y. Positive feedback loop of IL-1beta/Akt/RARalpha/Akt signaling mediates oncogenic property of raralpha in gastric carcinoma. Oncotarget 2017, 8, 6718–6729. [Google Scholar] [PubMed]
- Huang, F.Y.; Chan, A.O.; Lo, R.C.; Rashid, A.; Wong, D.K.; Cho, C.H.; Lai, C.L.; Yuen, M.F. Characterization of interleukin-1beta in helicobacter pylori-induced gastric inflammation and DNA methylation in interleukin-1 receptor type 1 knockout (IL-1R1-/-) mice. Eur. J. Cancer 2013, 49, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Sauter, K.A.; Wood, L.J.; Wong, J.; Iordanov, M.; Magun, B.E. Doxorubicin and daunorubicin induce processing and release of interleukin-1beta through activation of the nlrp3 inflammasome. Cancer Biol. Ther. 2011, 11, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Smith, L.B.; Magun, E.A.; Engstrom, T.; Kelley-Howard, K.; Jandhyala, D.M.; Thorpe, C.M.; Magun, B.E.; Wood, L.J. Small molecule kinase inhibitors block the ZAK-dependent inflammatory effects of doxorubicin. Cancer Biol. Ther. 2013, 14, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulos, C.; El Sanadi, C.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1beta via caspase-8 in dendritic cells. J. Immunol. 2013, 191, 4789–4803. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Faubel, S.; Edelstein, C.L. A pan caspase inhibitor decreases caspase-1, IL-1alpha and IL-1beta, and protects against necrosis of cisplatin-treated freshly isolated proximal tubules. Ren. Fail. 2015, 37, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, F.; Cao, X.; Zhai, Z.; GangHuang; Du, X.; Wang, Y.; Zhang, J.; Huang, Y.; Zhao, J.; et al. P2X7 receptor blockade protects against cisplatin-induced nephrotoxicity in mice by decreasing the activities of inflammasome components, oxidative stress and caspase-3. Toxicol. Appl. Pharmacol. 2014, 281, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Wu, C.; Gao, F.; Xiang, H.; Sun, N.; Peng, P.; Li, J.; Yuan, X.; Li, H.; Meng, X.; et al. Activation of NLRP3 inflammasome in peripheral nerve contributes to paclitaxel-induced neuropathic pain. Mol. Pain 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Ledeboer, A.; Jekich, B.M.; Sloane, E.M.; Mahoney, J.H.; Langer, S.J.; Milligan, E.D.; Martin, D.; Maier, S.F.; Johnson, K.W.; Leinwand, L.A.; et al. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav. Immun. 2007, 21, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin b release in myeloid-derived suppressor cells activates the nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Galluzzi, L.; Kroemer, G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat. Immunol. 2012, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Sota, J.; Vitale, A.; Insalaco, A.; Sfriso, P.; Lopalco, G.; Emmi, G.; Cattalini, M.; Manna, R.; Cimaz, R.; Priori, R.; et al. Safety profile of the interleukin-1 inhibitors anakinra and canakinumab in real-life clinical practice: A nationwide multicenter retrospective observational study. Clin. Rheumatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Anasagasti, M.J.; Olaso, E.; Calvo, F.; Mendoza, L.; Martin, J.J.; Bidaurrazaga, J.; Vidal-Vanaclocha, F. Interleukin 1-dependent and -independent mouse melanoma metastases. J. Natl. Cancer Inst. 1997, 89, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.T.; Bu, L.L.; Zhao, Y.Y.; Mao, L.; Deng, W.W.; Wu, T.F.; Zhang, W.F.; Sun, Z.J. CTLA4 blockade reduces immature myeloid cells in head and neck squamous cell carcinoma. Oncoimmunology 2016, 5, e1151594. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.W.; Mao, L.; Yu, G.T.; Bu, L.L.; Ma, S.R.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology 2016, 5, e1239005. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.R.; Leon, R.P.; Hall, M.K.; Schwertfeger, K.L. Interleukin-1beta and fibroblast growth factor receptor 1 cooperate to induce cyclooxygenase-2 during early mammary tumourigenesis. Breast Cancer Res. 2009, 11, R21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Guo, S.; Gonzalez-Perez, R.R. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br. J. Cancer 2011, 104, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and notch interleukin-1 leptin crosstalk outcome in cancer. World J. Methodol. 2016, 6, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via nlrp3/IL-1beta. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anti-Tumor Agent | Doses | IL-1β Enhancement Via | Effects | IL-1β Production | Incubation | Cell Type | Detection Method | Ref | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene Expression | Inflamma- Some | ? | Synergistic | Additive | Amounts | Pro-IL-1β | IL-1β | ||||||
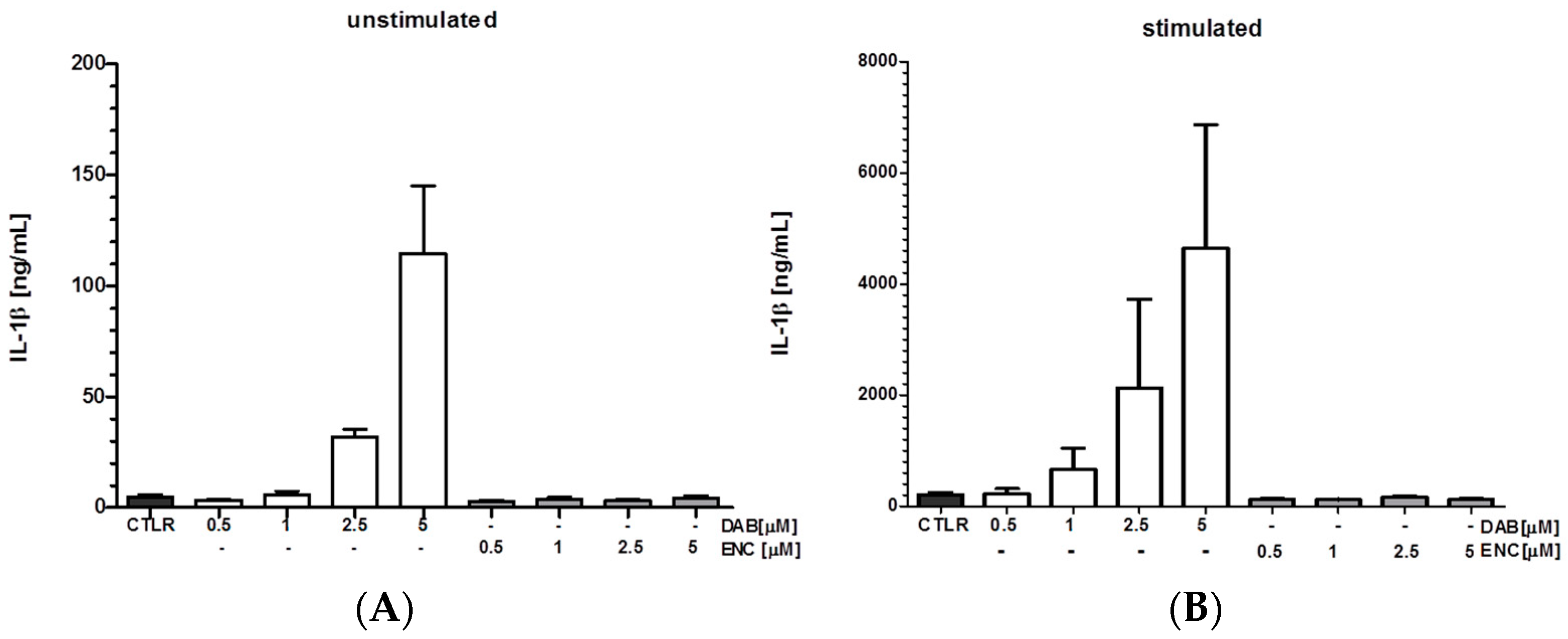
| Dabrafenib | 0.1; 1; 2.5; 5 μM | x | NLRC4 | - | - | MTX | ~10 ng/mL | 24 h | hDC, mBMDM, splenic mye. cell pop | qPCR, FACS | [6] | ||
| Vemurafenib | 0.1; 1; 2.5; 5 μM | - | NLRC5 | - | - | - | ~1 ng/mL | 24 h | mBMDM, splenic DC | qPCR, FACS | [6] | ||
| Doxorubicin | 2.5; 10; 25; 100 μM | - | NLRP3 | - | - | - | max. 700 pg (10 μM) | 8 h | mBMDM | ELISA | [281] | ||
| 5 μM | - | NLRP3 | - | - | - | ↑↑↑ | 18 h | mBMDM | WB | [273] | |||
| 5 μM | - | - | x | - | - | ~45 fold | x | 2, 12, 24 h | mBMDM, mouse blood | FACS, qPCR | [282] | ||
| 10 μM | - | NLRP3 | - | - | - | 6.5 pg/mL IL-1β | 48 h | Hmeso/H2373 MM cells | ELISA/WB | [276] | |||
| 5 μM | - | NLRP3 | - | - | - | ~2300 pg/mL IL-1β | x | 2, 2–18 h | mBMDC | ELISA/WB | [283] | ||
| Daunorubicin | 0.1; 0.25; 1; 2.5 μM | - | NLRP3 | - | - | - | 230 pg/mL | 8 h | mBMDM | ELISA | [281] | ||
| Melphalan | 2.8 μM | - | NLRP3 | - | - | - | ↑↑↑ | 18 h | mBMDM | WB | [273] | ||
| Cisplatin | 17 μM | - | NLRP3 | - | - | - | ↑↑ | 18 h | mBMDM | WB | [273] | ||
| 100 μM | - | NLRP3 | - | - | - | 2.5 pg/mL IL-1β | 48 h | Hmeso/H2373MM cells | ELISA/WB | [276] | |||
| 50 μM | - | NLRP3 | - | - | - | 13.2 nmol/mg/min | ? | proximal tubulus cells | ELISA | [284] | |||
| 20 mg/kgKG I.p | - | x | - | - | ~13 pg/mg | 72 h | mouse kidney tissue | ELISA | [285] | ||||
| Vincrisitin | 0.4 μM | - | NLRP3 | - | - | - | ↑↑ | 18 h | mBMDM | WB | [273] | ||
| Etoposide | 33 μM | - | NLRP3 | - | - | - | ↑↑ | 18 h | mBMDM | WB | [273] | ||
| Paclitaxel | 1 μM | - | NLRP3 | - | - | - | ↑ | 18 h | mBMDM | WB | [273] | ||
| 200 nM/L 25/50 mg/kgKG | - | x | - | - | ~0.35 pg/mL | x | 24 h, 6, 12, 24, 48 h | blood | ELISA, RT-PCR | [278] | |||
| 2 mg/kg | - | NLRP3 | - | - | - | ~220%. | x | 1/2/3 weeks | macrophages | RT-PCR | [286] | ||
| 4 mg/kg | - | x | - | - | ~1.7 fold | x | 36 days | dorsal root ganglia | RT-PCR | [287] | |||
| Methotrexate | 2.3 μM | - | NLRP3 | - | - | - | ↑ | 18 h | mBMDM | WB | [273] | ||
| Cytarabine | 2 μM | - | NLRP3 | - | - | - | ↑ | 18 h | mBMDM | WB | [273] | ||
| 5-Flu | 1 μM | - | NLRP3 | - | - | - | ~60 pg/mL | 24, 48, 72 h | mMDSC | ELISA | [288] | ||
| Gemcitabine | 27 nM | - | NLRP3 | - | - | - | ~70 pg/mL | 24, 48, 72 h | mMDSC | ELISA | [288] | ||
| Doxo. & Vincr | 5 μM & 0.4 μM | - | NLRP3 | - | + | - | ↑↑↑ | 18 h | mBMDM | WB | [273] | ||
| Cispl. & Vincr | 17 μM & 0.4 μM | - | NLRP3 | - | - | + | ↑↑ | 18 h | mBMDM | WB | [273] | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta—A Friend or Foe in Malignancies? Int. J. Mol. Sci. 2018, 19, 2155. https://doi.org/10.3390/ijms19082155
Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 Beta—A Friend or Foe in Malignancies? International Journal of Molecular Sciences. 2018; 19(8):2155. https://doi.org/10.3390/ijms19082155
Chicago/Turabian StyleBent, Rebekka, Lorna Moll, Stephan Grabbe, and Matthias Bros. 2018. "Interleukin-1 Beta—A Friend or Foe in Malignancies?" International Journal of Molecular Sciences 19, no. 8: 2155. https://doi.org/10.3390/ijms19082155
APA StyleBent, R., Moll, L., Grabbe, S., & Bros, M. (2018). Interleukin-1 Beta—A Friend or Foe in Malignancies? International Journal of Molecular Sciences, 19(8), 2155. https://doi.org/10.3390/ijms19082155