Multi-Acting Mitochondria-Targeted Platinum(IV) Prodrugs of Kiteplatin with α-Lipoic Acid in the Axial Positions


 ,
,  ,
, 

Abstract
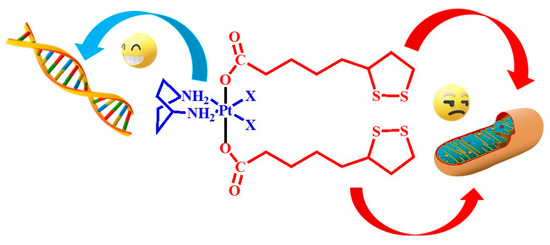
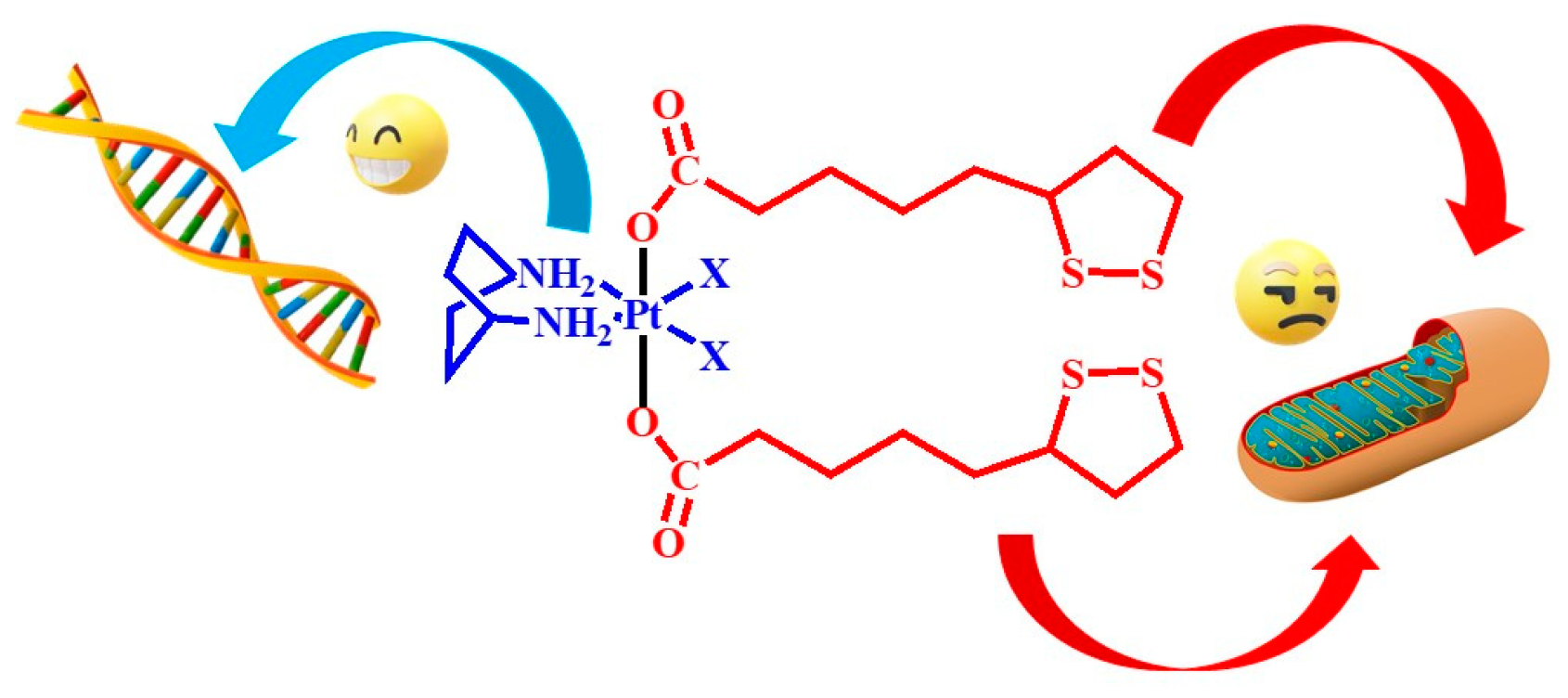
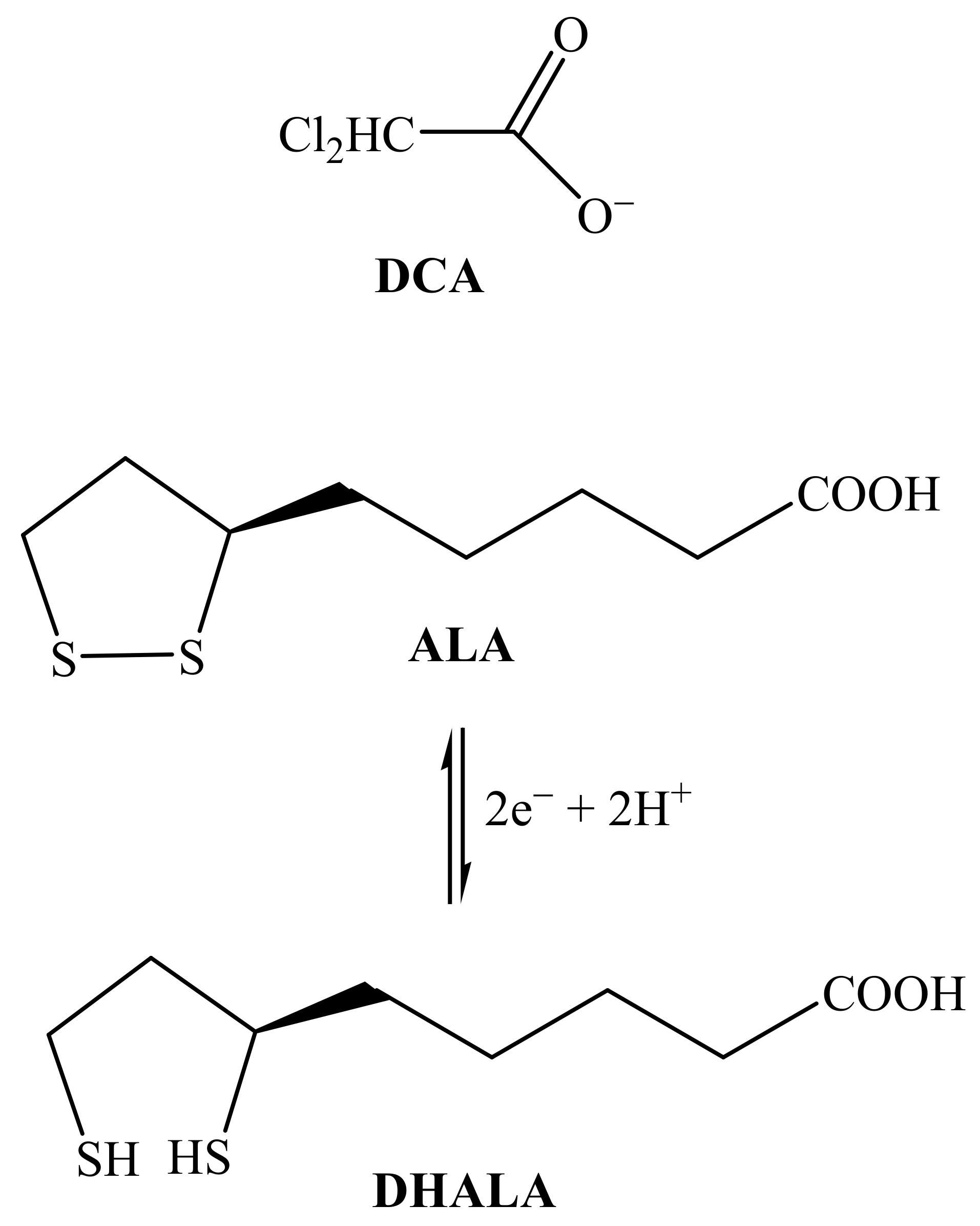
1. Introduction
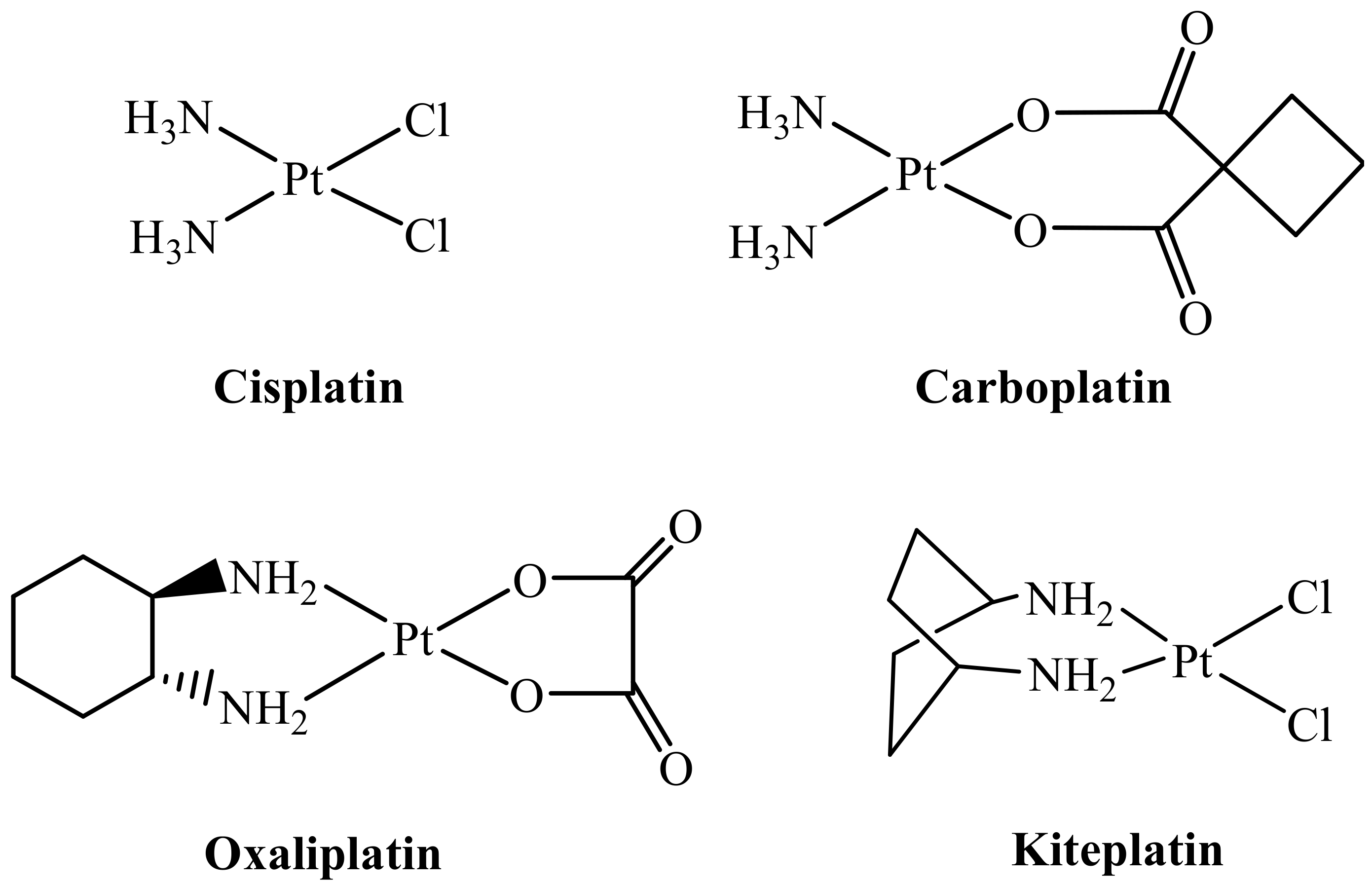
2. Results and Discussion
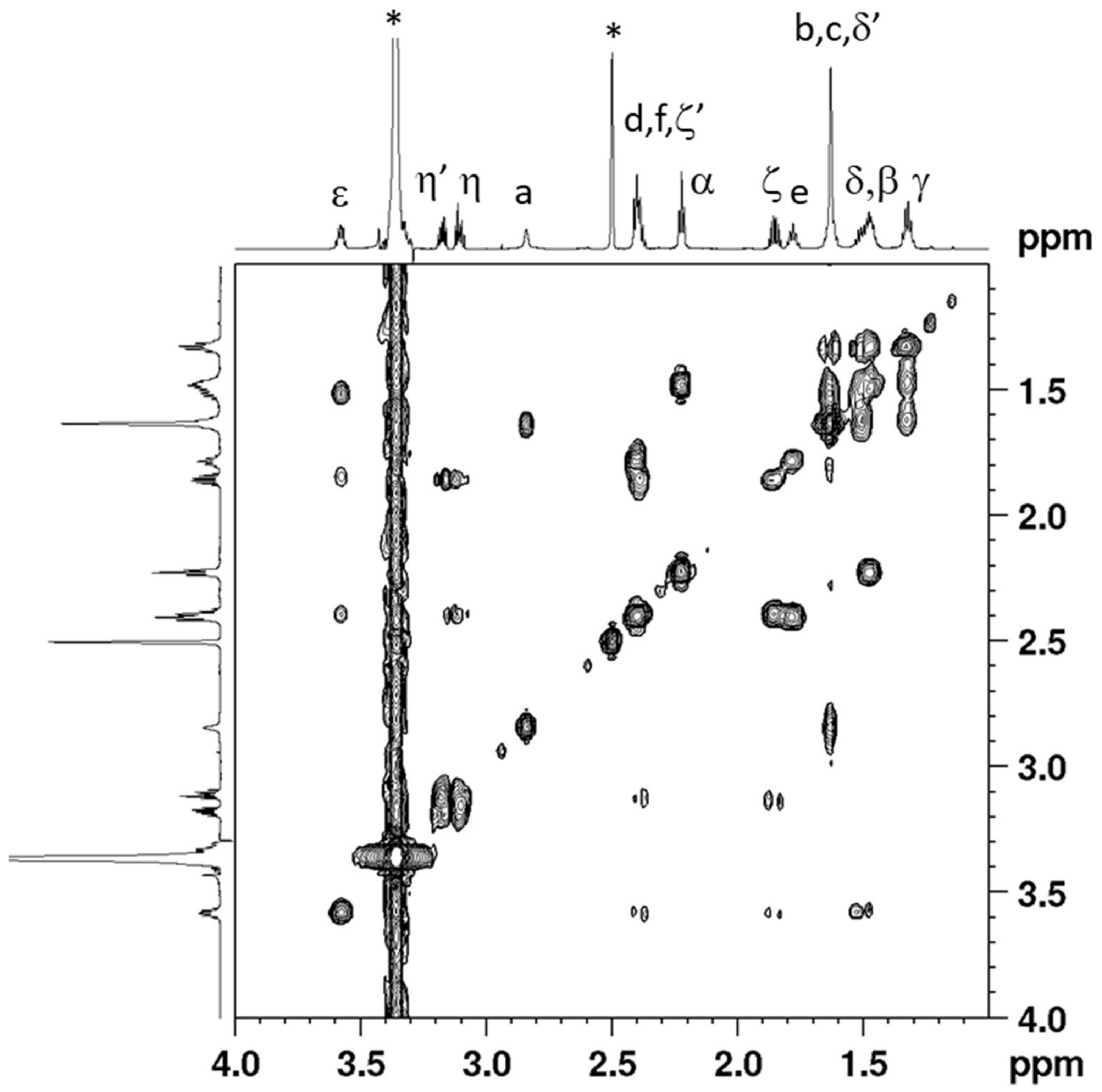
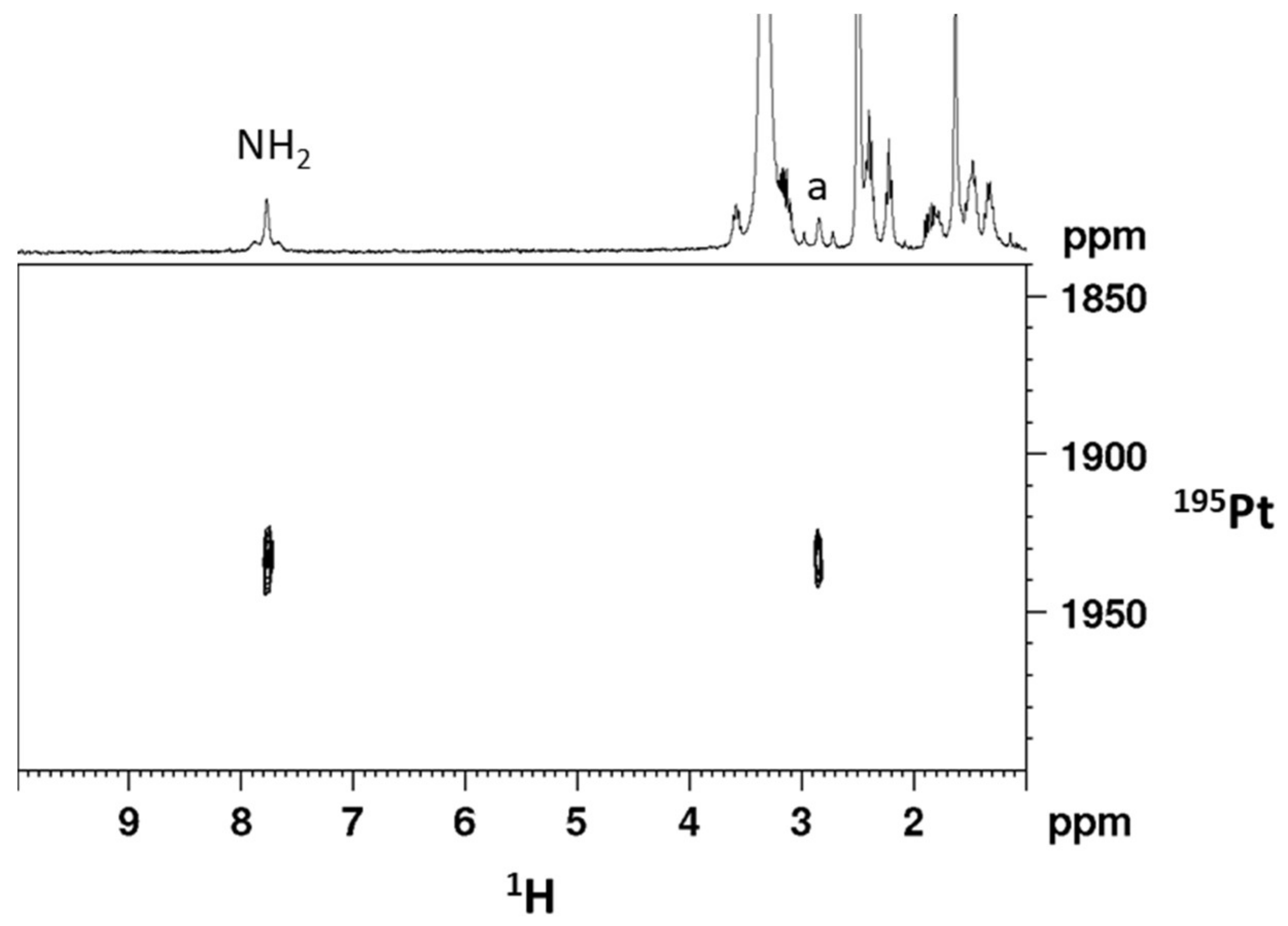
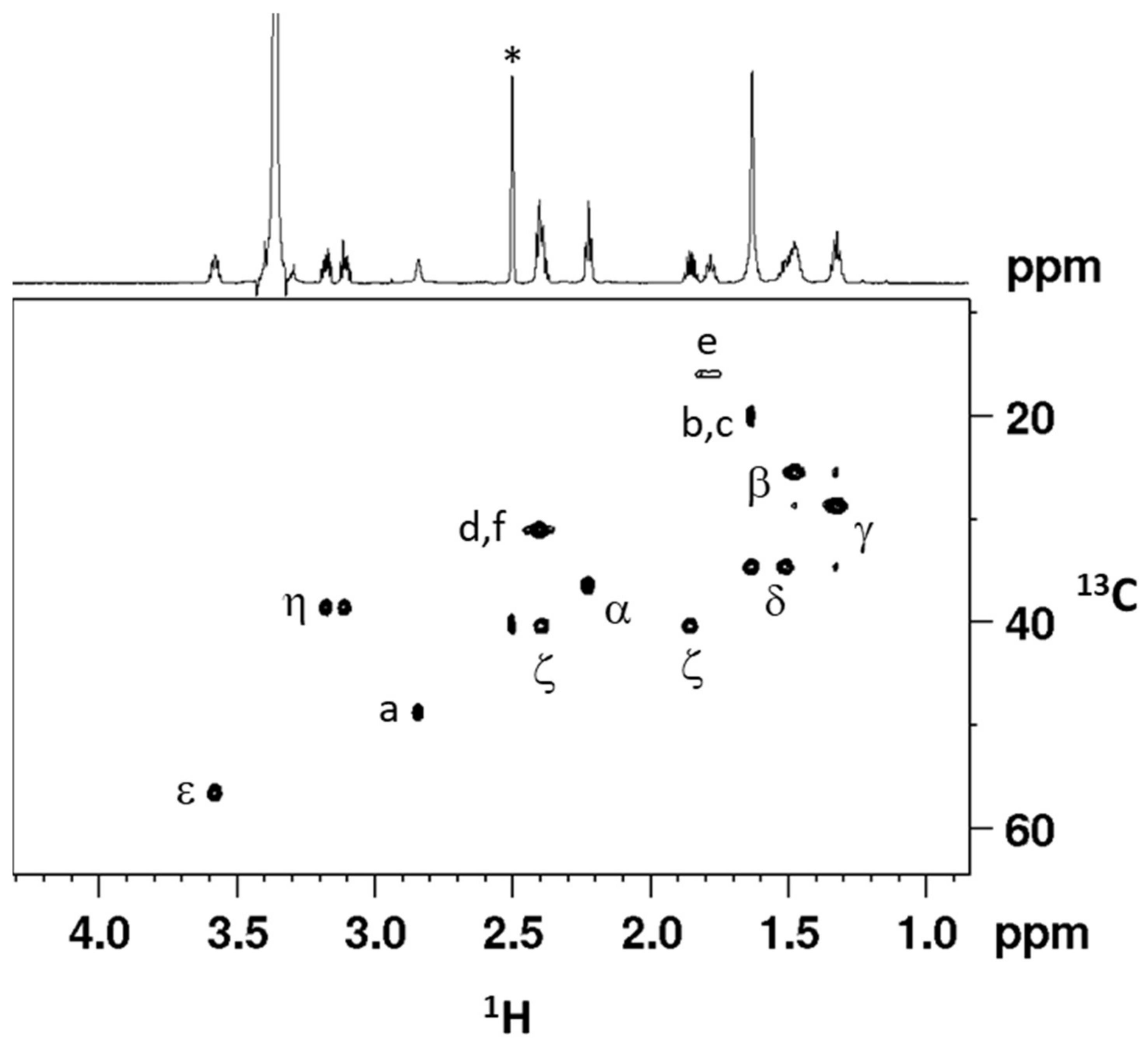
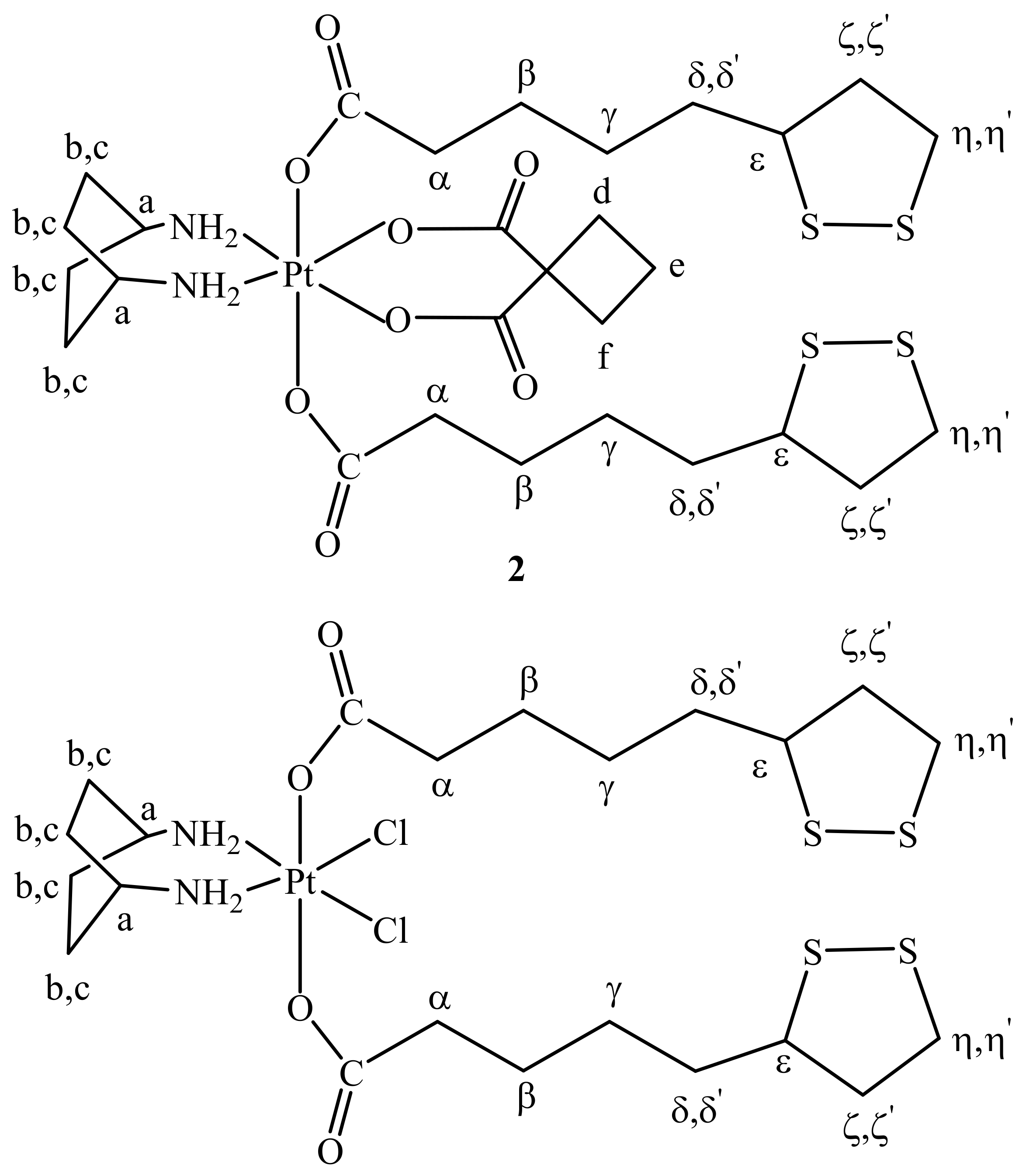
2.1. Synthesis and Characterization
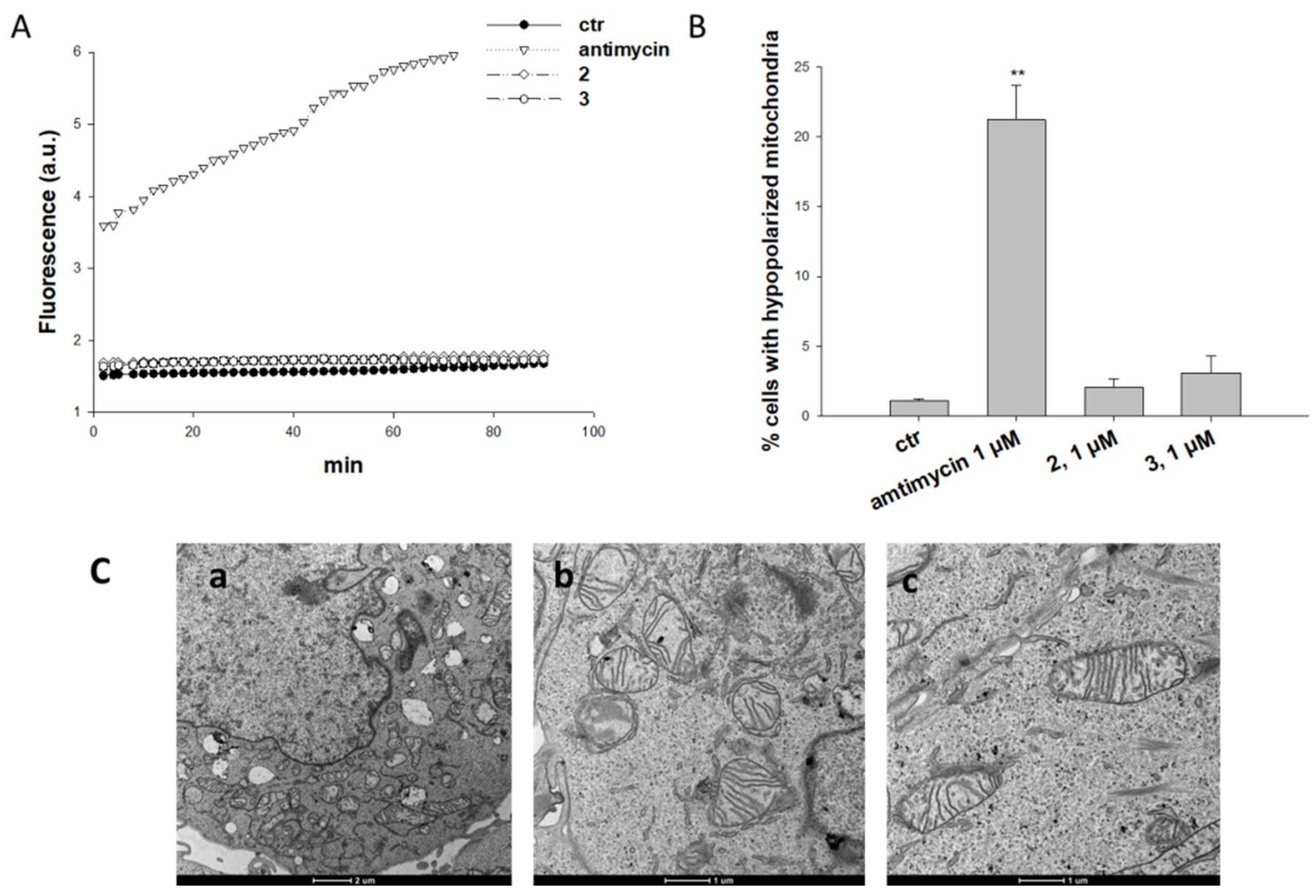
2.2. Biological Assays
3. Materials and Methods
3.1. Chemicals, Instrumentation, and Platinum Complexes Precursors
3.2. Synthesis of cis,trans,cis-[Pt(CBDCA)(OH)2(cis-1,4-DACH)] (1)
3.3. Synthesis of cis,trans,cis-[Pt(CBDCA)(ALA)2(cis-1,4-DACH)] (ALA = (±)-α-Lipoic Acid) (2)
3.4. Synthesis of cis,trans,cis-[PtCl2(ALA)2(cis-1,4-DACH)] (3)
3.5. Experiments with Cultured Human Cells
3.6. Cell Culture Studies
3.7. Cytotoxicity Assays
3.8. Spheroid Cultures
3.9. ROS Production
3.10. Mitochondrial Membrane Potential (ΔΨ)
3.11. Transmission Electron Microscopy (TEM) Analyses
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALA | (±)-α-Lipoic acid |
| CBDCA | 1,1-cyclobutanedicarboxylate |
| CCCP | Carbonylcyanide m-chlorophenyl hydrazone |
| CDDP | cisplatin |
| CM–H2DCFDA | 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester |
| COSY | correlation spectroscopy |
| DCA | dichloroacetate |
| DCC | dicyclohexylcarbodiimmide |
| DACH | diaminocyclohexane |
| DHALA | dihydro α-lipoic acid |
| DCU | dicyclohexylurea |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | dimethylsulfoxide |
| ESI-MS | Electrospray Ionisation Mass Spectrometry |
| FCS | Fetal calf serum |
| HSQC | Heteronuclear single quantum coherence spectroscopy |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| OXP | oxaliplatin |
| PBS | phosphate buffer saline |
| ROS | reactive oxygen species |
| RPMI | Roswell Park Memorial Institute |
| SDS | sodium dodecyl sulfate |
| TEM | Transmission electron microscopy |
References
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Ueber den Stoffwechsel der Carcinomzelle. Biochem. Z. 1924, 152, 309–344. [Google Scholar]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.G.; Sidhu, S.; Patel, M.S. R-lipoic acid inhibits mammalian pyruvate dehydrogenase kinase. Free Radic. Res. 2004, 38, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Novotny, L.; Rauko, P.; Cojocel, C. Alpha-Lipoic acid: The potential for use in cancer therapy. Neoplasma 2008, 55, 81–86. [Google Scholar] [PubMed]
- Bilska, A.; Włodek, L. Lipoic acid—The drug of the future? Pharmacol. Rep. 2005, 57, 570–577. [Google Scholar] [PubMed]
- Biewenga, G.P.; Haenen, G.R.; Bast, A. The pharmacology of the antioxidant lipoic acid. Gen. Pharmacol. 1997, 29, 315–331. [Google Scholar] [CrossRef]
- Shay, K.P.; Moreau, R.F.; Smith, E.J.; Smith, A.R.; Hagen, T.M. Alpha-lipoic acid as a dietary supplement: Molecular mechanisms and therapeutic potential. Biochim. Biophys. Acta 2009, 1790, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Handelman, G.J.; Han, D.; Tritschler, H.; Packer, L. Alpha-lipoic acid reduction by mammalian cells to the dithiol form, and release into the culture medium. Biochem. Pharmacol. 1994, 47, 1725–1730. [Google Scholar] [CrossRef]
- Packer, L.; Witt, E.H.; Tritschler, H.J. Alpha-Lipoic acid as a biological antioxidant. Free Radic. Biol. Med. 1995, 19, 227–250. [Google Scholar] [CrossRef]
- Gackowski, D.; Banaszkiewicz, Z.; Rozalski, R.; Jawien, A.; Olinski, R. Persistent oxidative stress in colorectal carcinoma patients. Int. J. Cancer 2002, 101, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Van de Mark, K.; Chen, J.S.; Steliou, K.; Perrine, S.P.; Faller, D.V. α-Lipoic acid induces p27Kip-dependent cell cycle arrest in non-transformed cell lines and apoptosis in tumor cell lines. J. Cell. Physiol. 2003, 194, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Sashwati, R.; Packer, L. Fas mediated apoptosis of human Jurkat T-cells: Intracellular events and potentiation by redox-active α-lipoic acid. Cell Death Differ. 1999, 6, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.H.; Campbell, J.S.; Stephenson, A.B.; Franklin, C.C.; Chaisson, M.; Poot, M.; Kavanagh, T.J.; Rabinovitch, P.S.; Fausto, N. Disruption of redox homeostasis in tumor necrosis factor-induced apoptosis in a murine hepatocyte cell line. Am. J. Pathol. 2000, 157, 221–236. [Google Scholar] [CrossRef]
- Piotrowski, P.; Wierzbicka, K.; Smiałek, M. Neuronal death in the rat hippocampus in experimental diabetes and cerebral ischaemia treated with antioxidants. Folia Neuropathol. 2001, 39, 147–154. [Google Scholar] [PubMed]
- Wenzel, U.; Nickel, A.; Daniel, H. α-Lipoic acid induces apoptosis in human colon cancer cells by increasing mitochondrial respiration with a concomitant O2·− generation. Apoptosis 2005, 10, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Pack, R.A.; Hardy, K.; Madigan, M.C.; Hunt, N.H. Differential effects of the antioxidant alpha-lipoic acid on the proliferation of mitogen-stimulated peripheral blood lymphocytes and leukaemic T cells. Mol. Immunol. 2002, 38, 733–745. [Google Scholar] [CrossRef]
- Na, M.H.; Seo, E.Y.; Kim, W.K. Effects of alpha-lipoic acid on cell proliferation and apoptosis in MDA-MB-231 human breast cells. Nutr. Res. Pract. 2009, 3, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Lippert, B. Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug; Verlag Helvetica Chimica Acta: Zürich, Switzerland, 1999; pp. 29–69. ISBN 9783906390420. [Google Scholar]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Arnesano, F.; Natile, G. Mechanistic insight into the cellular uptake and processing of cisplatin 30 years after its approval by FDA. Coord. Chem. Rev. 2009, 253, 2070–2081. [Google Scholar] [CrossRef]
- Margiotta, N.; Savino, S.; Marzano, C.; Pacifico, C.; Hoeschele, J.D.; Gandin, V.; Natile, G. Cytotoxicity-boosting of kiteplatin by Pt(IV) prodrugs with axial benzoate ligands. J. Inorg. Biochem. 2016, 160, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, N.; Savino, S.; Denora, N.; Marzano, C.; Laquintana, V.; Cutrignelli, A.; Hoeschele, J.D.; Gandin, V.; Natile, G. Encapsulation of lipophilic kiteplatin Pt(IV) prodrugs in PLGA-PEG micelles. Dalton Trans. 2016, 45, 13070–13081. [Google Scholar] [CrossRef] [PubMed]
- Savino, S.; Gandin, V.; Hoeschele, J.D.; Marzano, C.; Natile, G.; Margiotta, N. Dual-acting antitumor Pt(IV) prodrugs of kiteplatin with dichloroacetate axial ligands. Dalton Trans. 2018, 47, 7144–7158. [Google Scholar] [CrossRef] [PubMed]
- Ranaldo, R.; Margiotta, N.; Intini, F.P.; Pacifico, C.; Natile, G. Conformer distribution in (cis-1,4-DACH)bis(guanosine-5′-phosphate) platinum(II) adducts: A reliable model for DNA adducts of antitumoral cisplatin. Inorg. Chem. 2008, 47, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Kasparkova, J.; Suchankova, T.; Halamikova, A.; Zerzankova, L.; Vrana, O.; Margiotta, N.; Natile, G.; Brabec, V. Cytotoxicity, cellular uptake, glutathione and DNA interactions of an antitumor large-ring PtII chelate complex incorporating the cis-1,4-diaminocyclohexane carrier ligand. Biochem. Pharmacol. 2010, 79, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, N.; Marzano, C.; Gandin, V.; Osella, D.; Ravera, M.; Gabano, E.; Platts, J.A.; Petruzzella, E.; Hoeschele, J.D.; Natile, G. Revisiting [PtCl2(cis-1,4-DACH)]: An underestimated antitumor drug with potential application to the treatment of oxaliplatin-refractory colorectal cancer. J. Med. Chem. 2012, 55, 7182–7192. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Malina, J.; Margiotta, N.; Natile, G.; Kasparkova, J. Thermodynamic and mechanistic insights into translesion DNA synthesis catalyzed by Y-family DNA polymerase across a bulky double-base lesion of an antitumor platinum drug. Chem. Eur. J. 2012, 18, 15439–15448. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cho, H.-J.; Sagong, B.; Kim, S.-J.; Lee, J.-T.; So, H.-S.; Lee, I.-K.; Kim, U.-K.; Lee, K.-Y.; Choo, Y.-S. Alpha-lipoic acid protects against cisplatin-induced ototoxicity via the regulation of MAPKs and proinflammatory cytokines. Biochem. Biophys. Res. Commun. 2014, 449, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Somani, S.M.; Husain, K.; Whitworth, C.; Trammell, G.L.; Malafa, M.; Rybak, L.P. Dose-dependent protection by lipoic acid against cisplatin-induced nephrotoxicity in rats: Antioxidant defense system. Pharmacol. Toxicol. 2008, 86, 234–241. [Google Scholar] [CrossRef]
- Feuerecker, B.; Pirsig, S.; Seidl, C.; Aichler, M.; Feuchtinger, A.; Bruchelt, G.; Senekowitsch-Schmidtke, R. Lipoic acid inhibits cell proliferation of tumor cells in vitro and in vivo. Cancer Biol. Ther. 2012, 13, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Siemeling, U.; Bretthauer, F.; Bruhn, C.; Fellinger, T.-P.; Tong, W.-L.; Chan, M.C.W. Gold Nanoparticles Bearing an α-Lipoic Acid-based Ligand Shell: Synthesis, Model Complexes and Studies Concerning Phosphorescent Platinum(II)-Functionalisation. Z. Naturforsch. B 2010, 65. [Google Scholar] [CrossRef]
- Lal, M.; Palepu, N. Platinum Compound. WO2004/006859, 22 January 2004. [Google Scholar]
- Ang, W.H.; Pilet, S.; Scopelliti, R.; Bussy, F.; Juillerat-Jeanneret, L.; Dyson, P.J. Synthesis and characterization of Platinum(IV) anticancer drugs with functionalized aromatic carboxylate ligands: Influence of the ligands on drug efficacies and uptake. J. Med. Chem. 2005, 48, 8060–8069. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Papa, E.; Luini, M.; Monti, E.; Gariboldi, M.B.; Ravera, M.; Gabano, E.; Gaviglio, L.; Osella, D. Antiproliferative Pt(IV) complexes: Synthesis, biological activity, and quantitative structure–activity relationship modeling. J. Biol. Inorg. Chem. 2010, 15, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Petruzzella, E.; Margiotta, N.; Ravera, M.; Natile, G. NMR investigation of the spontaneous thermal- and/or photoinduced reduction of trans dihydroxido Pt(IV) derivatives. Inorg. Chem. 2013, 52, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Pregosin, P.S. Platinum-195 nuclear magnetic resonance. Coord. Chem. Rev. 1982, 44, 247–291. [Google Scholar] [CrossRef]
- Gabano, E.; Marengo, E.; Bobba, M.; Robotti, E.; Cassino, C.; Botta, M.; Osella, D. 195Pt NMR spectroscopy: A chemometric approach. Coord. Chem. Rev. 2006, 250, 2158–2174. [Google Scholar] [CrossRef]
- Kim, J. Bin Three-dimensional tissue culture models in cancer biology. Semin. Cancer Biol. 2005, 15, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Shamsuddin, S.; Santillan, C.C.; Stark, J.L.; Whitmire, K.H.; Siddik, Z.H.; Khokhar, A.R. Synthesis, characterization, and antitumor activity of new platinum(IV) trans-carboxylate complexes: Crystal structure of [Pt(cis-1,4-DACH)trans-(acetate)2Cl2]. J. Inorg. Biochem. 1998, 71, 29–35. [Google Scholar] [CrossRef]
- Shamsuddin, S.; Takahashi, I.; Siddik, Z.H.; Khokhar, A.R. Synthesis, characterization, and antitumor activity of a series of novel cisplatin analogs with cis-1,4-diaminocyclohexane as nonleaving amine group. J. Inorg. Biochem. 1996, 61, 291–301. [Google Scholar] [CrossRef]
- Liu, F.; Wang, M.; Wang, Z.; Zhang, X. Polymerized surface micelles formed under mild conditions. Chem. Commun. 2006, 1610. [Google Scholar] [CrossRef] [PubMed]
- Curci, A.; Denora, N.; Iacobazzi, R.M.; Ditaranto, N.; Hoeschele, J.D.; Margiotta, N.; Natile, G. Synthesis, characterization, and in vitro cytotoxicity of a Kiteplatin-Ibuprofen Pt(IV) prodrug. Inorganica Chim. Acta 2018, 472, 221–228. [Google Scholar] [CrossRef]
- Savino, S.; Denora, N.; Iacobazzi, R.M.; Porcelli, L.; Azzariti, A.; Natile, G.; Margiotta, N. Synthesis, characterization, and cytotoxicity of the first oxaliplatin Pt(IV) derivative having a TSPO ligand in the axial position. Int. J. Mol. Sci. 2016, 17, 1010. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) ± S.D. | |||||
|---|---|---|---|---|---|---|
| 2008 | MCF-7 | A431 | HCT-15 | H157 | Average | |
| [Pt(CBDCA)(cis-1,4-DACH)] | 25.47 ± 4.11 | 20.58 ± 3.15 | 13.56 ± 2.18 | 30.58 ± 5.25 | 19.21 ± 4.15 | 21.9 |
| 2 | 3.15 ± 0.85 | 3.05 ± 0.89 | 2.25 ± 0.52 | 2.31 ± 0.56 | 3.44 ± 0.97 | 2.8 |
| 3 | 0.34 ± 0.09 | 1.12 ± 0.57 | 0.10 ± 0.04 | 0.61 ± 0.15 | 0.99 ± 0.31 | 0.6 |
| CDDP | 2.22 ± 1.02 | 10.58 ± 0.82 | 2.10 ± 0.87 | 15.28 ± 2.63 | 2.12 ± 0.89 | 6.5 |
| OXP | 1.65 ± 0.46 | 4.52 ± 0.95 | 3.71 ± 0.76 | 1.15 ± 0.43 | 5.99 ± 1.85 | 3.4 |
| Kiteplatin | 1.89 ± 1.04 | 3.10 ± 1.42 | 3.95 ± 1.11 | 2.66 ± 0.95 | 2.08 ± 0.66 | 2.7 |
| IC50 (µM) ± S.D. | ||||||
|---|---|---|---|---|---|---|
| [Pt(CBDCA)(cis-1,4-DACH)] | 2 | 3 | Kiteplatin | CDDP | OXP | |
| A431 | 98.9 ± 6.8 | 58.4 ± 3.8 | 30.2 ± 5.1 | 65.2 ± 5.8 | 71.1 ± 3.9 | 65.3 ± 5.2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savino, S.; Marzano, C.; Gandin, V.; Hoeschele, J.D.; Natile, G.; Margiotta, N. Multi-Acting Mitochondria-Targeted Platinum(IV) Prodrugs of Kiteplatin with α-Lipoic Acid in the Axial Positions. Int. J. Mol. Sci. 2018, 19, 2050. https://doi.org/10.3390/ijms19072050
Savino S, Marzano C, Gandin V, Hoeschele JD, Natile G, Margiotta N. Multi-Acting Mitochondria-Targeted Platinum(IV) Prodrugs of Kiteplatin with α-Lipoic Acid in the Axial Positions. International Journal of Molecular Sciences. 2018; 19(7):2050. https://doi.org/10.3390/ijms19072050
Chicago/Turabian StyleSavino, Salvatore, Cristina Marzano, Valentina Gandin, James D. Hoeschele, Giovanni Natile, and Nicola Margiotta. 2018. "Multi-Acting Mitochondria-Targeted Platinum(IV) Prodrugs of Kiteplatin with α-Lipoic Acid in the Axial Positions" International Journal of Molecular Sciences 19, no. 7: 2050. https://doi.org/10.3390/ijms19072050
APA StyleSavino, S., Marzano, C., Gandin, V., Hoeschele, J. D., Natile, G., & Margiotta, N. (2018). Multi-Acting Mitochondria-Targeted Platinum(IV) Prodrugs of Kiteplatin with α-Lipoic Acid in the Axial Positions. International Journal of Molecular Sciences, 19(7), 2050. https://doi.org/10.3390/ijms19072050