The Molecular Mechanisms of Mutations in Actin and Myosin that Cause Inherited Myopathy


Abstract
1. Introduction
2. From Clinic to Fundamental Studies
3. How Mutations Cause Abnormal Function
4. How We Can Investigate Mutations That Cause Myopathies
- -
- Thick and thin filament assembly from monomers
- -
- Force transmission from cross bridge to Z-band
- -
- Force generation by contractile motor function
- -
- Regulation of contractility
5. From a Molecular Phenotype to an Explanation of Disease
5.1. MYH7 R403Q HCM Mutation Reduces Contractility
5.2. MYBPC3 HCM Mutations Are Mild and Late Onset
5.3. DCM Mutations Cause a Decrease in Ca2+-Sensitivity
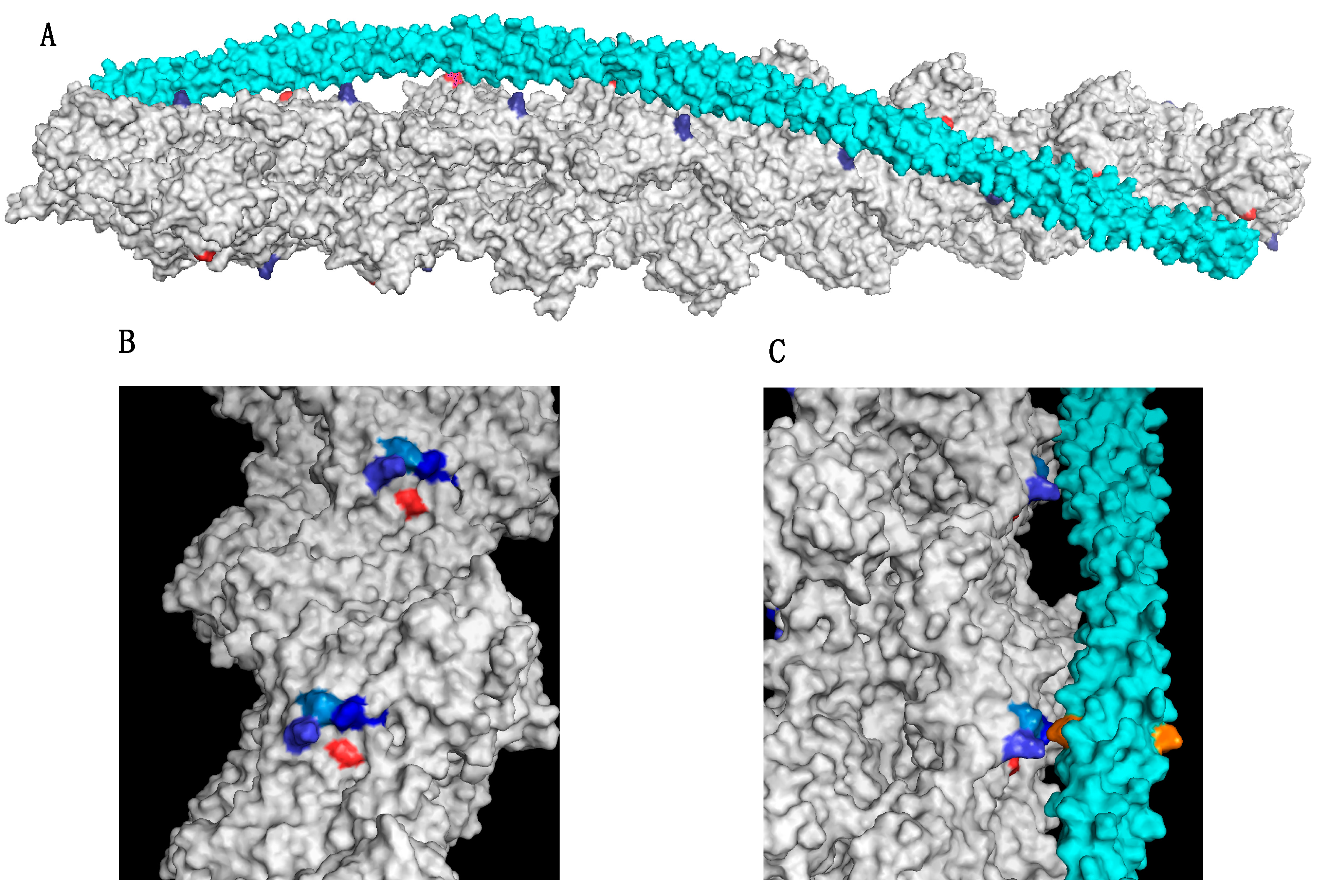
5.4. Skeletal Muscle Myopathy Phenotype due to ACTA1 Mutations Can Be Predicted from Actin’s Structure
6. A Triad System
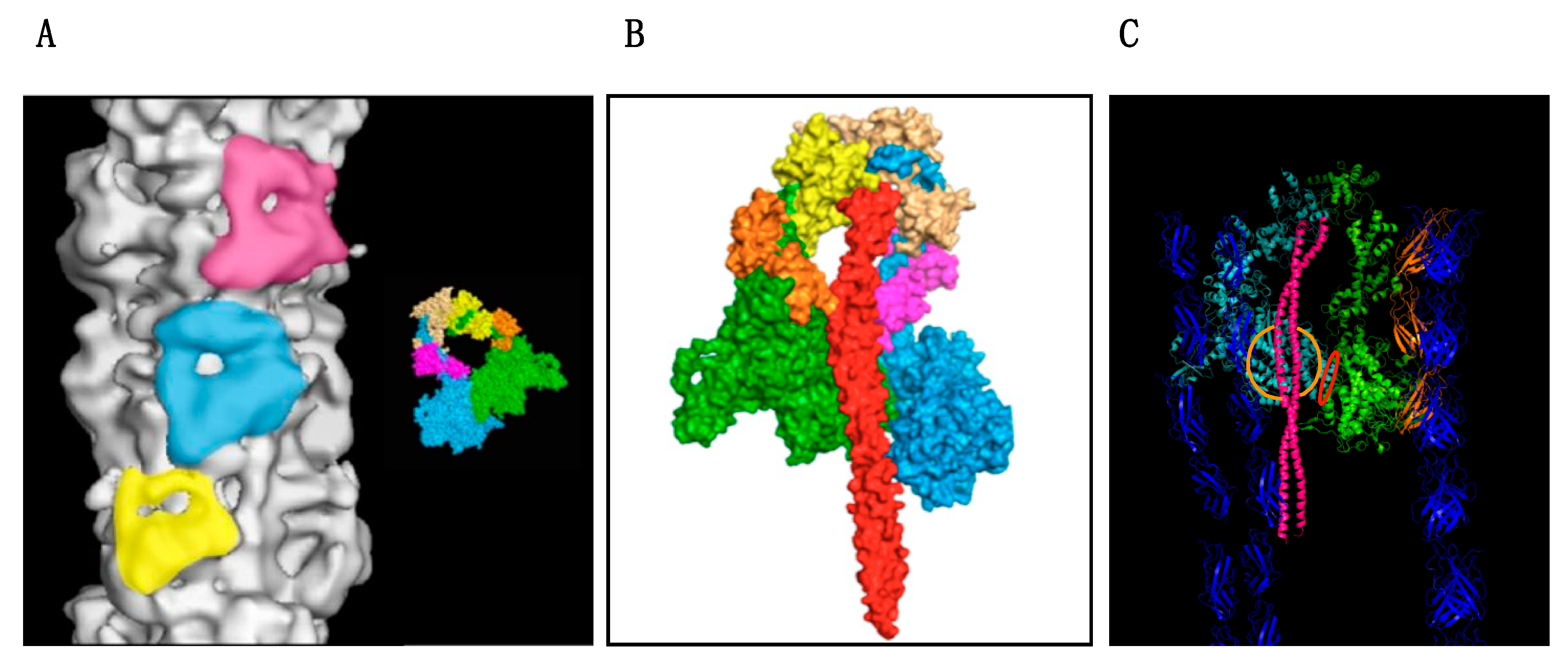
7. The Myosin Mesa and Interacting Heads Motif Model of HCM
The Interacting Heads Motif (IHM)
8. Unifying Hypothesis for Myosin and MyBP-C mutations Causing HCM
Acknowledgments
Conflicts of Interest
References
- Geeves, M.A.; Hitchcock-DeGregori, S.E.; Gunning, P.W. A systematic nomenclature for mammalian tropomyosin isoforms. J. Muscle Res. Cell Motil. 2015, 36, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, E.S.; Drummond, D.R.; Sparrow, J.C. Molecular genetics of actin function. Biochem. J. 1993, 286, 657–671. [Google Scholar] [CrossRef]
- Tajsharghi, H.; Oldfors, A. Myosinopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Laing, N.G.; Dye, D.E.; Wallgren-Pettersson, C.; Richard, G.; Monnier, N.; Lillis, S.; Winder, T.L.; Lochmüller, H.; Graziano, C.; Mitrani-Rosenbaum, S.; Twomey, D.; et al. Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1). Hum. Mutat. 2009, 30, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.B.; Marston, S.B.; Redwood, C.S. Modulation of thin filament activation by breakdown or isoform switching of thin filament proteins: Physiological and pathological implications. Circ. Res. 2003, 93, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Colegrave, M.; Peckham, M. Structural Implications of beta-Cardiac Myosin Heavy Chain Mutations in Human Disease. Anat. Rec. (Hoboken) 2014, 297, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Geist, J.; Grogan, A.; Hu, L.-Y.R.; Kontrogianni-Konstantopoulos, A. Thick Filament Protein Network, Functions, and Disease Association; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Volume 282, pp. 631–709. [Google Scholar]
- Nowak, K.J.; Wattanasirichaigoon, D.; Goebel, H.H.; Wilce, M.; Pelin, K.; Donner, K.; Jacob, R.L.; Hubner, C.; Oexle, K.; Anderson, J.R.; et al. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat. Genet. 1999, 23, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Van Driest, S.L.; Jaeger, M.A.; Ommen, S.R.; Will, M.L.; Gersh, B.J.; Tajik, A.J.; Ackerman, M.J. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.G.; Seidman, C. The genetic basis for cardiomyopathy: From mutation identification to mechanistic paradigms. Cell 2001, 104, 557–567. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; McKenna, W.J. Hypertrophic cardiomyopathy. Lancet 2004, 363, 1881–1891. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, C.; Owen, T.; Lawal, S.; Cao, S.; Pandey, S.; Yang, H.Y.; Song, W.; Wilkinson, R.; Alvarez-Laviada, A.; Gehmlich, K.; et al. Age and strain related aberrant Ca(2+) release is associated with sudden cardiac death in the ACTC E99K mouse model of hypertrophic cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2017, 313, H1213–H1226. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Yang, X.P.; Carretero, O.A.; Nakagawa, P.; D’Ambrosio, M.; Leung, P.; Xu, J.; Peterson, E.L.; Gonzalez, G.E.; Harding, P.; et al. Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6J mice. Exp. Physiol. 2011, 96, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.F.; Ilkovski, B.; Cooper, S.; Valova, V.A.; Robinson, P.J.; Nonaka, I.; Feng, J.J.; Marston, S.; North, K. The pathogenesis of ACTA1-related congenital fiber type disproportion. Ann. Neurol. 2007, 61, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Dyer, E.; Stuckey, D.; Copeland, O.; Leung, M.; Bayliss, C.; Messer, A.E.; Wilkinson, R.; Tremoleda, J.; Schneider, M.; et al. Molecular mechanism of the Glu99lys mutation in cardiac actin (ACTC gene) that causes apical hypertrophy in man and mouse. J. Biol. Chem. 2011, 286, 27582–27593. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.B. How do mutations in contractile proteins cause the primary familial cardiomyopathies? J. Cardiovasc. Transl. Res. 2011, 4, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Nier, V.; Schultz, I.; Brenner, B.; Forssmann, W.; Raida, M. Variability in the ratio of mutant to wildtype myosin heavy chain present in the soleus muscle of patients with familial hypertrophic cardiomyopathy. A new approach for the quantification of mutant to wildtype protein. FEBS Lett. 1999, 461, 246–252. [Google Scholar] [CrossRef]
- Marston, S.; Copeland, O.; Gehmlich, K.; Schlossarek, S.; Carrrier, L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? J. Muscle Res. Cell Motil. 2011, 33, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Schlossarek, S.; Mearini, G.; Carrier, L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: Mechanisms and therapeutic opportunities. J. Mol. Cell Cardiol. 2011, 50, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.; Ravenscroft, G.; Jackaman, C.; Filipovska, A.; Davies, S.M.; Lim, E.M.; Squire, S.E.; Potter, A.C.; Baker, E.; Clément, S.; Sewry, C.; et al. Rescue of skeletal muscle alpha-actin-null mice by cardiac (fetal) alpha-actin. J. Cell Biol. 2009, 185, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.J.; Sewry, C.A.; Navarro, C.; Squier, W.; Reina, C.; Ricoy, J.R.; Jayawant, S.S.; Childs, A.-M.; Dobbie, J.A.; Appleton, R.E.; et al. Nemaline myopathy caused by absence of alpha-skeletal muscle actin. Ann. Neurol. 2007, 61, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Akkari, A.P.; Nowak, K.J.; Beckman, K.; Walker, K.R.; Schachat, F.; Laing, N.G. Production of human skeletal alpha-actin proteins by the baculovirus expression system. Biochem. Biophys. Res. Commun. 2003, 307, 74–79. [Google Scholar] [CrossRef]
- Joel, P.B.; Fagnant, P.M.; Trybus, K.M. Expression of a nonpolymerizable actin mutant in Sf9 cells. Biochemistry 2004, 43, 11554–11559. [Google Scholar] [CrossRef] [PubMed]
- Bookwalter, C.S.; Trybus, K.M. Functional consequences of a mutation in an expressed human alpha-cardiac actin at a site implicated in familial hypertrophic cardiomyopathy. J. Biol. Chem. 2006, 281, 16777–16784. [Google Scholar] [CrossRef] [PubMed]
- Debold, E.P.; Saber, W.; Cheema, Y.; Bookwalter, C.S.; Trybus, K.M.; Warshaw, D.M.; Vanburen, P. Human actin mutations associated with hypertrophic and dilated cardiomyopathies demonstrate distinct thin filament regulatory properties in vitro. J. Mol. Cell Cardiol. 2010, 48, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Matsson, H.; Eason, J.; Bookwalter, C.S.; Klar, J.; Gustavsson, P.; Sunnegardh, J.; Enell, H.; Jonzon, A.; Vikkula, M.; Gutierrez, I.; et al. Alpha-cardiac actin mutations produce atrial septal defects. Hum. Mol. Genet. 2007, 17, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Sommese, R.F.; Nag, S.; Sutton, S.; Miller, S.M.; Spudich, J.A.; Ruppel, K.M. Effects of Troponin T Cardiomyopathy Mutations on the Calcium Sensitivity of the Regulated Thin Filament and the Actomyosin Cross-Bridge Kinetics of Human β-Cardiac Myosin. PLoS ONE 2013, 8, e83403. [Google Scholar] [CrossRef] [PubMed]
- Resnicow, D.I.; Deacon, J.C.; Warrick, H.M.; Spudich, J.A.; Leinwand, L.A. Functional diversity among a family of human skeletal muscle myosin motors. Proc. Natl. Acad. Sci. USA 2010, 107, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Vikhorev, P.G.; Kashyap, M.N.; Rowlands, C.; Ferenczi, M.A.; Woledge, R.C.; MacLeod, K.; Marston, S.; Curtin, N.A. Mechanical and energetic properties of papillary muscle from ACTC E99K transgenic mouse models of hypertrophic cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 2013, 304, H1513–H1524. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Dyer, E.; Stuckey, D.; Leung, M.-C.; Memo, M.; Mansfield, C.; Ferenczi, M.; Liu, K.; Redwood, C.; Nowak, K.; et al. Investigation of a transgenic mouse model of familial dilated cardiomyopathy. J. Mol. Cell Cardiol. 2010, 49, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.-A.T.; Joya, J.E.; Kee, A.J.; Domazetovska, A.; Yang, N.; Hook, J.W.; Lemckert, F.A.; Kettle, E.; Valova, V.A.; Robinson, P.J.; et al. Hypertrophy and dietary tyrosine ameliorate the phenotypes of a mouse model of severe nemaline myopathy. Brain J. Neurol. 2011, 134 Pt 12, 3516–3529. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, G.; Jackaman, C.; Bringans, S.; Papadimitriou, J.M.; Griffiths, L.M.; McNamara, E.; Bakker, A.J.; Davies, K.E.; Laing, N.G.; Nowak, K.J. Mouse models of dominant ACTA1 disease recapitulate human disease and provide insight into therapies. Brain 2011, 134 Pt 4, 1101–1115. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F.; Chen, S.; Patel, R.; Tsybouleva, N.; Lutucuta, S.; Kopelen, H.A.; Zoghbi, W.A.; Quinones, M.A.; Roberts, R.; Marian, A.J. Evolution of expression of cardiac phenotypes over a 4-year period in the beta-myosin heavy chain-Q403 transgenic rabbit model of human hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2004, 36, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.M.; Palmer, B.M.; Wang, Y.; Wang, Y.; Teekakirikul, P.; Teekakirikul, P.; Hinson, J.T.; Hinson, J.T.; Fatkin, D.; Fatkin, D.; et al. Myofilament mechanical performance is enhanced by R403Q myosin in mouse myocardium independent of sex. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H1939–H1947. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Kazmierczak, K.; Rojas, A.I.; Wang, Y.; Szczesna-Cordary, D. The R21C Mutation in Cardiac Troponin I Imposes Differences in Contractile Force Generation between the Left and Right Ventricles of Knock-In Mice. BioMed. Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Dumka, D.; Talent, J.; Akopova, I.; Guzman, G.; Szczesna-Cordary, D.; Borejdo, J. E22K mutation of RLC that causes familial hypertrophic cardiomyopathy in heterozygous mouse myocardium: Effect on cross-bridge kinetics. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H2098–H2106. [Google Scholar] [CrossRef] [PubMed]
- Kerrick, W.G.; Kazmierczak, K.; Xu, Y.; Wang, Y.; Szczesna-Cordary, D. Malignant familial hypertrophic cardiomyopathy D166V mutation in the ventricular myosin regulatory light chain causes profound effects in skinned and intact papillary muscle fibers from transgenic mice. FASEB J. 2009, 23, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Gollapudi, S.K.; Tardiff, J.C.; Chandra, M. The functional effect of dilated cardiomyopathy mutation (R144W) in mouse cardiac troponin T is differently affected by alpha- and beta-myosin heavy chain isoforms. Am. J. Physiol.-Heart Circ. Physiol. 2015, 308, H884–H893. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.; Mirza, M.; Abdulrazzak, H.; Sewry, C. Functional characterisation of a mutant actin (Met132Val) from a patient with nemaline myopathy. Neuromuscul. Disord. 2004, 14, 167–174. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.; Graziano, C.; Pacileo, G.; Petrini, S.; Nowak, K.J.; Boldrini, R.; Jacques, A.; Feng, J.J.; Porfirio, B.; Sewry, C.A.; et al. Fatal hypertrophic cardiomyopathy and nemaline myopathy associated with ACTA1 K336E mutation. Neuromuscul. Disord. 2006, 16, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Jayawant, S.; Squier, W.; Muntoni, F.; Sewry, C.A.; Manzur, A.; Quinlivan, R.; Lillis, S.; Jungbluth, H.; Sparrow, J.C.; et al. Nemaline myopathy with stiffness and hypertonia associated with an ACTA1 mutation. Neurology 2012, 78, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Jacques, A.; Briceno, N.; Messer, A.; Gallon, C.; Jalizadeh, S.; Garcia, E.; Kikonda-Kanda, G.; Goddard, J.; Harding, S.; Watkins, H.; et al. The molecular phenotype of human cardiac myosin associated with hypertrophic obstructive cardiomyopathy. Cardiovasc. Res. 2008, 79, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Jacques, A.; Hoskins, A.; Kentish, J.; Marston, S.B. From genotype to phenotype: A longitudinal study of a patient with hypertrophic cardiomyopathy due to a mutation in the MYBPC3 gene. J. Muscle Res. Cell Motil. 2009, 29, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.; Montgiraud, C.; Munster, A.B.; Copeland, O.; Choi, O.; Dos Remedios, C.; Messer, A.E.; Ehler, E.; Knoll, R. OBSCN Mutations Associated with Dilated Cardiomyopathy and Haploinsufficiency. PLoS ONE 2015, 10, e0138568. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Smoktunowicz, N.; Munster, A.B.; Copeland, O.; Kostin, S.; Montgiraud, C.; Messer, A.E.; Toliat, M.R.; Li, A.; Remedios, C.G.; et al. Abnormal contractility in human heart myofibrils from patients with dilated cardiomyopathy due to mutations in TTN and contractile protein genes. Nat. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.-H.; Biesiadecki, B.J.; Ziolo, M.T.; Davis, J.P.; Janssen, P.M.L. Myofilament Calcium Sensitivity: Role in Regulation of In vivo Cardiac Contraction and Relaxation. Front. Physiol. 2016, 7 Pt 1, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Cuda, G.; Fananazapir, L.; Zhu, W.S.; Sellers, J.R.; Epstein, N.D. Skeletal muscle expression and abnormal function of beta-myosin in hypertrophic cardiopmyopathy. J. Clin. Investig. 1993, 91, 2861–2865. [Google Scholar] [CrossRef] [PubMed]
- Sata, M.; Ikebe, M. Functional analysis of the mutations in the human cardiac beta-myosin that are responsible for familial hypertrophic cardiomyopathy. Implication for the clinical outcome. J. Clin. Investig. 1996, 98, 2866–2873. [Google Scholar] [CrossRef] [PubMed]
- Fujita, H.; Sugiura, S.; Monomura, S.; Omata, M.; Sugi, H.; Sutoh, K. Characterization of mutant myosins of Dictiostelium discoideum equivalent to human familial hypertrophic cardiomyopathy. J. Clin. Investig. 1997, 99, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Palmiter, K.A.; Tyska, M.J.; Haeberle, J.R.; Alpert, N.R.; Fananapazir, L.; Warshaw, D.M. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J. Muscle Res. Cell Motil. 2000, 21, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Tyska, M.J.; Hayes, E.; Giewat, M.; Seidman, C.E.; Seidman, J.G.; Warshaw, D.M. Single molecule mechanics of R403Q cardiac myosin isolated from mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 2000, 86, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Belus, A.; Piroddi, N.; Scellini, B.; Tesi, C.; D'Amati, G.; Girolami, F.; Yacoub, M.; Cecchi, F.; Olivotto, I.; Poggesi, C. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. J. Physiol. 2008, 586 Pt 15, 3639–3644. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, S.; Ashley, E.; Leinwand, L.; Spudich, J. Insights into Human Œ≤-Cardiac Myosin Function from Single Molecule and Single Cell Studies. J. Cardiovasc. Transl. Res. 2009, 2, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, E.; Seidman, C.; Seidman, J.G.; LeWinter, M.; Maughan, D. Altered crossbridge kinetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 1999, 84, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Shimizu, M.; Ino, H.; Matsuyama, T.; Yamaguchi, M.; Terai, H.; Hayashi, K.; Mabuchi, T.; Kiyama, M.; Sakata, K.; et al. A novel missense mutation in the myosin binding protein-C gene is responsible for hypertrophic cardiomyopathy with left ventricular dysfunction and dilation in elderly patients. J. Am. Coll. Cardiol. 2003, 41, 781–786. [Google Scholar] [CrossRef]
- Fay, W.P.; Taliercio, C.P.; Ilstrup, D.M.; Tajik, A.J.; Gersh, B.J. Natural history of hypertrophic cardiomyopathy in the elderly. J. Am. Coll. Cardiol. 1990, 16, 821–826. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Vasile, V.C.; Ommen, S.R.; Will, M.L.; Tajik, A.J.; Gersh, B.J.; Ackerman, M.J. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2004, 44, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Memo, M.; Leung, M.-C.; Ward, D.G.; dos Remedios, C.; Morimoto, S.; Zhang, L.; Ravenscroft, G.; McNamara, E.; Nowak, K.J.; Marston, S.B.; et al. Mutations in thin Filament Proteins that Cause Familial Dilated Cardiomyopathy Uncouple Troponin I Phosphorylation from Changes in Myofibrillar Ca2+-Sensitivity. Cardiovasc. Res. 2013, 99, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.; Marston, S.; Willott, R.; Ashley, C.; Mogensen, J.; McKenna, W.; Robinson, P.; Redwood, C.; Watkins, H. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J. Biol. Chem. 2005, 280, 28498–28506. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Griffiths, P.J.; Watkins, H.; Redwood, C.S. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 2007, 101, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Mirza, M.; Knott, A.; Abdulrazzak, H.; Willott, R.; Marston, S.; Watkins, H.; Redwood, C. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J. Biol. Chem. 2002, 277, 40710–40716. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.N.; Potter, J.D. Sarcomeric protein mutations in dilated cardiomyopathy. Heart Fail. Rev. 2005, 10, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Messer, A.; Marston, S. Investigating the role of uncoupling of Troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of Cardiomyopathy. Front. Physiol. 2014, 5, 315. [Google Scholar] [CrossRef] [PubMed]
- Du, C.K.; Morimoto, S.; Nishii, K.; Minakami, R.; Ohta, M.; Tadano, N.; Lu, Q.W.; Wang, Y.Y.; Zhan, D.Y.; Mochizuki, M.; et al. Knock-In Mouse Model of Dilated Cardiomyopathy Caused by Troponin Mutation. Circ. Res. 2007, 101, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Dyer, E.; Jacques, A.; Hoskins, A.; Ward, D.; Gallon, C.; Messer, A.; Kaski, J.; Burch, M.; Kentish, J.; Marston, S. Functional Analysis of a Unique Troponin C Mutation, Gly159Asp that Causes Familial Dilated Cardiomyopathy, Studied in Explanted Heart Muscle. Circ. Heart Fail. 2009, 2, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Biesiadecki, B.J.; Kobayashi, T.; Walker, J.S.; John Solaro, R.; de Tombe, P.P. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 2007, 100, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Lakdawala, N.K.; Dellefave, L.; Redwood, C.S.; Sparks, E.; Cirino, A.L.; Depalma, S.; Colan, S.D.; Funke, B.; Zimmerman, R.S.; Robinson, P.; et al. Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: The distinctive natural history of sarcomeric dilated cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Vikhorev, P.G.; Song, W.; Wilkinson, R.; Copeland, O.; Messer, A.E.; Ferenczi, M.A.; Marston, S.B. The dilated cardiomyopathy-causing mutation ACTC E361G in cardiac muscle myofibrils specifically abolishes modulation of Ca(2+) regulation by phosphorylation of troponin I. Biophys. J. 2014, 107, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.C.; Nowak, K.J.; Durling, H.J.; Beggs, A.H.; Wallgren-Pettersson, C.; Romero, N.; Nonaka, I.; Laing, N.G. Muscle disease caused by mutations in the skeletal muscle alpha-actin gene (ACTA1). Neuromuscul. Disord. 2003, 13, 519–531. [Google Scholar] [CrossRef]
- Feng, J.-J.; Marston, S. Genotype-phenotype correlations in ACTA1 mutations that cause congenital myopathies. Neuromuscul. Disord. 2009, 19, 6–16. [Google Scholar] [CrossRef] [PubMed]
- McKillop, D.F.A.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment-1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef]
- Vibert, P.; Craig, R.; Lehman, W. Steric-model for activation of muscle thin filamnets. J. Mol. Biol. 1997, 266, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.R.; Campbell, S.G.; Lehman, W. Structural determinants of muscle thin filament cooperativity. Arch Biochem. Biophys. 2016, 594, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Lehman, W. Switching Muscles On and Off in Steps: The McKillop-Geeves Three-State Model of Muscle Regulation. Biophys. J. 2017, 112, 2459–2466. [Google Scholar] [CrossRef] [PubMed]
- Razzaq, A. The Purification and in vitro Motility Analysis of Drosophila Melanogaster Act88F Mutant Actins. Ph.D. Thesis, University of York, New York, NY, USA, 1995. [Google Scholar]
- Bing, W.; Razzaq, A.; Sparrow, J.; Marston, S. Tropomyosin and troponin regulation of wild type and E93K mutant actin filaments from Drososhila flight muscle. Charge reversal on actin changes actin-tropomyosin from on to off state. J. Biol. Chem. 1998, 273, 15016–15021. [Google Scholar] [CrossRef] [PubMed]
- Li, X.E.; Tobacman, L.S.; Mun, J.Y.; Craig, R.; Fischer, S.; Lehman, W. Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys. J. 2011, 100, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Marston, S.; Memo, M.; Messer, A.; Papadaki, M.; Nowak, K.; McNamara, E.; Ong, R.; El-Mezgueldi, M.; Li, X.; Lehman, W. Mutations in repeating structural motifs of tropomyosin cause gain of function in skeletal muscle myopathy patients. Hum. Mol. Genet. 2013, 22, 4978–4987. [Google Scholar] [CrossRef] [PubMed]
- Donkervoort, S.; Papadaki, M.; de Winter, J.M.; Neu, M.B.; Kirschner, J.; Bolduc, V.; Yang, M.L.; Gibbons, M.A.; Hu, Y.; Dastgir, J.; et al. TPM3 deletions cause a hypercontractile congenital muscle stiffness phenotype. Ann. Neurol. 2015, 78, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Rynkiewicz, M.J.; Prum, T.; Hollenberg, S.; Kiani, F.A.; Fagnant, P.M.; Marston, S.B.; Trybus, K.M.; Fischer, S.; Moore, J.R.; Lehman, W. Tropomyosin Must Interact Weakly with Actin to Effectively Regulate Thin Filament Function. Biophys. J. 2017, 113, 2444–2451. [Google Scholar] [CrossRef] [PubMed]
- Orzechowski, M.; Fischer, S.; Moore, J.R.; Lehman, W.; Farman, G.P. Energy landscapes reveal the myopathic effects of tropomyosin mutations. Arch. Biochem. Biophys. 2014, 564, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Orzechowski, M.; Fischer, S.; Lehman, W. Influence of Actin Mutation on the Energy Landscape of Actin-Tropomyosin Filaments. Biophys. J. 2013, 104, 480a. [Google Scholar] [CrossRef]
- Mogensen, J.; Klausen, I.C.; Pedersen, A.K.; Egeblad, H.; Bross, P.; Kruse, T.A.; Gregersen, N.; Hansen, P.S.; Baandrup, U.; Borglum, A.D. Alpha-cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J. Clin. Investig. 1999, 103, R39–R43. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, M.C.; Schmidt, W.; Rynkiewicz, M.J.; Agarwal, K.; Gao, J.; Katz, J.; Lehman, W.; Cammarato, A. Distortion of the Actin A-Triad Results in Contractile Disinhibition and Cardiomyopathy. Cell. Rep. 2017, 20, 2612–2625. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Sweitzer, N.K.; McDonough, B.; Maron, B.J.; Casey, S.A.; Seidman, J.G.; Seidman, C.E.; Solomon, S.D. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation 2002, 105, 2992–2997. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A.; Aksel, T.; Bartholomew, S.R.; Nag, S.; Kawana, M.; Yu, E.C.; Sarkar, S.S.; Sung, J.; Sommese, R.F.; Sutton, S.; et al. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human β-cardiac myosin. J. Exp. Biol. 2016, 219 Pt 2, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Marston, S. Why is there a limit to the changes in myofilament Ca2+-sensitivity associated with myopathy causing mutations? Front. Physiol. 2016, 7, 415. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A. Hypertrophic and Dilated Cardiomyopathy: Four Decades of Basic Research on Muscle Lead to Potential Therapeutic Approaches to These Devastating Genetic Diseases. Biophys. J. 2014, 106, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Crilley, J.G.; Boehm, E.A.; Blair, E.; Rajagopalan, B.; Blamire, A.M.; Styles, P.; McKenna, W.J.; Ostman-Smith, I.; Clarke, K.; Watkins, H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. J. Am. Coll. Cardiol. 2003, 41, 1776–1782. [Google Scholar] [CrossRef]
- Ashrafian, H.; Redwood, C.; Blair, E.; Watkins, H. Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet. 2003, 19, 263–268. [Google Scholar] [CrossRef]
- Fraser, I.D.; Marston, S.B. In vitro motility analysis of actin-tropomyosin regulation by troponin and calcium. The thin filament is switched as a single cooperative unit. J. Biol. Chem. 1995, 270, 7836–7841. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.S.; Geeves, M.A. The muscle thin filament as a classical cooperative/allosteric regulatory system. J. Mol. Biol. 1998, 277, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S. The key regulatory switch of the muscle thin filament: Ca2+ or myosin heads? J. Muscle Res. Cell. Motil. 1994, 15, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Homburger, J.R.; Green, E.M.; Caleshu, C.; Sunitha, M.S.; Taylor, R.E.; Ruppel, K.M.; Metpally, R.P.R.; Colan, S.D.; Michels, M.; Day, S.M.; et al. Multidimensional structure-function relationships in human β-cardiac myosin from population-scale genetic variation. Proc. Natl. Acad. Sci. USA 2016, 113, 6701–6706. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A. The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 2015, 43, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Trivedi, D.V.; Sarkar, S.S.; Adhikari, A.S.; Sunitha, M.S.; Sutton, S.; Ruppel, K.M.; Spudich, J.A. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat. Struct. Mol. Biol. 2017, 24, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, D.V.; Adhikari, A.S.; Sarkar, S.S.; Ruppel, K.M.; Spudich, J.A. Hypertrophic cardiomyopathy and the myosin mesa: Viewing an old disease in a new light. Bio. Rev. 2017, 282, 9204–9222. [Google Scholar] [CrossRef] [PubMed]
- Wendt, T.; Taylor, D.; Trybus, K.M.; Taylor, K. Three-dimensional image reconstruction of dephosphorylated smooth muscle heavy meromyosin reveals asymmetry in the interaction between myosin heads and placement of subfragment 2. Proc. Natl. Acad. Sci. USA 2001, 98, 4361–4366. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Sulbarán, G.; Yang, S.; Mun, J.Y.; Alamo, L.; Pinto, A.; Sato, O.; Ikebe, M.; Liu, X.; Korn, E.D.; Sarsoza, F.; et al. Interacting-heads motif has been conserved as a mechanism of myosin II inhibition since before the origin of animals. Proc. Natl. Acad. Sci. USA 2018, 115, E1991–E2000. [Google Scholar] [CrossRef] [PubMed]
- Al-Khayat, H.A.; Kensler, R.W.; Squire, J.M.; Marston, S.B.; Morris, E.P. Atomic model of the human cardiac muscle myosin filament. Proc. Natl. Acad. Sci. USA 2013, 110, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Komatsu, S.; Ikebe, M.; Craig, R. Head-head and head-tail interaction: A general mechanism for switching off myosin II activity in cells. Mol. Biol. Cell 2008, 19, 3234–3242. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.W.; Li, A.; Lal, S.; Bos, J.M.; Harris, S.P.; van der Velden, J.; Ackerman, M.J.; Cooke, R.; dos Remedios, C.G. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS ONE 2017, 12, e0180064. [Google Scholar] [CrossRef] [PubMed]
- Hooijman, P.; Stewart, M.A.; Cooke, R. A new state of cardiac myosin with very slow ATP turnover: A potential cardioprotective mechanism in the heart. Biophys. J. 2011, 100, 1969–1976. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.W.; Li, A.; dos Remedios, C.G.; Cooke, R. The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys. Rev. 2014, 7, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Toepfer, C.; Caorsi, V.; Kampourakis, T.; Sikkel, M.B.; West, T.G.; Leung, M.-C.; Al-Saud, S.A.; MacLeod, K.T.; Lyon, A.R.; Marston, S.B.; et al. Myosin Regulatory Light Chain (RLC) Phosphorylation Change as a Modulator of Cardiac Muscle Contraction in Disease. J. Biol. Chem. 2013, 288, 13446–13454. [Google Scholar] [CrossRef] [PubMed]
- Kampourakis, T.; Sun, Y.-B.; Irving, M. Myosin light chain phosphorylation enhances contraction of heart muscle via structural changes in both thick and thin filaments. Proc. Natl. Acad. Sci. USA 2016, 113, E3039–E3047. [Google Scholar] [CrossRef] [PubMed]
- Ratti, J.; Rostkova, E.; Gautel, M.; Pfuhl, M. Structure and interactions of myosin-binding protein C domain C0: Cardiac-specific regulation of myosin at its neck? J. Biol. Chem. 2011, 286, 12650–12658. [Google Scholar] [CrossRef] [PubMed]
- Alamo, L.; Ware, J.S.; Pinto, A.; Gillilan, R.E.; Seidman, J.G.; Seidman, C.E.; Padrón, R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife 2017, 6, 318. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Wong, F.L.; Morck, M.; Ruppel, K.M.; et al. Mavacamten stabilizes a folded-back sequestered super-relaxed state of β-cardiac myosin. bioRxiv 2018, 1–19. [Google Scholar] [CrossRef]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.A.; Markova, S.; Ueda, Y.; Kim, J.B.; Pascoe, P.J.; Evanchik, M.J.; Green, E.M.; Harris, S.P. A Small Molecule Inhibitor of Sarcomere Contractility Acutely Relieves Left Ventricular Outflow Tract Obstruction in Feline Hypertrophic Cardiomyopathy. PLoS ONE 2016, 11, e0168407. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Protein | Gene | Protein Name | Predominant Expression in Muscle | Myopathy | Number of Mutations |
|---|---|---|---|---|---|
| Myosin heavy chain | MYH1 | MyHC-2X | Fast-twitch skeletal muscle (Type IIx) | ||
| MYH2 | MyHC-2A | Fast-twitch skeletal muscle (Type IIx/IIa) | “myopathy”, inclusion body myopathy, distal and proximal myopathy, opthalmoplegia | 15 | |
| MYH3 | MyHC-embryonic | Embryo | Distal arthrogryposis types 1, 2A, 2B, 8, Freeman-Sheldon, Sheldon-Hall syndrome | 33 | |
| MYH4 | MyHC-2B | Fast-twitch skeletal muscle (Type IIb) | 0 | ||
| MYH6 | α-MyHC | Atria | Hypertrophic cardiomyopathy, dilated cardiomyopathy, atrial-septal defect, other congenital defects | 33 | |
| MYH7 | β-MyHC | Cardiac ventricles; slow-twitch skeletal muscle (Type I) | Hypertrophic cardiomyopathy, Dilated cardiomyopathy, left ventricular non-compaction, Laing distal myopathy, Scapuloperineal and limb girdle syndromes | >800 | |
| MYH8 | MyHC-perinatal | Fetal skeletal muscle | Distal arthrogryposis DA7 | 1 | |
| Essential light chain (Alkaline light chain) | MYL1 | MLC1f, MLC3f | Fast-twitch skeletal muscle | ||
| MYL3 | VLC1, MLC1V | Cardiac ventricles; slow-twitch skeletal muscle | Hypertrophic cardiomyopathy, Dilated cardiomyopathy | 21 | |
| MYL4 | ALC1 | Atria; embryonic cardiac ventricles and skeletal muscle | Atrial fibrillation | 1 | |
| Regulatory light chain | MYL2 | MLC-2 | Heart; skeletal muscle | Hypertrophic cardiomyopathy | 24 |
| MYL7 | MYL2A, MLC-2a | Atrial; embryo | 0 | ||
| MYLPF | MLC2B, MLC-2f | Fast-twitch skeletal muscle | 0 | ||
| Actin | ACTA1 | Skeletal actin | Skeletal muscle | Nemaline myopathy, actin myopathy, congenital fibre-type disproportion, stiff patient | 235 |
| ACTC | Cardiac actin | Cardiac muscle, embryonic skeletal muscle | Hypertrophic cardiomyopathy, dilated cardiomyopathy, left ventricular non-compaction, atrial-septal defect | 40 | |
| ACTA2 | Smooth muscle actin | Vascular smooth muscle | Aneurism | 1 |
| Mutation | Ratio EC50 mut/wt for Native Thin Filaments | Ref. | Ratio EC50 mut/wt for Synthetic System | Ref. |
|---|---|---|---|---|
| TNNT2 DK210 (homozygous) | 1.6 | [67] | 0.65 Recombinant troponin, Asα-tropomyosin | [62] |
| 2.2 | [61] | |||
| TNNC1 G159D | 0.55 | [68] | 1.8 Recombinant troponin, ASα-tropomyosin | [62] |
| 0.99 Mouse heart fibres exchanged with recombinant TnC | [69] | |||
| 1.9 Human heart troponin, ASα-tropomyosin | [68] | |||
| TPM1 E54K | 1.0 | [61] | 1.7 Recombinant troponin, ASα-tropomyosin | [62] |
| 0.52 Recombinant troponin, ASα-tropomyosin | [63] | |||
| TPM1 D230N | 0.43 | [61] | 1.7 Recombinant troponin, ASα-tropomyosin | [70] |
| 1.7 Skeletal muscle troponin, native αtropomyosin | [61] | |||
| ACTC E361G | 0.95 | [33] | 3.3 Skeletal muscle troponin, native α-tropomyosin | [33] |
| 0.45 cardiac myofibrils | [71] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marston, S. The Molecular Mechanisms of Mutations in Actin and Myosin that Cause Inherited Myopathy. Int. J. Mol. Sci. 2018, 19, 2020. https://doi.org/10.3390/ijms19072020
Marston S. The Molecular Mechanisms of Mutations in Actin and Myosin that Cause Inherited Myopathy. International Journal of Molecular Sciences. 2018; 19(7):2020. https://doi.org/10.3390/ijms19072020
Chicago/Turabian StyleMarston, Steven. 2018. "The Molecular Mechanisms of Mutations in Actin and Myosin that Cause Inherited Myopathy" International Journal of Molecular Sciences 19, no. 7: 2020. https://doi.org/10.3390/ijms19072020
APA StyleMarston, S. (2018). The Molecular Mechanisms of Mutations in Actin and Myosin that Cause Inherited Myopathy. International Journal of Molecular Sciences, 19(7), 2020. https://doi.org/10.3390/ijms19072020