GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling


Abstract
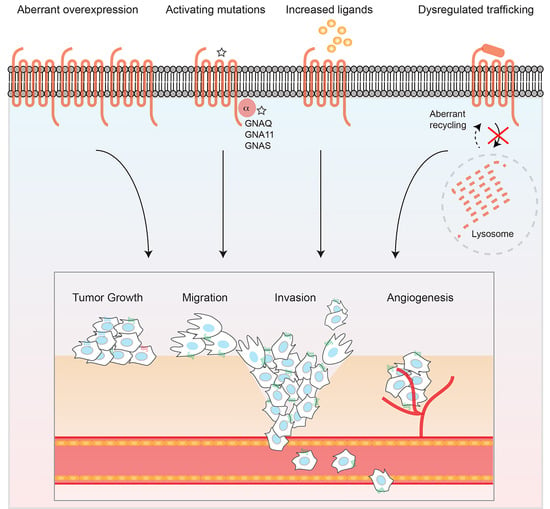
1. Introduction
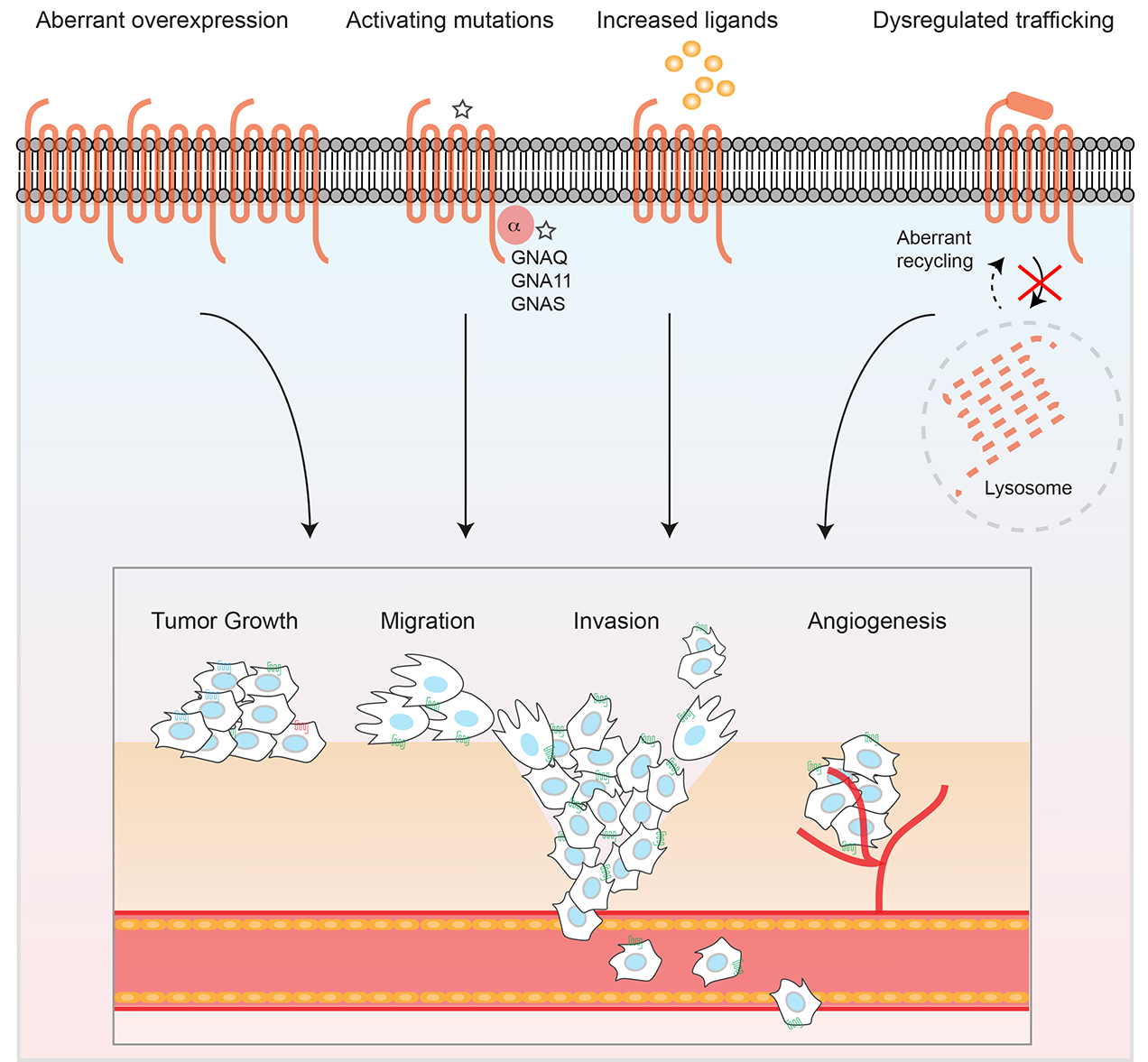
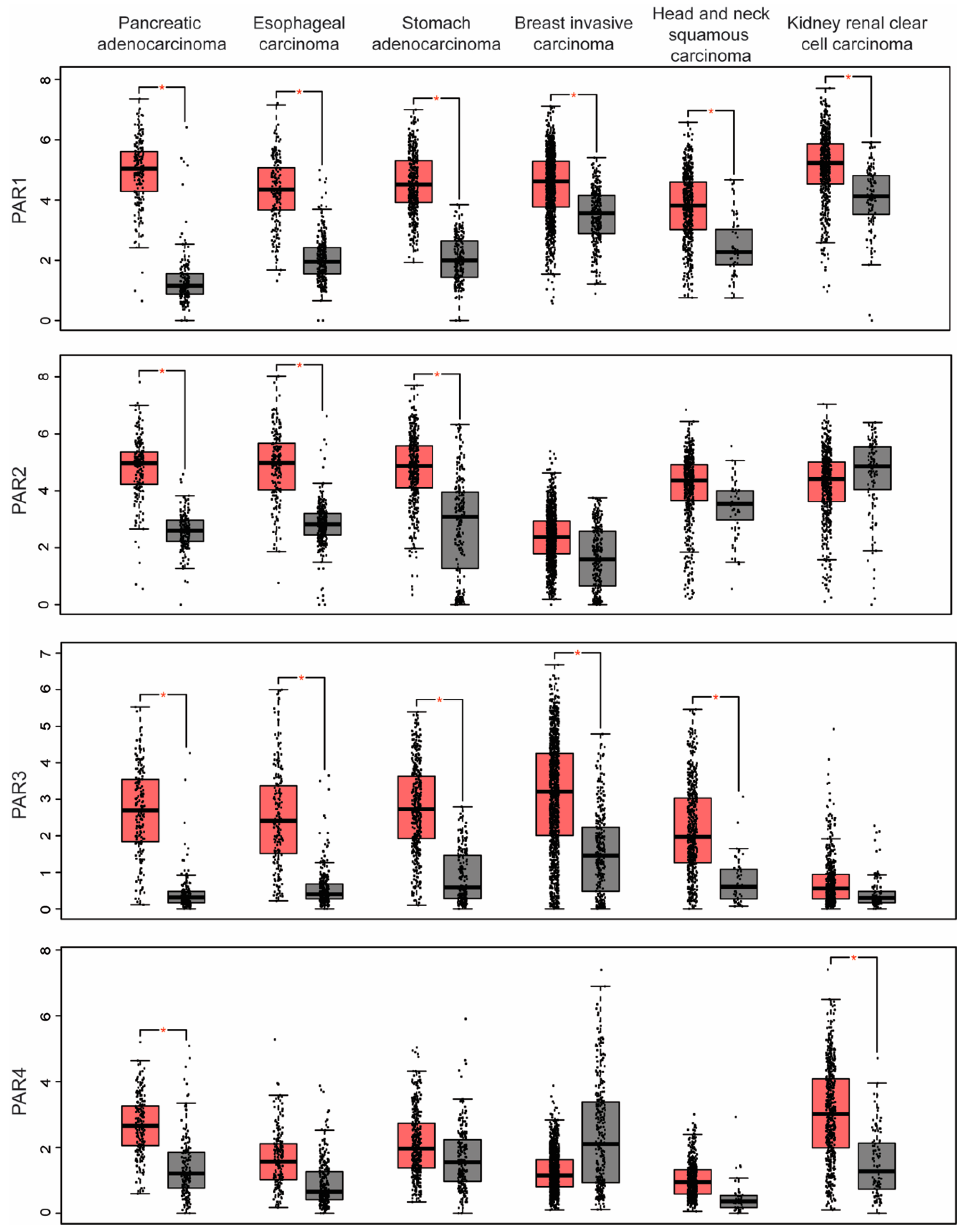
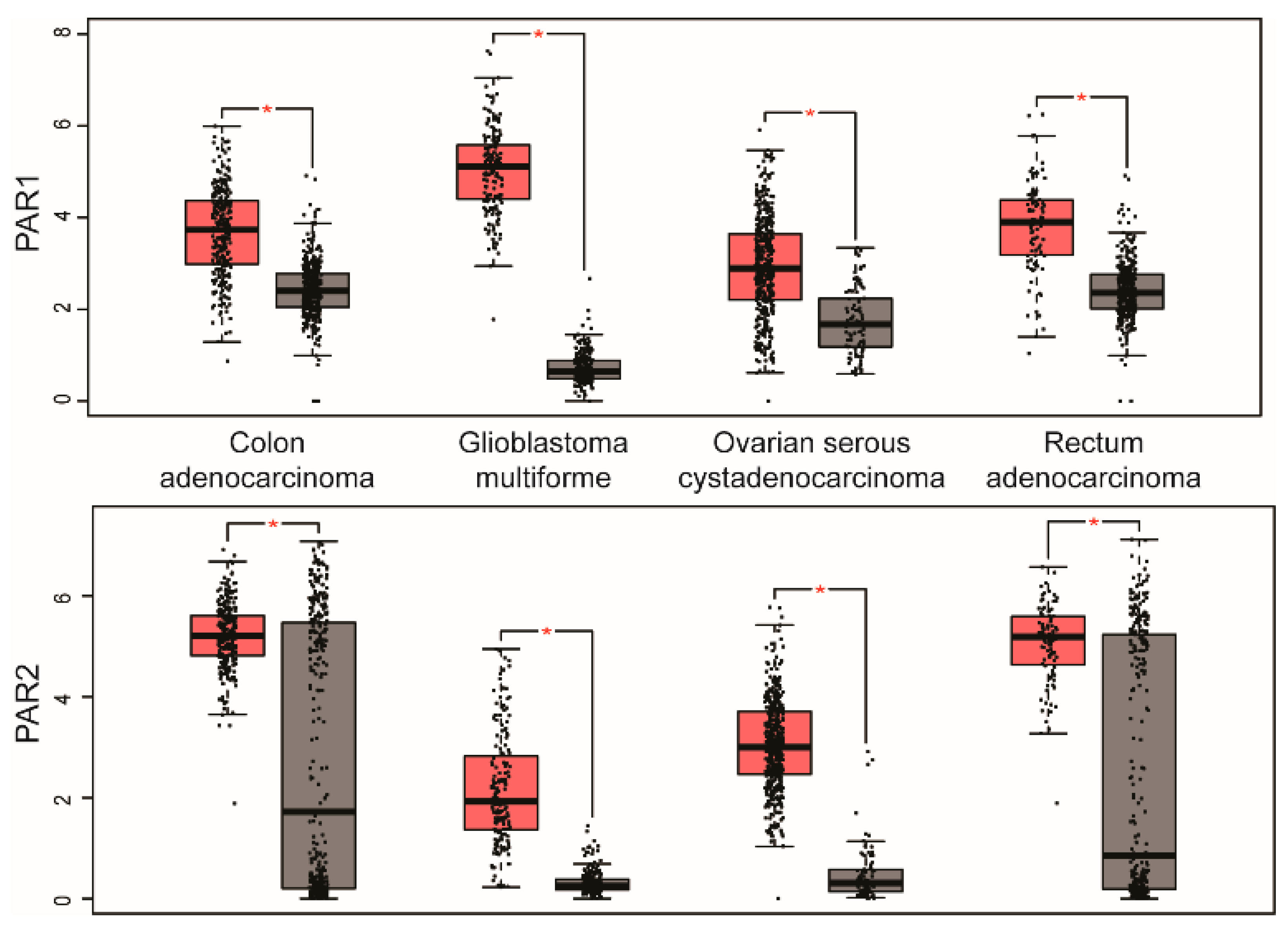
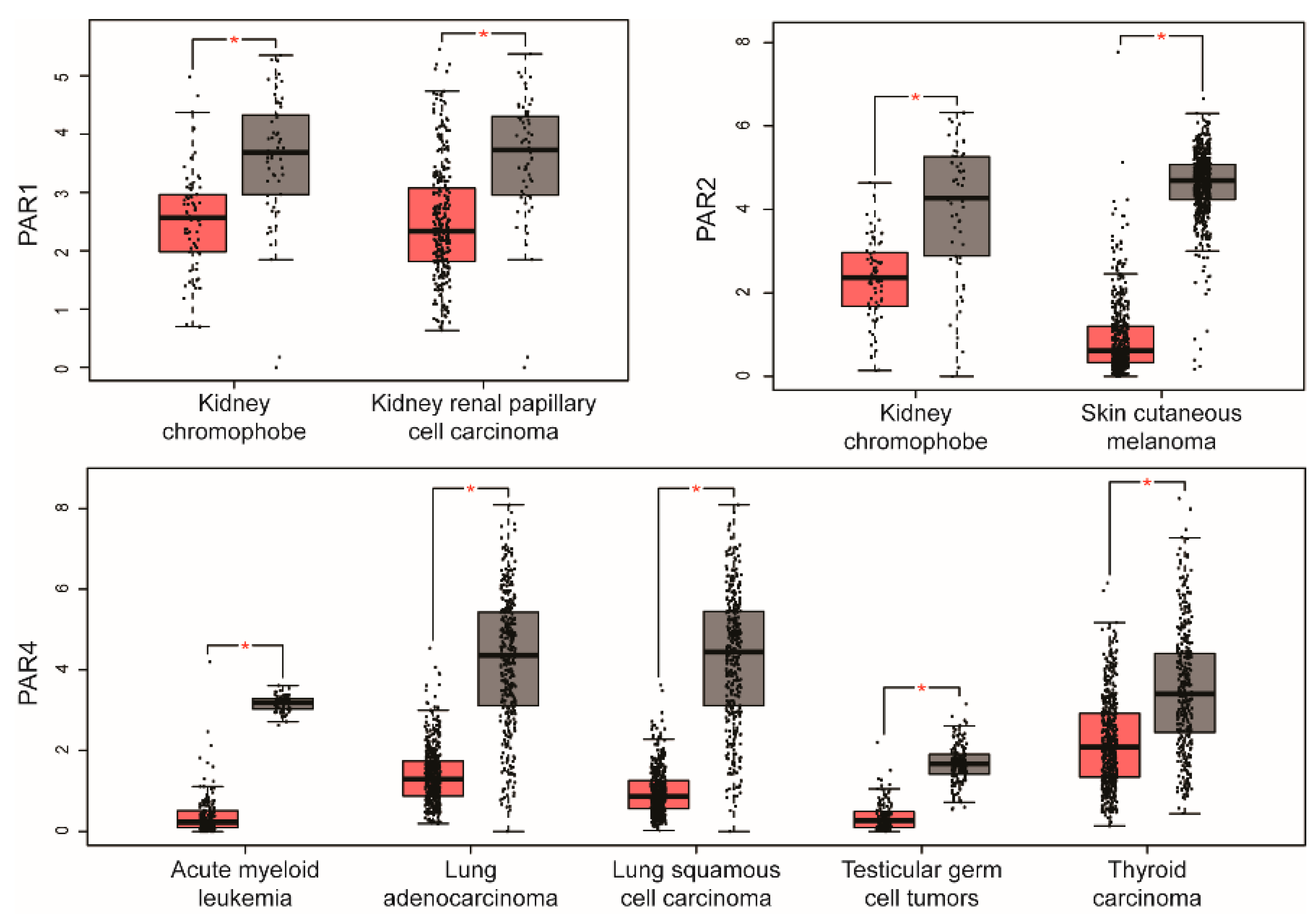
2. PAR Expression in Human Cancer
3. Internalization and Recycling of PARs and Implications in Cancer
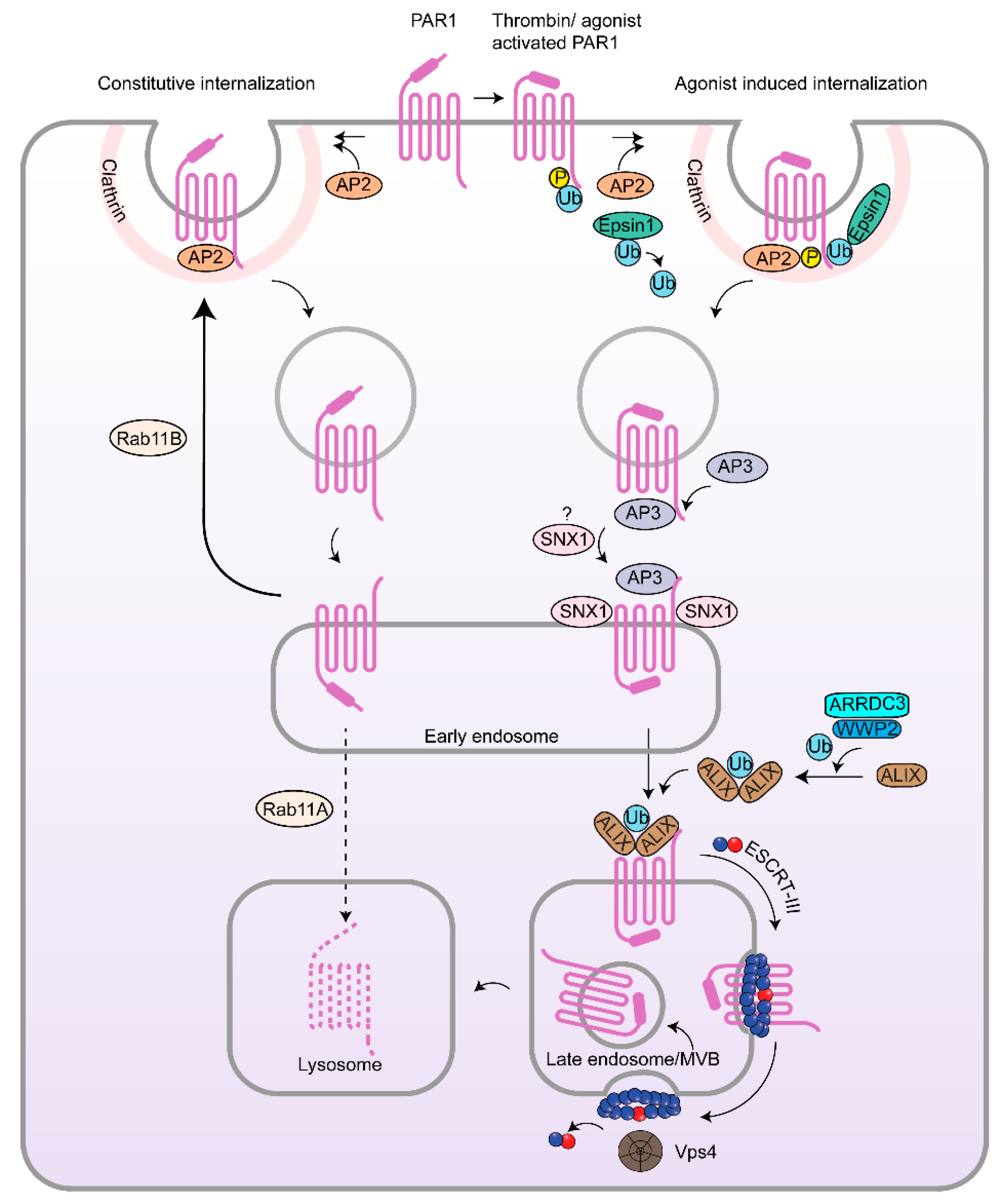
3.1. Clathrin-Mediated Endocytosis of PARs
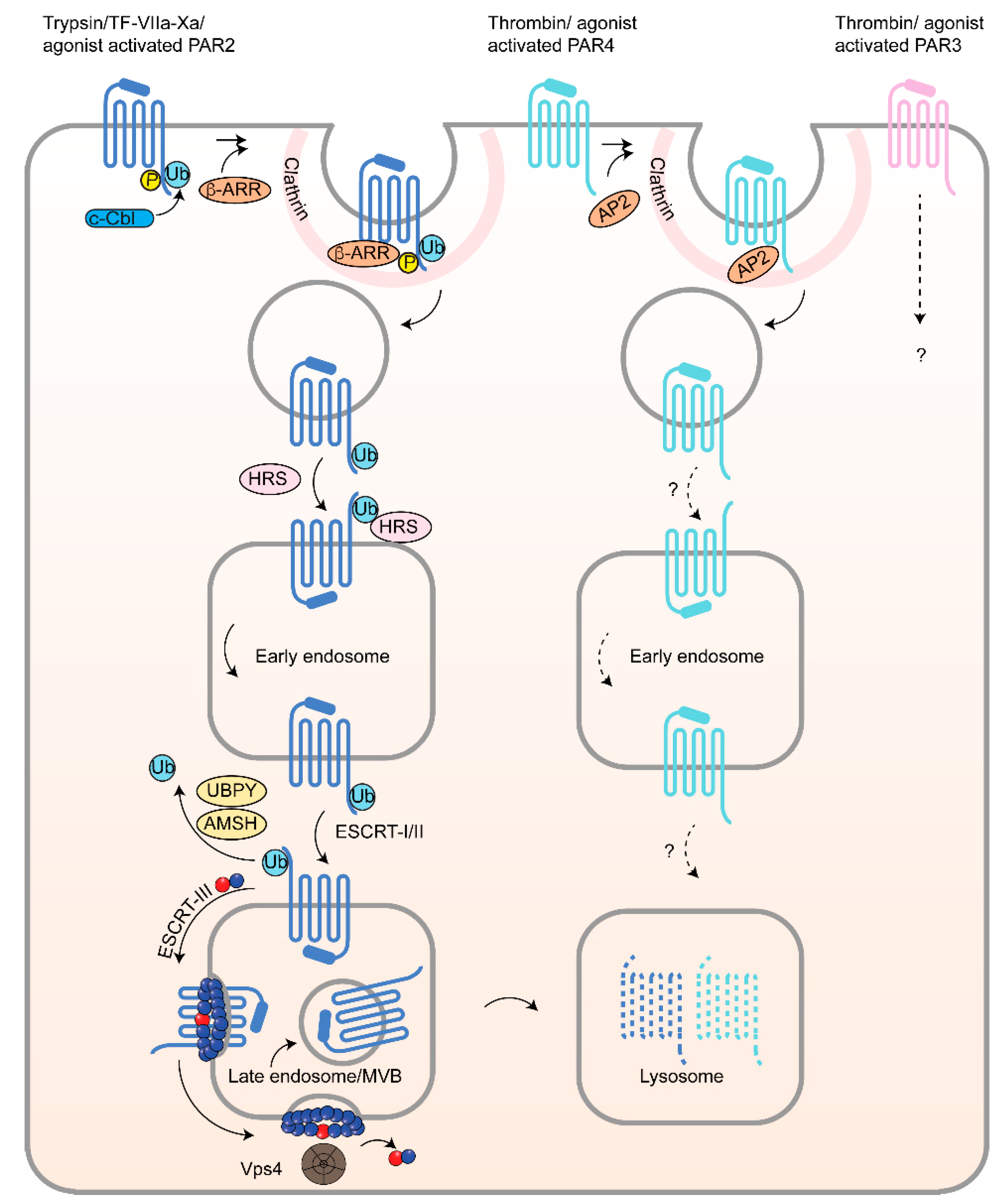
3.2. β-Arrestins, Epsins and Adaptor Protein Complex-2 (AP-2) and PARs in Cancer
3.3. Rab11, PAR Recycling and Implications in Cancer
4. Lysosomal Sorting of PARs and Dysregulation in Cancer
4.1. Endosomal Sorting of PAR1 by Sorting Nexin 1 (SNX1) and Adaptor Protein-3 (AP-3) and Role in Cancer
4.2. Atypical Ubiquitin-Independent Lysosomal Sorting of PAR1 Mediated by ALIX and ARRDC3
4.3. ESCRT-III/Charged MVB Protein 4 (CHMP4) and Vps4-Dependent Sorting of PAR1
4.4. PAR2 Lysosomal Sorting Requires Ubiquitination and Canonical ESCRT
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Singh, A.; Nunes, J.J.; Ateeq, B. Role and therapeutic potential of g-protein coupled receptors in breast cancer progression and metastases. Eur. J. Pharmacol. 2015, 763, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G protein-coupled receptors in cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Vazquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of g proteins and g-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. Gpcrs as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Wei, X.; Cardenas-Navia, I.; Teer, J.K.; Lin, J.C.; Walia, V.; Gartner, J.; Jiang, J.; Cherukuri, P.F.; Molinolo, A.; et al. Exon capture analysis of g protein-coupled receptors identifies activating mutations in grm3 in melanoma. Nat. Genet. 2011, 43, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Kuzumaki, N.; Suzuki, A.; Narita, M.; Hosoya, T.; Nagasawa, A.; Imai, S.; Yamamizu, K.; Morita, H.; Suzuki, T.; Okada, Y.; et al. Multiple analyses of g-protein coupled receptor (gpcr) expression in the development of gefitinib-resistance in transforming non-small-cell lung cancer. PLoS ONE 2012, 7, e44368. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of yap by the gnaq uveal melanoma oncogene through a trio-regulated rho gtpase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Jin, M.S.; Miyagi, E.; Hirahara, F.; Nakamura, Y.; Piao, J.H.; Asai, A.; Yoshida, A.; Tsuchiya, E.; Ruf, W.; et al. Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Res. 2006, 66, 9453–9460. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Covic, L.; Agarwal, A.; Jacques, S.; Sherifi, S.; Kuliopulos, A. Par1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 2005, 120, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Kuliopulos, A.; Covic, L.; Seeley, S.K.; Sheridan, P.J.; Helin, J.; Costello, C.E. Plasmin desensitization of the par1 thrombin receptor: Kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry 1999, 38, 4572–4585. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Vu, T.K.; Wheaton, V.I.; Hung, D.T.; Charo, I.; Coughlin, S.R. Domains specifying thrombin-receptor interaction. Nature 1991, 353, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Ricks, T.K.; Trejo, J. Protease-activated receptor signalling, endocytic sorting and dysregulation in cancer. J. Cell Sci. 2007, 120, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, N.J.; Aguilar, B.; Smith, T.H.; Le, P.; Soohoo, A.L.; Puthenveedu, M.A.; Nizet, V.; Trejo, J. Ubiquitin plays an atypical role in gpcr-induced p38 map kinase activation on endosomes. J. Cell Biol. 2015, 210, 1117–1131. [Google Scholar] [CrossRef] [PubMed]
- DeFea, K.A.; Zalevsky, J.; Thoma, M.S.; Dery, O.; Mullins, R.D.; Bunnett, N.W. Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated erk1/2. J. Cell Biol. 2000, 148, 1267–1281. [Google Scholar] [CrossRef] [PubMed]
- Stalheim, L.; Ding, Y.; Gullapalli, A.; Paing, M.M.; Wolfe, B.L.; Morris, D.R.; Trejo, J. Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol. Pharmacol. 2005, 67, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. Gepia: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, J.N.; Patterson, M.M.; Malik, A.B. Protease-activated receptor-3 (par3) regulates par1 signaling by receptor dimerization. Proc. Natl. Acad. Sci. USA 2007, 104, 5662–5667. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.; Moeller, B.J.; Juneja, J.; Booden, M.A.; Der, C.J.; Daaka, Y.; Dewhirst, M.W.; Fields, T.A.; Casey, P.J. The g12 family of heterotrimeric g proteins promotes breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 8173–8178. [Google Scholar] [CrossRef] [PubMed]
- Leger, A.J.; Jacques, S.L.; Badar, J.; Kaneider, N.C.; Derian, C.K.; Andrade-Gordon, P.; Covic, L.; Kuliopulos, A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation 2006, 113, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.H.; Li, J.G.; Dores, M.R.; Trejo, J. Protease-activated receptor-4 and purinergic receptor p2y12 dimerize, co-internalize, and activate akt signaling via endosomal recruitment of beta-arrestin. J. Biol. Chem. 2017, 292, 13867–13878. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, A.P.; Smith, T.H.; Trejo, J. Cofactoring and dimerization of proteinase-activated receptors. Pharmacol. Rev. 2013, 65, 1198–1213. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.J.; Prevost, N.; Molino, M.; Hollinger, M.K.; Woolkalis, M.J.; Woulfe, D.S.; Brass, L.F. Thrombin responses in human endothelial cells. Contributions from receptors other than par1 include the transactivation of par2 by thrombin-cleaved par1. J. Biol. Chem. 2000, 275, 13502–13509. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Gangadharan, B.; Brass, L.F.; Ruf, W.; Mueller, B.M. Protease-activated receptors (par1 and par2) contribute to tumor cell motility and metastasis. Mol. Cancer Res. 2004, 2, 395–402. [Google Scholar] [PubMed]
- Grimsey, N.J.; Coronel, L.J.; Cordova, I.C.; Trejo, J. Recycling and endosomal sorting of protease-activated receptor-1 is distinctly regulated by rab11a and rab11b proteins. J. Biol. Chem. 2016, 291, 2223–2236. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.; Ishii, K.; Coughlin, S.R.; Kobilka, B.K. Intracellular targeting and trafficking of thrombin receptors. A novel mechanism for resensitization of a g protein-coupled receptor. J. Biol. Chem. 1994, 269, 27719–27726. [Google Scholar] [PubMed]
- Ishii, K.; Hein, L.; Kobilka, B.; Coughlin, S.R. Kinetics of thrombin receptor cleavage on intact cells. Relation to signaling. J. Biol. Chem. 1993, 268, 9780–9786. [Google Scholar] [PubMed]
- Horvat, R.; Palade, G.E. The functional thrombin receptor is associated with the plasmalemma and a large endosomal network in cultured human umbilical vein endothelial cells. J. Cell Sci. 1995, 108 Pt 3, 1155–1164. [Google Scholar] [PubMed]
- Ohno, H.; Stewart, J.; Fournier, M.C.; Bosshart, H.; Rhee, I.; Miyatake, S.; Saito, T.; Gallusser, A.; Kirchhausen, T.; Bonifacino, J.S. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 1995, 269, 1872–1875. [Google Scholar] [CrossRef] [PubMed]
- Paing, M.M.; Johnston, C.A.; Siderovski, D.P.; Trejo, J. Clathrin adaptor ap2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol. Cell. Biol. 2006, 26, 3231–3242. [Google Scholar] [CrossRef] [PubMed]
- Paing, M.M.; Stutts, A.B.; Kohout, T.A.; Lefkowitz, R.J.; Trejo, J. Beta-arrestins regulate protease-activated receptor-1 desensitization but not internalization or down-regulation. J. Biol. Chem. 2002, 277, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Damke, H.; Baba, T.; Warnock, D.E.; Schmid, S.L. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 1994, 127, 915–934. [Google Scholar] [CrossRef] [PubMed]
- Marks, B.; Stowell, M.H.; Vallis, Y.; Mills, I.G.; Gibson, A.; Hopkins, C.R.; McMahon, H.T. Gtpase activity of dynamin and resulting conformation change are essential for endocytosis. Nature 2001, 410, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dores, M.R.; Grimsey, N.; Canto, I.; Barker, B.L.; Trejo, J. Adaptor protein complex-2 (ap-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J. Biol. Chem. 2011, 286, 40760–40770. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, B.L.; Marchese, A.; Trejo, J. Ubiquitination differentially regulates clathrin-dependent internalization of protease-activated receptor-1. J. Cell Biol. 2007, 177, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Paing, M.M.; Trejo, J. Termination of protease-activated receptor-1 signaling by beta-arrestins is independent of receptor phosphorylation. J. Biol. Chem. 2004, 279, 10020–10031. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.; Altschuler, Y.; Fu, H.W.; Mostov, K.E.; Coughlin, S.R. Protease-activated receptor-1 down-regulation: A mutant hela cell line suggests novel requirements for par1 phosphorylation and recruitment to clathrin-coated pits. J. Biol. Chem. 2000, 275, 31255–31265. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.C.; Katzmann, D.J.; Schnell, J.D.; Sutanto, M.; Emr, S.D.; Hicke, L. Epsins and vps27p/hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nat. Cell Biol. 2002, 4, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Chini, B.; Parenti, M. G-protein-coupled receptors, cholesterol and palmitoylation: Facts about fats. J. Mol. Endocrinol. 2009, 42, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, T.; Tsutsumi, R.; Noritake, J.; Fukata, Y.; Fukata, M. Dynamic protein palmitoylation in cellular signaling. Prog. Lipid Res. 2009, 48, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.E.; Deschenes, R.J. Palmitoylation: Policing protein stability and traffic. Nat. Rev. Mol. Cell. Biol. 2007, 8, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Qanbar, R.; Bouvier, M. Role of palmitoylation/depalmitoylation reactions in g-protein-coupled receptor function. Pharmacol. Ther. 2003, 97, 1–33. [Google Scholar] [CrossRef]
- Canto, I.; Trejo, J. Palmitoylation of protease-activated receptor-1 regulates adaptor protein complex-2 and -3 interaction with tyrosine-based motifs and endocytic sorting. J. Biol. Chem. 2013, 288, 15900–15912. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.H.; Coronel, L.J.; Li, J.G.; Dores, M.R.; Nieman, M.T.; Trejo, J. Protease-activated receptor-4 signaling and trafficking is regulated by the clathrin adaptor protein complex-2 independent of beta-arrestins. J. Biol. Chem. 2016, 291, 18453–18464. [Google Scholar] [CrossRef] [PubMed]
- Ricks, T.K.; Trejo, J. Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J. Biol. Chem. 2009, 284, 34444–34457. [Google Scholar] [CrossRef] [PubMed]
- Dery, O.; Thoma, M.S.; Wong, H.; Grady, E.F.; Bunnett, N.W. Trafficking of proteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescent protein. Beta-arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 1999, 274, 18524–18535. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Rajagopal, S. The beta-arrestins: Multifunctional regulators of g protein-coupled receptors. J. Biol. Chem. 2016, 291, 8969–8977. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Rastogi, S.; Pillai, S.; Ordonez-Ercan, D.; Morris, M.; Haura, E.; Chellappan, S. Nicotine induces cell proliferation by beta-arrestin-mediated activation of src and rb-raf-1 pathways. J. Clin. Investig. 2006, 116, 2208–2217. [Google Scholar] [CrossRef] [PubMed]
- Suleymanova, N.; Crudden, C.; Shibano, T.; Worrall, C.; Oprea, I.; Tica, A.; Calin, G.A.; Girnita, A.; Girnita, L. Functional antagonism of beta-arrestin isoforms balance igf-1r expression and signalling with distinct cancer-related biological outcomes. Oncogene 2017, 36, 5734–5744. [Google Scholar] [CrossRef] [PubMed]
- Ge, L.; Shenoy, S.K.; Lefkowitz, R.J.; DeFea, K. Constitutive protease-activated receptor-2-mediated migration of mda mb-231 breast cancer cells requires both beta-arrestin-1 and -2. J. Biol. Chem. 2004, 279, 55419–55424. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, F.G.; Gorden, D.L.; Matta, P.; Shi, Q.; Matrisian, L.M.; DuBois, R.N. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Flaceliere, M.; Grillet, F.; Dantec, C.; Desvignes, J.P.; Pannequin, J.; Severac, D.; Dubois, E.; Bibeau, F.; Escriou, V.; et al. Essential requirement for beta-arrestin2 in mouse intestinal tumors with elevated wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 3047–3052. [Google Scholar] [CrossRef] [PubMed]
- Rosano, L.; Cianfrocca, R.; Masi, S.; Spinella, F.; Di Castro, V.; Biroccio, A.; Salvati, E.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. Beta-arrestin links endothelin a receptor to beta-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc. Natl. Acad. Sci. USA 2009, 106, 2806–2811. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S.K.; Han, S.; Zhao, Y.L.; Hara, M.R.; Oliver, T.; Cao, Y.; Dewhirst, M.W. Beta-arrestin1 mediates metastatic growth of breast cancer cells by facilitating hif-1-dependent vegf expression. Oncogene 2012, 31, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Fereshteh, M.; Ito, T.; Kovacs, J.J.; Zhao, C.; Kwon, H.Y.; Tornini, V.; Konuma, T.; Chen, M.; Lefkowitz, R.J.; Reya, T. Beta-arrestin2 mediates the initiation and progression of myeloid leukemia. Proc. Natl. Acad. Sci. USA 2012, 109, 12532–12537. [Google Scholar] [CrossRef] [PubMed]
- Di Modugno, F.; Caprara, V.; Chellini, L.; Tocci, P.; Spadaro, F.; Ferrandina, G.; Sacconi, A.; Blandino, G.; Nistico, P.; Bagnato, A.; et al. Hmena is a key regulator in endothelin-1/beta-arrestin1-induced invadopodial function and metastatic process. Proc. Natl. Acad. Sci. USA 2018, 115, 3132–3137. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Trevino, J.; Rawal, B.; Singh, S.; Kovacs, M.; Li, X.; Schell, M.; Haura, E.; Bepler, G.; Chellappan, S. Beta-arrestin-1 mediates nicotine-induced metastasis through e2f1 target genes that modulate epithelial-mesenchymal transition. Cancer Res. 2015, 75, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Tessneer, K.L.; Pasula, S.; Cai, X.; Dong, Y.; Liu, X.; Yu, L.; Hahn, S.; McManus, J.; Chen, Y.; Chang, B.; et al. Endocytic adaptor protein epsin is elevated in prostate cancer and required for cancer progression. ISRN Oncol. 2013, 2013, 420597. [Google Scholar] [CrossRef] [PubMed]
- Pasula, S.; Cai, X.; Dong, Y.; Messa, M.; McManus, J.; Chang, B.; Liu, X.; Zhu, H.; Mansat, R.S.; Yoon, S.J.; et al. Endothelial epsin deficiency decreases tumor growth by enhancing vegf signaling. J. Clin. Investig. 2012, 122, 4424–4438. [Google Scholar] [CrossRef] [PubMed]
- Puthenveedu, M.A.; Lauffer, B.; Temkin, P.; Vistein, R.; Carlton, P.; Thorn, K.; Taunton, J.; Weiner, O.D.; Parton, R.G.; von Zastrow, M. Sequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomains. Cell 2010, 143, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Hanyaloglu, A.C.; von Zastrow, M. Regulation of gpcrs by endocytic membrane trafficking and its potential implications. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 537–568. [Google Scholar] [CrossRef] [PubMed]
- Seachrist, J.L.; Ferguson, S.S. Regulation of g protein-coupled receptor endocytosis and trafficking by rab gtpases. Life Sci. 2003, 74, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab gtpases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell. Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Bohm, S.K.; Khitin, L.M.; Grady, E.F.; Aponte, G.; Payan, D.G.; Bunnett, N.W. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J. Biol. Chem. 1996, 271, 22003–22016. [Google Scholar] [CrossRef] [PubMed]
- Roosterman, D.; Schmidlin, F.; Bunnett, N.W. Rab5a and rab11a mediate agonist-induced trafficking of protease-activated receptor 2. Am. J. Physiol. Cell Physiol. 2003, 284, C1319–C1329. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Wei, W.C.; Huang, S.H.; Shih, C.M.; Hsu, C.P.; Chang, K.J.; Chao, W.T. Rab11 regulates e-cadherin expression and induces cell transformation in colorectal carcinoma. BMC Cancer 2014, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, X.; Li, H.; Chen, H.; Liu, H. Rab11a sustains gsk3beta/wnt/beta-catenin signaling to enhance cancer progression in pancreatic cancer. Tumour Biol. 2016, 37, 13821–13829. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, Z.; Wang, H.; Cao, Z.; Zhao, Y.; Gong, C.; Ma, L.; Wang, X.; Hu, X.; Chen, S. Microrna-320a inhibits proliferation and invasion of breast cancer cells by targeting rab11a. Am. J. Cancer Res. 2015, 5, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, G.; Fan, Z.; Liu, T. Tumor-suppressive microrna-452 inhibits migration and invasion of breast cancer cells by directly targeting rab11a. Oncol. Lett. 2017, 14, 2559–2565. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Fu, L.; Zhao, Y.; Du, Y.; Li, Q.; Qiu, X.; Wang, E. Rab11a promotes proliferation and invasion through regulation of yap in non-small cell lung cancer. Oncotarget 2017, 8, 27800–27811. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Regalado, A.; Guzman-Hernandez, M.L.; Ramirez-Rangel, I.; Robles-Molina, E.; Balla, T.; Vazquez-Prado, J.; Reyes-Cruz, G. G protein-coupled receptor-promoted trafficking of gbeta1gamma2 leads to akt activation at endosomes via a mechanism mediated by gbeta1gamma2-rab11a interaction. Mol. Biol. Cell 2008, 19, 4188–4200. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.; Hammes, S.R.; Coughlin, S.R. Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc. Natl. Acad. Sci. USA 1998, 95, 13698–13702. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.; Coughlin, S.R. The cytoplasmic tails of protease-activated receptor-1 and substance p receptor specify sorting to lysosomes versus recycling. J. Biol. Chem. 1999, 274, 2216–2224. [Google Scholar] [CrossRef] [PubMed]
- Booden, M.A.; Eckert, L.B.; Der, C.J.; Trejo, J. Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol. Cell. Biol. 2004, 24, 1990–1999. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Cuevas, B.D.; Russo, A.; Johnson, G.L.; Trejo, J. Persistent transactivation of egfr and erbb2/her2 by protease-activated receptor-1 promotes breast carcinoma cell invasion. Oncogene 2008, 27, 4434–4445. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, A.K.S.; Pan, W.A.; Lin, H.; Trejo, J. The alpha-arrestin arrdc3 suppresses breast carcinoma invasion by regulating g protein-coupled receptor lysosomal sorting and signaling. J. Biol. Chem. 2018, 293, 3350–3362. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Paing, M.M.; Temple, B.R.; Trejo, J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 601–629. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Trejo, J. Atypical regulation of g protein-coupled receptor intracellular trafficking by ubiquitination. Curr. Opin. Cell Biol. 2014, 27, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Chen, B.; Lin, H.; Soh, U.J.; Paing, M.M.; Montagne, W.A.; Meerloo, T.; Trejo, J. Alix binds a ypx(3)l motif of the gpcr par1 and mediates ubiquitin-independent escrt-iii/mvb sorting. J. Cell Biol. 2012, 197, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Haft, C.R.; de la Luz Sierra, M.; Barr, V.A.; Haft, D.H.; Taylor, S.I. Identification of a family of sorting nexin molecules and characterization of their association with receptors. Mol. Cell. Biol. 1998, 18, 7278–7287. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Lazar, C.S.; Tronchere, H.; Sato, T.; Meerloo, T.; Yeo, M.; Songyang, Z.; Emr, S.D.; Gill, G.N. Endosomal localization and function of sorting nexin 1. Proc. Natl. Acad. Sci. USA 2002, 99, 6767–6772. [Google Scholar] [CrossRef] [PubMed]
- Haft, C.R.; de la Luz Sierra, M.; Bafford, R.; Lesniak, M.A.; Barr, V.A.; Taylor, S.I. Human orthologs of yeast vacuolar protein sorting proteins vps26, 29, and 35: Assembly into multimeric complexes. Mol. Biol. Cell 2000, 11, 4105–4116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Szabo, K.; Haft, C.R.; Trejo, J. Down-regulation of protease-activated receptor-1 is regulated by sorting nexin 1. Mol. Biol. Cell 2002, 13, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, A.; Wolfe, B.L.; Griffin, C.T.; Magnuson, T.; Trejo, J. An essential role for snx1 in lysosomal sorting of protease-activated receptor-1: Evidence for retromer-, hrs-, and tsg101-independent functions of sorting nexins. Mol. Biol. Cell 2006, 17, 1228–1238. [Google Scholar] [CrossRef] [PubMed]
- Heydorn, A.; Sondergaard, B.P.; Ersboll, B.; Holst, B.; Nielsen, F.C.; Haft, C.R.; Whistler, J.; Schwartz, T.W. A library of 7tm receptor c-terminal tails. Interactions with the proposed post-endocytic sorting proteins erm-binding phosphoprotein 50 (ebp50), n-ethylmaleimide-sensitive factor (nsf), sorting nexin 1 (snx1), and g protein-coupled receptor-associated sorting protein (gasp). J. Biol. Chem. 2004, 279, 54291–54303. [Google Scholar] [PubMed]
- Nguyen, L.N.; Holdren, M.S.; Nguyen, A.P.; Furuya, M.H.; Bianchini, M.; Levy, E.; Mordoh, J.; Liu, A.; Guncay, G.D.; Campbell, J.S.; et al. Sorting nexin 1 down-regulation promotes colon tumorigenesis. Clin. Cancer Res. 2006, 12, 6952–6959. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Feng, Y.; Xue, Y.; Hu, Y.; Wang, Q.; Zhou, L.; Liu, Z.; Zhang, J.; Yin, Y.; Gu, B.; et al. Down-regulation of snx1 predicts poor prognosis and contributes to drug resistance in colorectal cancer. Tumour. Biol. 2016, 37, 6619–6625. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, S.; Wang, Q.; Liang, L.; Ni, S.; Wang, L.; Sheng, W.; He, X.; Du, X. Microrna-95 promotes cell proliferation and targets sorting nexin 1 in human colorectal carcinoma. Cancer Res. 2011, 71, 2582–2589. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, S.; Hang, W.; Huang, H.; Ma, H. Mir-95 induces proliferation and chemo- or radioresistance through directly targeting sorting nexin1 (snx1) in non-small cell lung cancer. Biomed. Pharmacother. 2014, 68, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Q.; Zhang, M.; Zhang, Y.; Li, F.; Lei, P. Analysis for the mechanism between the small cell lung cancer and non-small cell lung cancer combing the mirna and mrna expression profiles. Thorac. Cancer 2015, 6, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Peden, A.A.; Oorschot, V.; Hesser, B.A.; Austin, C.D.; Scheller, R.H.; Klumperman, J. Localization of the ap-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J. Cell Biol. 2004, 164, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Paing, M.M.; Lin, H.; Montagne, W.A.; Marchese, A.; Trejo, J. Ap-3 regulates par1 ubiquitin-independent mvb/lysosomal sorting via an alix-mediated pathway. Mol. Biol. Cell 2012, 23, 3612–3623. [Google Scholar] [CrossRef] [PubMed]
- Rozenfeld, R.; Devi, L.A. Regulation of cb1 cannabinoid receptor trafficking by the adaptor protein ap-3. FASEB J. 2008, 22, 2311–2322. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.; Lizardi-Ortiz, J.E.; Westphalen, R.I.; Brandstetter, M.; Hemmings, H.C., Jr.; Sulzer, D.; Flajolet, M.; Greengard, P. Agap1/ap-3-dependent endocytic recycling of m5 muscarinic receptors promotes dopamine release. EMBO J. 2010, 29, 2813–2826. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Bouley, R.; Lin, H.Y.; Bechoua, S.; Sun, T.X.; Del Re, E.; Shioda, T.; Raychowdhury, M.K.; Lu, H.A.; Abou-Samra, A.B.; et al. Alix (aip1) is a vasopressin receptor (v2r)-interacting protein that increases lysosomal degradation of the v2r. Am. J. Physiol. Renal. Physiol. 2007, 292, F1303–F1313. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Liu, B.; Jose-Lafuente, M.; Chibalina, M.V.; Grierson, A.; Maclean, A.; Nasir, J. Alg-2 interacting protein aip1: A novel link between d1 and d3 signalling. Eur. J. Neurosci. 2008, 27, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Grimsey, N.J.; Mendez, F.; Trejo, J. Alix regulates the ubiquitin-independent lysosomal sorting of the p2y1 purinergic receptor via a ypx3l motif. PLoS ONE 2016, 11, e0157587. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.L. Reciprocal regulation of signaling and endocytosis: Implications for the evolving cancer cell. J. Cell Biol. 2017, 216, 2623–2632. [Google Scholar] [CrossRef] [PubMed]
- Dores, M.R.; Lin, H.; N, J.G.; Mendez, F.; Trejo, J. The alpha-arrestin arrdc3 mediates alix ubiquitination and g protein-coupled receptor lysosomal sorting. Mol. Biol. Cell 2015, 26, 4660–4673. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.E. On the origins of arrestin and rhodopsin. BMC Evol. Biol. 2008, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; O’Hayre, M.; Gutkind, J.S.; Hurley, J.H. Structural and biochemical basis for ubiquitin ligase recruitment by arrestin-related domain-containing protein-3 (arrdc3). J. Biol. Chem. 2014, 289, 4743–4752. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Martin-Serrano, J. Multiple interactions between the escrt machinery and arrestin-related proteins: Implications for ppxy-dependent budding. J. Virol. 2011, 85, 3546–3556. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Brou, C. Alpha-arrestins—New players in notch and gpcr signaling pathways in mammals. J. Cell Sci. 2014, 127, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Adelaide, J.; Finetti, P.; Bekhouche, I.; Repellini, L.; Geneix, J.; Sircoulomb, F.; Charafe-Jauffret, E.; Cervera, N.; Desplans, J.; Parzy, D.; et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007, 67, 11565–11575. [Google Scholar] [CrossRef] [PubMed]
- Draheim, K.M.; Chen, H.B.; Tao, Q.; Moore, N.; Roche, M.; Lyle, S. Arrdc3 suppresses breast cancer progression by negatively regulating integrin beta4. Oncogene 2010, 29, 5032–5047. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Pruitt, K.; Chung, J. Epigenetic silencing of arrdc3 expression in basal-like breast cancer cells. Sci. Rep. 2014, 4, 3846. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lin, Z.Y.; Xie, J.J.; Jiang, F.N.; Chen, C.J.; Li, J.X.; Zhou, X.; Zhong, W.D. Arrdc3 inhibits the progression of human prostate cancer through arrdc3-itgbeta4 pathway. Curr. Mol. Med. 2017, 17, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yang, P.N.; Chen, J.; Zhou, X.Y.; Liu, Q.J.; Li, H.J.; Li, C.L. Promoter hypermethylation may be an important mechanism of the transcriptional inactivation of arrdc3, gata5, and elp3 in invasive ductal breast carcinoma. Mol. Cell. Biochem. 2014, 396, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Xu, C.; Fang, Z.; Li, Y.; Liu, H.; Wang, Y.; Xu, C.; Sun, Y. Androgen receptor regulated microrna mir-182-5p promotes prostate cancer progression by targeting the arrdc3/itgb4 pathway. Biochem. Biophys. Res. Commun. 2016, 474, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. Wwp2 is an e3 ubiquitin ligase for pten. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wan, L.; Liu, J.; Yuan, Z.; Zhang, J.; Guo, J.; Malumbres, M.; Liu, J.; Zou, W.; Wei, W. Cdh1 inhibits wwp2-mediated ubiquitination of pten to suppress tumorigenesis in an apc-independent manner. Cell Discov. 2016, 2, 15044. [Google Scholar] [CrossRef] [PubMed]
- Clements, A.E.; Bravo, V.; Koivisto, C.; Cohn, D.E.; Leone, G. Wwp2 and its association with pten in endometrial cancer. Gynecol. Oncol. Rep. 2015, 13, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, C.; Nakashima, D.; Kasamatsu, A.; Unozawa, M.; Shida-Sakazume, T.; Higo, M.; Ogawara, K.; Yokoe, H.; Shiiba, M.; Tanzawa, H.; et al. Wwp2 is overexpressed in human oral cancer, determining tumor size and poor prognosis in patients: Downregulation of wwp2 inhibits the akt signaling and tumor growth in mice. Oncoscience 2014, 1, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.Y.; Huang, Y.J.; Tang, J.D.; Li, G.; Jiang, P.Q.; Wu, H.T. Silencing of hypoxia-inducible factor-1alpha promotes thyroid cancer cell apoptosis and inhibits invasion by downregulating wwp2, wwp9, vegf and vegfr2. Exp. Ther. Med. 2016, 12, 3735–3741. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.G.; Stoeck, A.; Guan, B.; Wu, R.C.; Zhu, H.; Blackshaw, S.; Shih Ie, M.; Wang, T.L. Notch3 interactome analysis identified wwp2 as a negative regulator of notch3 signaling in ovarian cancer. PLoS Genet. 2014, 10, e1004751. [Google Scholar] [CrossRef] [PubMed]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by escrt complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the escrt machinery: It’s all in the neck. Nat. Rev. Mol. Cell. Biol. 2010, 11, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jiang, D.; Chen, Y.; Wei, L.; Zhang, S.; Zhao, F.; Ni, R.; Lu, C.; Wan, C. High chmp4b expression is associated with accelerated cell proliferation and resistance to doxorubicin in hepatocellular carcinoma. Tumour. Biol. 2015, 36, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Li, X.; Chen, J.L.; Sun, X.; Cooper, F.N.; Chen, Y.R.; Zhang, W.; Chung, Y.; Li, A.; Cheng, C.T.; et al. Identification of an aaa atpase vps4b-dependent pathway that modulates epidermal growth factor receptor abundance and signaling during hypoxia. Mol. Cell. Biol. 2012, 32, 1124–1138. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Cottrell, G.S.; Gehringer, D.; Schmidlin, F.; Grady, E.F.; Bunnett, N.W. C-cbl mediates ubiquitination, degradation, and down-regulation of human protease-activated receptor 2. J. Biol. Chem. 2005, 280, 16076–16087. [Google Scholar] [CrossRef] [PubMed]
- Levkowitz, G.; Waterman, H.; Zamir, E.; Kam, Z.; Oved, S.; Langdon, W.Y.; Beguinot, L.; Geiger, B.; Yarden, Y. C-cbl/sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 1998, 12, 3663–3674. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Mullane-Robinson, K.P.; Lill, N.L.; Douillard, P.; Band, H. Cbl-mediated negative regulation of platelet-derived growth factor receptor-dependent cell proliferation. A critical role for cbl tyrosine kinase-binding domain. J. Biol. Chem. 1999, 274, 16619–16628. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Wang, Y.; Dominguez, M.G.; Yeung, Y.G.; Murphy, M.A.; Bowtell, D.D.; Stanley, E.R. The cbl protooncoprotein stimulates csf-1 receptor multiubiquitination and endocytosis, and attenuates macrophage proliferation. EMBO J. 1999, 18, 3616–3628. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, A.; Gilestro, G.F.; Lanzardo, S.; Comoglio, P.M.; Migone, N.; Giordano, S. The endophilin-cin85-cbl complex mediates ligand-dependent downregulation of c-met. Nature 2002, 416, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Liyasova, M.S.; Ma, K.; Lipkowitz, S. Molecular pathways: Cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin. Cancer Res. 2015, 21, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Bache, K.G.; Gillooly, D.J.; Madshus, I.H.; Stang, E.; Stenmark, H. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat. Cell Biol. 2002, 4, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Bishop, N.; Horman, A.; Woodman, P. Mammalian class e vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 2002, 157, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Hasdemir, B.; Bunnett, N.W.; Cottrell, G.S. Hepatocyte growth factor-regulated tyrosine kinase substrate (hrs) mediates post-endocytic trafficking of protease-activated receptor 2 and calcitonin receptor-like receptor. J. Biol. Chem. 2007, 282, 29646–29657. [Google Scholar] [CrossRef] [PubMed]
- Hasdemir, B.; Murphy, J.E.; Cottrell, G.S.; Bunnett, N.W. Endosomal deubiquitinating enzymes control ubiquitination and down-regulation of protease-activated receptor 2. J. Biol. Chem. 2009, 284, 28453–28466. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima, M.; Tanaka, N.; Aoki, J.; Tanaka, Y.; Murata, K.; Kyuuma, M.; Kobayashi, H.; Ishii, N.; Yaegashi, N.; Sugamura, K. Inhibition of tumor growth and metastasis by depletion of vesicular sorting protein hrs: Its regulatory role on e-cadherin and beta-catenin. Cancer Res. 2007, 67, 5162–5171. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PARs | Cancers with Upregulated PARs | Cancers with Downregulated PARs |
|---|---|---|
| PAR1 | Breast invasive carcinoma Colon adenocarcinoma Lymphoid neoplasm diffuse large B-cell lymphoma Esophageal carcinoma Glioblastoma multiforme Head and neck squamous cell carcinoma Kidney renal clear cell carcinoma Brain lower grade glioma Ovarian serous cystadenocarcinoma Pancreatic adenocarcinoma Rectum adenocarcinoma Stomach adenocarcinoma Thymoma | Kidney chromophobe Kidney renal papillary cell carcinoma β |
| PAR2 | Cervical squamous cell carcinoma and endocervical adenocarcinomaColon adenocarcinoma Esophageal carcinoma Glioblastoma multiforme Acute myeloid leukemia Lung adenocarcinoma Lung squamous cell carcinoma Ovarian serous cystadenocarcinoma Pancreatic adenocarcinoma Prostate adenocarcinoma Rectum adenocarcinoma Stomach adenocarcinoma Testicular germ cell tumors Uterine corpus endometrial carcinoma Uterine carcinosarcoma | Kidney chromophobe Skin cutaneous melanoma |
| PAR3 | Breast invasive carcinoma Esophageal carcinoma Head and neck squamous cell carcinoma Pancreatic adenocarcinoma Stomach adenocarcinoma | ND |
| PAR4 | Kidney renal clear cell carcinoma Pancreatic adenocarcinoma | Acute myeloid leukemia Lung adenocarcinoma Lung squamous cell carcinoma Testicular germ cell tumors Thyroid carcinoma |
| Adaptors | Cancers with Upregulated Adaptors | Cancers with Downregulated Adaptors |
|---|---|---|
| PDCD6IP (ALIX) | Glioblastoma multiforme Brain lower grade glioma Pancreatic adenocarcinoma | Adrenocortical carcinoma Uterine corpus endometrial carcinoma Uterine carcinosarcoma |
| STAMBP (AMSH) | Cholangio carcinoma Lymphoid neoplasm diffuse large B-cell lymphoma Glioblastoma multiforme Brain lower grade glioma Lung squamous carcinoma Pancreatic adenocarcinoma Stomach adenocarcinoma Thymoma | ND |
| AP2A1 (AP-2) | Pancreatic adenocarcinoma | Testicular germ cell tumor Uterine corpus endometrial carcinoma |
| AP3B1 (AP-3) | Cholangio carcinoma Lymphoid neoplasm diffuse large B-cell lymphoma Glioblastoma multiforme Brain lower grade glioma Liver hepatocellular carcinoma Pancreatic adenocarcinoma Thymoma | ND |
| ARRDC3 | Glioblastoma multiforme Brain lower grade glioma Pancreatic adenocarcinoma | Breast invasive carcinoma Kidney chromophobe Ovarian serous cystadenocarcinoma Pheochromocytoma and paraganglioma |
| ARRB1 (β-arrestin-1) | Acute myeloid leukemia | Adrenocortical carcinoma Bladder urothelial carcinoma Cervical squamous cell carcinoma Glioblastoma multiforme Kidney chromophobe Lung squamous carcinoma Uterine corpus endometrial carcinoma |
| ARRB2 (β-arrestin-2) | Kidney chromophobe Kidney renal clear cell carcinoma Acute myeloid leukemia Pancreatic adenocarcinoma | Adrenocortical carcinoma Bladder urothelial carcinoma Lung adenocarcinoma Lung squamous cell carcinoma Thymoma |
| CBL | Cholangio carcinoma Brain lower grade glioma Pancreatic adenocarcinoma | Testicular germ cell tumors Uterine corpus endometrial carcinoma |
| CHMP4B | Cholangio carcinoma Lymphoid neoplasm diffuse large B-cell lymphoma Liver hepatocellular carcinoma Pancreatic adenocarcinoma Thymoma | ND |
| EPN1 (Epsin-1) | Lymphoid neoplasm diffuse large B-cell lymphoma Thymoma | Testicular germ cell tumor |
| HGS (Hrs) | Cholangio carcinoma Pancreatic adenocarcinoma Thymoma | Testicular germ cell tumors |
| RAB11A | Cholangio carcinoma Colon adenocarcinoma Lymphoid neoplasm diffuse large B-cell lymphoma Glioblastoma multiforme Ovarian serous cystadenocarcinoma Pancreatic adenocarcinoma Prostate adenocarcinoma Rectal adenocarcinoma Stomach adenocarcinoma Thymoma | Acute myeloid leukemia |
| RAB11B | Cholangio carcinoma Thymoma | ND |
| SNX1 | Glioblastoma multiforme Brain lower grade glioma Pancreatic adenocarcinoma Thymoma | ND |
| TSG101 | Cholangio carcinoma Lymphoid neoplasm diffuse large B-cell lymphoma Glioblastoma multiforme Brain lower grade glioma Pancreatic adenocarcinoma Thymoma | ND |
| USP8 | Brain lower grade glioma Pancreatic adenocarcinoma Thymoma | ND |
| WWP2 | Cholangio carcinoma Acute myeloid leukemia | Uterine corpus endometrial carcinoma Uterine carcinosarcoma |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arakaki, A.K.S.; Pan, W.-A.; Trejo, J. GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling. Int. J. Mol. Sci. 2018, 19, 1886. https://doi.org/10.3390/ijms19071886
Arakaki AKS, Pan W-A, Trejo J. GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling. International Journal of Molecular Sciences. 2018; 19(7):1886. https://doi.org/10.3390/ijms19071886
Chicago/Turabian StyleArakaki, Aleena K. S., Wen-An Pan, and JoAnn Trejo. 2018. "GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling" International Journal of Molecular Sciences 19, no. 7: 1886. https://doi.org/10.3390/ijms19071886
APA StyleArakaki, A. K. S., Pan, W.-A., & Trejo, J. (2018). GPCRs in Cancer: Protease-Activated Receptors, Endocytic Adaptors and Signaling. International Journal of Molecular Sciences, 19(7), 1886. https://doi.org/10.3390/ijms19071886