Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update

 ,
, 

{kind=link}
Abstract
1. Introduction
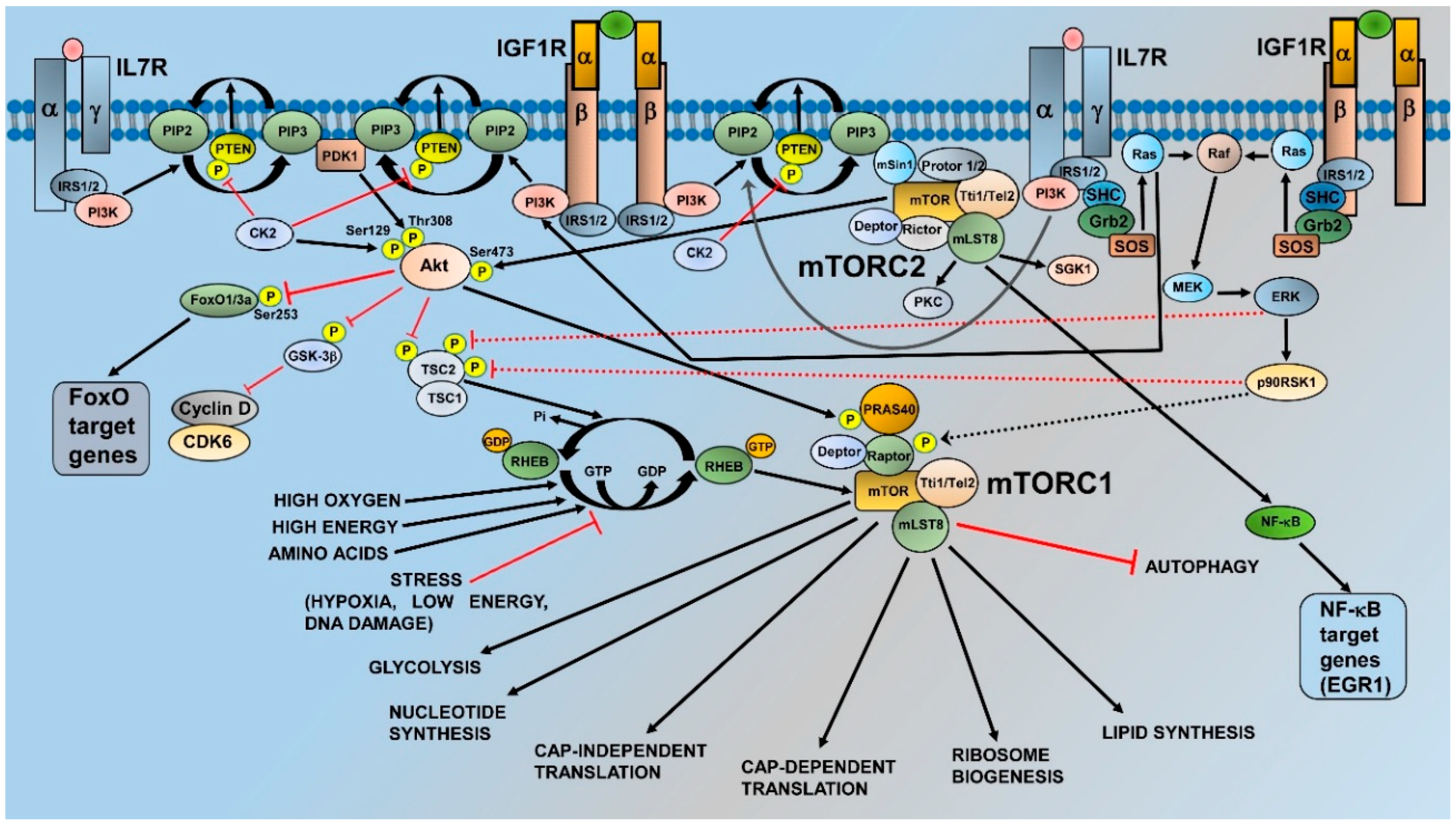
2. mTOR and Its Complexes: Structure, Activation, and Functions
3. Activation of mTORC1 and mTORC2 in T-ALL Cells
3.1. Phosphatase and Tensin Deleted on Chromsome 10
3.2. NOTCH1 Signaling
3.3. RAS Signaling
3.4. RTK Signaling
3.5. IL7 Signaling
3.6. Integrins and Chemokines
3.7. PI3K Activating Mutations
3.8. mTOR Mutations
4. Roles of mTORC1 and mTORC2 in T-ALL
5. Therapeutic Targeting of mTORC1 and mTORC2 in T-ALL Cells: Preclinical Studies
5.1. Allosteric mTOR Inhibitors
5.2. Dual PI3K/mTOR Inhibitors
5.3. TORKIs
6. Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4E-BP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| Bcl-2 | B-cell lymphoma-2 |
| BIM | Bcl-2-like protein 11 |
| CCR | C-C chemokine receptor |
| CDK | Cyclin dependent kinase |
| CK2 | Casein kinase 2 |
| c-Myc | c-Myelocytomatosis oncogene protein |
| Cn | Calcineurin |
| CVAD | Cyclophosphamide, vincristine, adriamycin, dexamethasone |
| CXCR | C-X-C chemokine receptor |
| DCs | Dendritic cells |
| DEP | Dishevelled, Egl-10 and Pleckstrin |
| Deptor | DEP domain-containing mTOR-interacting protein |
| DNMT | DNA methyltransferase |
| EGR1 | Early Growth Response 1 |
| eIF4E | Eukaryotic translation initiation factor 4E |
| ERK | Extracellular signal-regulated kinase |
| ETP | Early T-cell-progenitor |
| EZH2 | Enhancer of Zeste 2 |
| FAT | FRAP, ATM, TRRAP |
| FoxO1/3a | Forkhead box O1/3a |
| GAP | GTP-ase activing protein |
| GCs | Glucocorticoids |
| GSI | γ-secretase inhibitor |
| Grb2 | Growth factor receptor-bound protein 2 |
| GSK3β | Glycogen synthase kinase 3β |
| H-RAS | Harvey-RAS |
| HER2 | human epidermal growth factor receptor 2 |
| HES1 | Hairy and enhancer of split-1 |
| HSP90 | Heat shock protein 90 |
| JAK3 | Janus kinase 3 |
| ICN1 | Intracellular polypeptide of NOTCH1 |
| IGF-1 | Insulin-like growth factor-1 |
| IGF1R | Insulin-like growth factor-1 receptor |
| IL | Interleukin |
| IL7R | Interleukin 7 receptor |
| IRS1/2 | Insulin receptor substrate 1/2 |
| K-RAS | Kirsten-RAS |
| MAML1 | Mastermind like transcriptional coactivator 1 |
| MCL-1 | Myeloid leukemia cell differentiation |
| MEK | Mitogen-activated protein kinase kinase |
| mLST8 | Mammalian lethal with SEC13 protein 8 |
| MNKs | Mitogen-activated protein kinase interacting kinases |
| mSin1 | Mammalian Stress-activated protein kinase interacting protein 1 |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | mTOR complex 1 |
| N-RAS | Neuroblastoma-RAS |
| NF-κb | Nuclear factor κ-light-chain-enhancer of activated B cells |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| NSG | NOD-SCID IL2Rγnull |
| NTRK2 | Neurotrophic tyrosine receptor kinase |
| p70S6K1 | p70 Ribosomal protein S6 kinase 1 |
| p90RSK1 | p90 ribosomal S6 kinase 1 |
| PDK1 | Phosphoinositide-dependent kinase 1 |
| PH | Pleckstrin homology |
| PI3K | Phosphatidylinositol 3-kinase |
| PIKK | PI3K-related kinase |
| PIP2 | Phosphatidylinositol 4,5 bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5 trisphosphate |
| PKC | Protein kinase C |
| Pol | RNA polymerase |
| PP2A | Protein phosphatase 2A |
| PPP | Prednisone poor responders |
| PRAS40 | Proline-rich Akt substrate 1 40 |
| Protor | Protein observed with Rictor |
| PTEN | Phosphatase and tensin deleted on chromosome 10 |
| PUMA | p53 upregulated modulator of apoptosis |
| Raf | Rapidly accelerated fibrosarcoma |
| Raptor | Regulatory associated protein of mTOR |
| RAS | Rat sarcoma |
| RAS-GAP | RAS GTPase activating protein |
| RASGRP1 | RAS guanine nucleotide-releasing protein 1 |
| Rb | Retinoblastoma protein |
| Rheb | RAS homolog enriched in brain |
| Rictor | Rapamycin independent companion of tor |
| RTK | Receptor tyrosine kinase |
| S6RP | S6 ribosomal protein |
| SGK1 | Serum and glucocorticoid-activated kinase 1 |
| SOS | Son of sevenless homolog |
| T-ALL | T-cell acute lymphoblastic leukemia |
| TORKIs | ATP-competitive mTOR kinase inhibitors |
| TSC1 | Tuberous scelerosis complex 1 |
| TSC2 | Tuberous sclerosis complex 2 |
| Tti1 | Tel2-interacting protein 1 |
References
- Paul, S.; Kantarjian, H.; Jabbour, E.J. Adult acute lymphoblastic leukemia. Mayo Clin. Proc. 2016, 91, 1645–1666. [Google Scholar] [CrossRef] [PubMed]
- Vadillo, E.; Dorantes-Acosta, E.; Pelayo, R.; Schnoor, M. T cell acute lymphoblastic leukemia (T-ALL): New insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018, 32, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Raetz, E.A.; Teachey, D.T. T-cell acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Gianfelici, V.; Chiaretti, S.; Demeyer, S.; Di Giacomo, F.; Messina, M.; La Starza, R.; Peragine, N.; Paoloni, F.; Geerdens, E.; Pierini, V.; et al. RNA sequencing unravels the genetics of refractory/relapsed T-cell acute lymphoblastic leukemia. Prognostic and therapeutic implications. Haematologica 2016, 101, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Richter-Pechanska, P.; Kunz, J.B.; Hof, J.; Zimmermann, M.; Rausch, T.; Bandapalli, O.R.; Orlova, E.; Scapinello, G.; Sagi, J.C.; Stanulla, M.; et al. Identification of a genetically defined ultra-high-risk group in relapsed pediatric T-lymphoblastic leukemia. Blood Cancer J. 2017, 7, e523. [Google Scholar] [CrossRef] [PubMed]
- Teepen, J.C.; van Leeuwen, F.E.; Tissing, W.J.; van Dulmen-den Broeder, E.; van den Heuvel-Eibrink, M.M.; van der Pal, H.J.; Loonen, J.J.; Bresters, D.; Versluys, B.; Neggers, S.; et al. Long-term risk of subsequent malignant neoplasms after treatment of childhood cancer in the DCOG LATER study cohort: Role of chemotherapy. J. Clin. Oncol. 2017, 35, 2288–2298. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.L.; Akkapeddi, P.; Alcobia, I.; Almeida, A.R.; Cardoso, B.A.; Fragoso, R.; Serafim, T.L.; Barata, J.T. From the outside, from within: Biological and therapeutic relevance of signal transduction in T-cell acute lymphoblastic leukemia. Cell. Signal. 2017, 38, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Evangelisti, C.; Chiarini, F.; Lonetti, A.; Buontempo, F.; Bressanin, D.; Cappellini, A.; Orsini, E.; McCubrey, J.A.; Martelli, A.M. Therapeutic potential of targeting mTOR in T-cell acute lymphoblastic leukemia. Int. J. Oncol. 2014, 45, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Khanna, A.; Bhushan, B.; Chauhan, P.S.; Saxena, S.; Gupta, D.K.; Siraj, F. High mTOR expression independently prognosticates poor clinical outcome to induction chemotherapy in acute lymphoblastic leukemia. Clin. Exp. Med. 2018, 18, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Lench, N.J.; Macadam, R.; Markham, A.F. The human gene encoding FKBP-rapamycin associated protein (FRAP) maps to chromosomal band 1p36.2. Hum. Genet. 1997, 99, 547–549. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Wang, L.; Harris, T.E.; Lawrence, J.C., Jr. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem. 2008, 283, 15619–15627. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Hong, S.; Suzuki, T.; Nada, S.; Mannan, A.M.; Wang, J.; Okada, M.; Guan, K.L.; Inoki, K. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J. Biol. Chem. 2011, 286, 32651–32660. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Alain, T.; Finestone, L.K.; Huang, B.P.; Rolfe, M.; Jiang, T.; Yao, Z.; Hernandez, G.; Bennett, C.F.; Proud, C.G. Pharmacological and genetic evaluation of proposed roles of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK), extracellular signal-regulated kinase (ERK), and p90(RSK) in the control of mTORC1 protein signaling by phorbol esters. J. Biol. Chem. 2011, 286, 27111–27122. [Google Scholar] [CrossRef] [PubMed]
- Carriere, A.; Cargnello, M.; Julien, L.A.; Gao, H.; Bonneil, E.; Thibault, P.; Roux, P.P. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 2008, 18, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Rad, E.; Murray, J.T.; Tee, A.R. Oncogenic signalling through mechanistic target of rapamycin (mTOR): A driver of metabolic transformation and cancer progression. Cancers 2018, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR pathways in cancer and autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-dependent activation of the mTORC2 kinase complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Sommer, E.M.; Sakamoto, K.; Wullschleger, S.; Alessi, D.R. Protor-1 is required for efficient mTORC2-mediated activation of SGK1 in the kidney. Biochem. J. 2011, 436, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 2017, 32, 807.e12–823.e12. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Franklin, R.A.; Montalto, G.; Cervello, M.; Libra, M.; Candido, S.; Malaponte, G.; et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: How mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012, 3, 1068–1111. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, N.; Lin, Y.; Travis, G.; Simpson, A.M.; McGowan, E.M.; Nassif, N.T. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol. Cancer 2018, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.D.; Cante-Barrett, K.; Pieters, R.; Meijerink, J.P. The relevance of PTEN-AKT in relation to NOTCH1-directed treatment strategies in T-cell acute lymphoblastic leukemia. Haematologica 2016, 101, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Tesio, M.; Trinquand, A.; Macintyre, E.; Asnafi, V. Oncogenic PTEN functions and models in T-cell malignancies. Oncogene 2016, 35, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.; Trinquand, A.; Chevret, S.; Ballerini, P.; Cayuela, J.M.; Grardel, N.; Touzart, A.; Brethon, B.; Lapillonne, H.; Schmitt, C.; et al. Oncogenetic mutations combined with MRD improve outcome prediction in pediatric T-cell acute lymphoblastic leukemia. Blood 2018, 131, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.; Yunes, J.A.; Cardoso, B.A.; Martins, L.R.; Jotta, P.Y.; Abecasis, M.; Nowill, A.E.; Leslie, N.R.; Cardoso, A.A.; Barata, J.T. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J. Clin. Investig. 2008, 118, 3762–3774. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, F.; McCubrey, J.A.; Orsini, E.; Ruzzene, M.; Cappellini, A.; Lonetti, A.; Evangelisti, C.; Chiarini, F.; Evangelisti, C.; Barata, J.T.; et al. Therapeutic targeting of CK2 in acute and chronic leukemias. Leukemia 2018, 32, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Evangelisti, C.; Melchionda, F.; et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2014, 28, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Di Maira, G.; Brustolon, F.; Pinna, L.A.; Ruzzene, M. Dephosphorylation and inactivation of Akt/PKB is counteracted by protein kinase CK2 in HEK 293T cells. Cell. Mol. Life Sci. 2009, 66, 3363–3373. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Yang, Y.; Chen, J.; Li, W.; Li, W.; Zhang, Q.; Mi, Y.; Goswami, R.S.; You, J.Q.; Lin, D.; et al. Regulation of PI3K signaling in T-cell acute lymphoblastic leukemia: A novel PTEN/Ikaros/miR-26b mechanism reveals a critical targetable role for PIK3CD. Leukemia 2017, 31, 2355–2364. [Google Scholar] [CrossRef] [PubMed]
- Yuzugullu, H.; Von, T.; Thorpe, L.M.; Walker, S.R.; Roberts, T.M.; Frank, D.A.; Zhao, J.J. NTRK2 activation cooperates with PTEN deficiency in T-ALL through activation of both the PI3K-AKT and JAK-STAT3 pathways. Cell Discov. 2016, 2, 16030. [Google Scholar] [CrossRef] [PubMed]
- Efimenko, E.; Dave, U.P.; Lebedeva, I.V.; Shen, Y.; Sanchez-Quintero, M.J.; Diolaiti, D.; Kung, A.; Lannutti, B.J.; Chen, J.; Realubit, R.; et al. PI3Kg/d and NOTCH1 cross-regulate pathways that define the T-cell acute lymphoblastic leukemia disease signature. Mol. Cancer Ther. 2017, 16, 2069–2082. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Sulis, M.L.; Cortina, M.; Real, P.J.; Barnes, K.; Ciofani, M.; Caparros, E.; Buteau, J.; Brown, K.; Perkins, S.L.; et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat. Med. 2007, 13, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Hales, E.C.; Orr, S.M.; Larson Gedman, A.; Taub, J.W.; Matherly, L.H. Notch1 receptor regulates AKT protein activation loop (Thr308) dephosphorylation through modulation of the PP2A phosphatase in phosphatase and tensin homolog (PTEN)-null T-cell acute lymphoblastic leukemia cells. J. Biol. Chem. 2013, 288, 22836–22848. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; Wolfe, A.L.; Oricchio, E.; Palomero, T.; de Keersmaecker, K.; McJunkin, K.; Zuber, J.; James, T.; Khan, A.A.; Leslie, C.S.; et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat. Cell Biol. 2010, 12, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Grebliunaite, R.; Feng, H.; Kozakewich, E.; Zhu, S.; Guo, F.; Payne, E.; Mansour, M.; Dahlberg, S.E.; Neuberg, D.S.; et al. Pten mediates Myc oncogene dependence in a conditional zebrafish model of T cell acute lymphoblastic leukemia. J. Exp. Med. 2011, 208, 1595–1603. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.M.; Weng, A.P.; Tibshirani, R.; Aster, J.C.; Utz, P.J. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood 2007, 110, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Ravitz, M.J.; Chen, L.; Lynch, M.; Schmidt, E.V. c-myc Repression of TSC2 contributes to control of translation initiation and Myc-induced transformation. Cancer Res. 2007, 67, 11209–11217. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Nam, K.T.; Cho, S.H.; Gudapati, P.; Hwang, Y.; Park, D.S.; Potter, R.; Chen, J.; Volanakis, E.; Boothby, M. Vital roles of mTOR complex 2 in Notch-driven thymocyte differentiation and leukemia. J. Exp. Med. 2012, 209, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Trimarchi, T.; Ruocco, M.G.; Reavie, L.; Cathelin, S.; Mar, B.G.; Klinakis, A.; Lukyanov, Y.; Tseng, J.C.; Sen, F.; et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature 2009, 459, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.A.; Hackl, C.; Moser, C.; Fichtner-Feigl, S.; Koehl, G.E.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim. Biophys. Acta 2010, 1803, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Guo, H.; Bu, J.; Zhou, M.; Cheng, H.; He, F.; Wang, J.; Wang, X.; Zhang, Y.; Wang, Q.; et al. Rictor/mammalian target of rapamycin 2 regulates the development of Notch1 induced murine T-cell acute lymphoblastic leukemia via forkhead box O3. Exp. Hematol. 2014, 42, 1031.e4–1040.e4. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Kume, T. Forkhead transcription factors regulate expression of the chemokine receptor CXCR4 in endothelial cells and CXCL12-induced cell migration. Biochem. Biophys. Res. Commun. 2008, 367, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Su, H.; Liu, C.; Wang, Z.; Huang, L.; Wang, Q.; Liu, S.; Chen, S.; Zhou, J.; Li, P.; et al. DEPTOR is a direct NOTCH1 target that promotes cell proliferation and survival in T-cell leukemia. Oncogene 2017, 36, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Varusai, T.M.; Nguyen, L.K. Dynamic modelling of the mTOR signalling network reveals complex emergent behaviours conferred by DEPTOR. Sci. Rep. 2018, 8, 643. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, J.; Chapuis, N.; Bardet, V.; Park, S.; Sujobert, P.; Willems, L.; Ifrah, N.; Dreyfus, F.; Mayeux, P.; Lacombe, C.; et al. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: Rationale for therapeutic inhibition of both pathways. Blood 2008, 111, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Kano, Y.; Cook, J.D.; Lee, J.E.; Ohh, M. New structural and functional insight into the regulation of Ras. Semin. Cell Dev. Biol. 2016, 58, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Danis, E.; Yamauchi, T.; Echanique, K.; Zhang, X.; Haladyna, J.N.; Riedel, S.S.; Zhu, N.; Xie, H.; Orkin, S.H.; Armstrong, S.A.; et al. Ezh2 Controls an early hematopoietic program and growth and survival signaling in early T cell precursor acute lymphoblastic leukemia. Cell Rep. 2016, 14, 1953–1965. [Google Scholar] [CrossRef] [PubMed]
- Kindler, T.; Cornejo, M.G.; Scholl, C.; Liu, J.; Leeman, D.S.; Haydu, J.E.; Frohling, S.; Lee, B.H.; Gilliland, D.G. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to g-secretase inhibitors. Blood 2008, 112, 3373–3382. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Du, J.; Liu, Y.; Meline, B.; Chang, Y.I.; Ranheim, E.A.; Wang, J.; Zhang, J. Notch1 gene mutations target KRAS G12D-expressing CD8+ cells and contribute to their leukemogenic transformation. J. Biol. Chem. 2013, 288, 18219–18227. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.D.; Hixon, J.A.; Andrews, C.; Porter, R.J.; Rodrigues, G.O.L.; Wu, X.; Back, T.; Czarra, K.; Michael, H.; Cam, M.; et al. Mutant IL-7Ra and mutant NRas are sufficient to induce murine T cell acute lymphoblastic leukemia. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Von Lintig, F.C.; Huvar, I.; Law, P.; Diccianni, M.B.; Yu, A.L.; Boss, G.R. Ras activation in normal white blood cells and childhood acute lymphoblastic leukemia. Clin. Cancer Res. 2000, 6, 1804–1810. [Google Scholar] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Yokota, S.; Nakao, M.; Horiike, S.; Seriu, T.; Iwai, T.; Kaneko, H.; Azuma, H.; Oka, T.; Takeda, T.; Watanabe, A.; et al. Mutational analysis of the N-ras gene in acute lymphoblastic leukemia: A study of 125 Japanese pediatric cases. Int. J. Hematol. 1998, 67, 379–387. [Google Scholar] [CrossRef]
- Perentesis, J.P.; Bhatia, S.; Boyle, E.; Shao, Y.; Shu, X.O.; Steinbuch, M.; Sather, H.N.; Gaynon, P.; Kiffmeyer, W.; Envall-Fox, J.; et al. RAS oncogene mutations and outcome of therapy for childhood acute lymphoblastic leukemia. Leukemia 2004, 18, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Wiemels, J.L.; Zhang, Y.; Chang, J.; Zheng, S.; Metayer, C.; Zhang, L.; Smith, M.T.; Ma, X.; Selvin, S.; Buffler, P.A.; et al. RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia 2005, 19, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Lamb, A.V.; O’Brien, S.; Ravandi, F.; Konopleva, M.; Jabbour, E.; Zuo, Z.; Jorgensen, J.; Lin, P.; Pierce, S.; et al. Early T-cell precursor acute lymphoblastic leukemia/lymphoma (ETP-ALL/LBL) in adolescents and adults: A high-risk subtype. Blood 2016, 127, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Hoshii, T.; Kasada, A.; Hatakeyama, T.; Ohtani, M.; Tadokoro, Y.; Naka, K.; Ikenoue, T.; Ikawa, T.; Kawamoto, H.; Fehling, H.J.; et al. Loss of mTOR complex 1 induces developmental blockage in early T-lymphopoiesis and eradicates T-cell acute lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3805–3810. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, C.; Ksionda, O.; Lemmens, E.; Coakley, K.; Yang, M.; Dail, M.; Harvey, R.C.; Govern, C.; Bakker, J.; Lenstra, T.L.; et al. Dysregulated RasGRP1 responds to cytokine receptor input in T cell leukemogenesis. Sci. Signal. 2013, 6, ra21. [Google Scholar] [CrossRef] [PubMed]
- Ksionda, O.; Melton, A.A.; Bache, J.; Tenhagen, M.; Bakker, J.; Harvey, R.; Winter, S.S.; Rubio, I.; Roose, J.P. RasGRP1 overexpression in T-ALL increases basal nucleotide exchange on Ras rendering the Ras/PI3K/Akt pathway responsive to protumorigenic cytokines. Oncogene 2016, 35, 3658–3668. [Google Scholar] [CrossRef] [PubMed]
- Biagi, C.; Astolfi, A.; Masetti, R.; Serravalle, S.; Franzoni, M.; Chiarini, F.; Melchionda, F.; Pession, A. Pediatric early T-cell precursor leukemia with NF1 deletion and high-sensitivity in vitro to tipifarnib. Leukemia 2010, 24, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, B.A.; Lapinski, P.E.; Oliver, J.A.; Ksionda, O.; Parada, L.F.; Zhu, Y.; Maillard, I.; Chiang, M.; Roose, J.; King, P.D. Cutting Edge: Codeletion of the Ras GTPase-activating proteins (RasGAPs) neurofibromin 1 and p120 RasGAP in T cells results in the development of T cell acute lymphoblastic leukemia. J. Immunol. 2015, 195, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Mues, M.; Roose, J.P. Distinct oncogenic Ras signals characterized by profound differences in flux through the RasGDP/RasGTP cycle. Small GTPases 2017, 8, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Ksionda, O.; Mues, M.; Wandler, A.M.; Donker, L.; Tenhagen, M.; Jun, J.; Ducker, G.S.; Matlawska-Wasowska, K.; Shannon, K.; Shokat, K.M.; et al. Comprehensive analysis of T cell leukemia signals reveals heterogeneity in the PI3 kinase-Akt pathway and limitations of PI3 kinase inhibitors as monotherapy. PLoS ONE 2018, 13, e0193849. [Google Scholar] [CrossRef] [PubMed]
- Gusscott, S.; Jenkins, C.E.; Lam, S.H.; Giambra, V.; Pollak, M.; Weng, A.P. IGF1R Derived PI3K/AKT signaling maintains growth in a subset of human T-cell acute lymphoblastic leukemias. PLoS ONE 2016, 11, e0161158. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Gusscott, S.; Wang, H.; Tseng, J.C.; Wai, C.; Nemirovsky, O.; Trumpp, A.; Pflumio, F.; Carboni, J.; Gottardis, M.; et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J. Exp. Med. 2011, 208, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P.; Fabbri, G.; Dalla-Favera, R.; Tsirigos, A.; Aifantis, I. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Gusscott, S.; Kuchenbauer, F.; Humphries, R.K.; Weng, A.P. Notch-mediated repression of miR-223 contributes to IGF1R regulation in T-ALL. Leuk. Res. 2012, 36, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Triplett, T.A.; Cardenas, K.T.; Lancaster, J.N.; Hu, Z.; Selden, H.J.; Jasso, G.J.; Balasubramanyam, S.; Chan, K.; Li, L.; Chen, X.; et al. Endogenous dendritic cells from the tumor microenvironment support T-ALL growth via IGF1R activation. Proc. Natl. Acad. Sci. USA 2016, 113, E1016–E1025. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Buijs-Gladdines, J.G.; Cante-Barrett, K.; Stubbs, A.P.; Vroegindeweij, E.M.; Smits, W.K.; van Marion, R.; Dinjens, W.N.; Horstmann, M.; Kuiper, R.P.; et al. IL-7 receptor mutations and steroid resistance in pediatric T cell acute lymphoblastic leukemia: A genome sequencing study. PLoS Med. 2016, 13, e1002200. [Google Scholar] [CrossRef] [PubMed]
- Melao, A.; Spit, M.; Cardoso, B.A.; Barata, J.T. Optimal interleukin-7 receptor-mediated signaling, cell cycle progression and viability of T-cell acute lymphoblastic leukemia cells rely on casein kinase 2 activity. Haematologica 2016, 101, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Cante-Barrett, K.; Spijkers-Hagelstein, J.A.; Buijs-Gladdines, J.G.; Uitdehaag, J.C.; Smits, W.K.; van der Zwet, J.; Buijsman, R.C.; Zaman, G.J.; Pieters, R.; Meijerink, J.P. MEK and PI3K-AKT inhibitors synergistically block activated IL7 receptor signaling in T-cell acute lymphoblastic leukemia. Leukemia 2016, 30, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garcia, S.; Garcia-Peydro, M.; Alcain, J.; Toribio, M.L. Notch1 and IL-7 receptor signalling in early T-cell development and leukaemia. Curr. Top. Microbiol. Immunol. 2012, 360, 47–73. [Google Scholar] [CrossRef] [PubMed]
- Keely, P.J. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia 2011, 16, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.X.; Zhou, C.H.; Zeng, H.; Zuo, D.Q.; Wang, Z.Y.; Yin, F.; Hua, Y.Q.; Cai, Z.D. The role of the CXCL12-CXCR4/CXCR7 axis in the progression and metastasis of bone sarcomas. Int. J. Mol. Med. 2013, 32, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Naci, D.; Aoudjit, F. a2b1 integrin promotes T cell survival and migration through the concomitant activation of ERK/Mcl-1 and p38 MAPK pathways. Cell. Signal. 2014, 26, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Piovan, E.; Tosello, V.; Amadori, A.; Zanovello, P. Chemotactic Cues for NOTCH1-Dependent Leukemia. Front. Immunol. 2018, 9, 633. [Google Scholar] [CrossRef] [PubMed]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Sanda, T.; Grebliunaite, R.; Carracedo, A.; Salmena, L.; Ahn, Y.; Dahlberg, S.; Neuberg, D.; Moreau, L.A.; Winter, S.S.; et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 2009, 114, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Buontempo, F.; McCubrey, J.A. Drug discovery targeting the mTOR pathway. Clin. Sci. (Lond.) 2018, 132, 543–568. [Google Scholar] [CrossRef] [PubMed]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.P.; Marshall, C.B.; Coric, T.; Shim, E.H.; Kirkman, R.; Ballestas, M.E.; Ikura, M.; Bjornsti, M.A.; Sudarshan, S. Point mutations of the mTOR-RHEB pathway in renal cell carcinoma. Oncotarget 2015, 6, 17895–17910. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Cardenas, A.; Chen, H.; Garcia, C.; Guo, W.; Bradner, J.; Wu, H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer Res. 2014, 74, 7048–7059. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Rhein, M.; Schiedlmeier, B.; Kustikova, O.; Rudolph, C.; Kamino, K.; Neumann, T.; Yang, M.; Wahlers, A.; Fehse, B.; et al. Remarkable leukemogenic potency and quality of a constitutively active neurotrophin receptor, DTrkA. Leukemia 2007, 21, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, A.; Holtmann, H.; Brugman, M.; Meyer, J.; Schauerte, C.; Zuber, J.; Steinemann, D.; Schlegelberger, B.; Li, Z.; Baum, C. Hyperactivation of mTORC1 and mTORC2 by multiple oncogenic events causes addiction to eIF4E-dependent mRNA translation in T-cell leukemia. Oncogene 2015, 34, 3593–3604. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, N.; Sonenberg, N. Signalling to eIF4E in cancer. Biochem. Soc. Trans. 2015, 43, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, J.; Wen, Q.; Luo, J.; Chu, S.; Chen, L.; Qing, Z.; Xie, G.; Xu, L.; Alnemah, M.M.; et al. 4EGI-1 induces apoptosis and enhances radiotherapy sensitivity in nasopharyngeal carcinoma cells via DR5 induction on 4E-BP1 dephosphorylation. Oncotarget 2016, 7, 21728–21741. [Google Scholar] [CrossRef] [PubMed]
- Kosciuczuk, E.M.; Saleiro, D.; Platanias, L.C. Dual targeting of eIF4E by blocking MNK and mTOR pathways in leukemia. Cytokine 2017, 89, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Martelli, A.M.; Cocco, L.; Libra, M.; Nicoletti, F.; Abrams, S.L.; McCubrey, J.A. The therapeutic potential of mTOR inhibitors in breast cancer. Br. J. Clin. Pharmacol. 2016, 82, 1189–1212. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Chiarini, F.; Evangelisti, C.; Cappellini, A.; Buontempo, F.; Bressanin, D.; Fini, M.; McCubrey, J.A. Two hits are better than one: Targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget 2012, 3, 371–394. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Vo, T.T.; Fruman, D.A. Targeting mTOR for the treatment of B cell malignancies. Br. J. Clin. Pharmacol. 2016, 82, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Barata, J.T.; Cardoso, A.A.; Nadler, L.M.; Boussiotis, V.A. Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27(kip1). Blood 2001, 98, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.; Barata, J.T.; Raderschall, E.; Sallan, S.E.; Carlesso, N.; Nadler, L.M.; Cardoso, A.A. Targeting of active mTOR inhibits primary leukemia T cells and synergizes with cytotoxic drugs and signaling inhibitors. Exp. Hematol. 2011, 39, 457.e3–472.e3. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Arakawa, H.; Tanaka, T.; Matsuda, K.; Tanikawa, C.; Mori, T.; Nishimori, H.; Tamai, K.; Tokino, T.; Nakamura, Y.; et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 2000, 102, 849–862. [Google Scholar] [CrossRef]
- Li, H.; Kong, X.; Cui, G.; Ren, C.; Fan, S.; Sun, L.; Zhang, Y.; Cao, R.; Li, Y.; Zhou, J. Rapamycin restores p14, p15 and p57 expression and inhibits the mTOR/p70S6K pathway in acute lymphoblastic leukemia cells. Int. J. Hematol. 2015, 102, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Huang, Y.T.; Huang, T.S.; Pang, W.; Zhu, J.J.; Liu, Y.F.; Tang, R.Z.; Zhao, C.R.; Yao, W.J.; Li, Y.S.; et al. The mammalian target of rapamycin and DNA methyltransferase 1 axis mediates vascular endothelial dysfunction in response to disturbed flow. Sci. Rep. 2017, 7, 14996. [Google Scholar] [CrossRef] [PubMed]
- Avellino, R.; Romano, S.; Parasole, R.; Bisogni, R.; Lamberti, A.; Poggi, V.; Venuta, S.; Romano, M.F. Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood 2005, 106, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.N.; Zhao, Y.M.; He, Y.; Wang, B.S.; Du, K.L.; Fu, S.; Hu, K.M.; Zhang, L.F.; Liu, L.Z.; Hu, Y.X.; et al. Rapamycin interacts synergistically with idarubicin to induce T-leukemia cell apoptosis in vitro and in a mesenchymal stem cell simulated drug-resistant microenvironment via Akt/mammalian target of rapamycin and extracellular signal-related kinase signaling pathways. Leuk. Lymphoma 2014, 55, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hua, C.; Cheng, H.; Wang, W.; Hao, S.; Xu, J.; Wang, X.; Gao, Y.; Zhu, X.; Cheng, T.; et al. Distinct sensitivity of CD8+ CD4− and CD8+ CD4+ leukemic cell subpopulations to cyclophosphamide and rapamycin in Notch1-induced T-ALL mouse model. Leuk. Res. 2013, 37, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Teachey, D.T.; Sheen, C.; Hall, J.; Ryan, T.; Brown, V.I.; Fish, J.; Reid, G.S.; Seif, A.E.; Norris, R.; Chang, Y.J.; et al. mTOR inhibitors are synergistic with methotrexate: An effective combination to treat acute lymphoblastic leukemia. Blood 2008, 112, 2020–2023. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Zhou, C.; Liu, H.; Gao, J.; Li, Q.; Mu, D.; Ma, Z. Rapamycin sensitizes T-ALL cells to dexamethasone-induced apoptosis. J. Exp. Clin. Cancer Res. 2010, 29, 150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ryu, Y.K.; Chen, T.Z.; Hall, C.P.; Webster, D.R.; Kang, M.H. Synergistic activity of rapamycin and dexamethasone in vitro and in vivo in acute lymphoblastic leukemia via cell-cycle arrest and apoptosis. Leuk. Res. 2012, 36, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Serafin, V.; Capuzzo, G.; Milani, G.; Minuzzo, S.A.; Pinazza, M.; Bortolozzi, R.; Bresolin, S.; Porcu, E.; Frasson, C.; Indraccolo, S.; et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood 2017, 130, 2750–2761. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Valsecchi, M.G.; Bartram, C.R.; Schrauder, A.; Panzer-Grumayer, R.; Moricke, A.; Parasole, R.; Zimmermann, M.; Dworzak, M.; Buldini, B.; et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: Results of the AIEOP-BFM-ALL 2000 study. Blood 2011, 118, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhou, C.Y.; Li, Q.; Gao, J.; Zhu, Y.P.; Gu, L.; Ma, Z.G. Rapamycin sensitizes glucocorticoid resistant acute lymphoblastic leukemia CEM-C1 cells to dexamethasone induced apoptosis through both mTOR suppression and up-regulation and activation of glucocorticoid receptor. Biomed. Environ. Sci. 2013, 26, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Cullion, K.; Draheim, K.M.; Hermance, N.; Tammam, J.; Sharma, V.M.; Ware, C.; Nikov, G.; Krishnamoorthy, V.; Majumder, P.K.; Kelliher, M.A. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009, 113, 6172–6181. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, S.; Ricciardi, M.R.; Allegretti, M.; Mirabilii, S.; Licchetta, R.; Bergamo, P.; Rinaldo, C.; Zeuner, A.; Foa, R.; Milella, M.; et al. Co-targeting of Bcl-2 and mTOR pathway triggers synergistic apoptosis in BH3 mimetics resistant acute lymphoblastic leukemia. Oncotarget 2015, 6, 32089–32103. [Google Scholar] [CrossRef] [PubMed]
- Akers, L.J.; Fang, W.; Levy, A.G.; Franklin, A.R.; Huang, P.; Zweidler-McKay, P.A. Targeting glycolysis in leukemia: A novel inhibitor 3-BrOP in combination with rapamycin. Leuk. Res. 2011, 35, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Alexe, G.; Roti, G.; Conway, A.S.; Furman, A.; Lee, E.S.; Place, A.E.; Kim, S.; Saran, C.; Modiste, R.; et al. Synergistic drug combinations with a CDK4/6 inhibitor in T-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2017, 23, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Chilosi, M.; Doglioni, C.; Yan, Z.; Lestani, M.; Menestrina, F.; Sorio, C.; Benedetti, A.; Vinante, F.; Pizzolo, G.; Inghirami, G. Differential expression of cyclin-dependent kinase 6 in cortical thymocytes and T-cell lymphoblastic lymphoma/leukemia. Am. J. Pathol. 1998, 152, 209–217. [Google Scholar] [PubMed]
- Mullighan, C.G.; Phillips, L.A.; Su, X.; Ma, J.; Miller, C.B.; Shurtleff, S.A.; Downing, J.R. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gounari, F.; Protopopov, A.; Khazaie, K.; von Boehmer, H. Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J. Exp. Med. 2008, 205, 2851–2861. [Google Scholar] [CrossRef] [PubMed]
- Joshi, I.; Minter, L.M.; Telfer, J.; Demarest, R.M.; Capobianco, A.J.; Aster, J.C.; Sicinski, P.; Fauq, A.; Golde, T.E.; Osborne, B.A. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood 2009, 113, 1689–1698. [Google Scholar] [CrossRef] [PubMed]
- Sawai, C.M.; Freund, J.; Oh, P.; Ndiaye-Lobry, D.; Bretz, J.C.; Strikoudis, A.; Genesca, L.; Trimarchi, T.; Kelliher, M.A.; Clark, M.; et al. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 2012, 22, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Kovatcheva, M.; Davis, L.E.; Tap, W.D.; Koff, A. CDK4/6 Inhibitors: The mechanism of action may not be as simple as once thought. Cancer Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Witkiewicz, A.K. The strange case of CDK4/6 inhibitors: Mechanisms, resistance, and combination strategies. Trends Cancer 2017, 3, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Yi, S.Y.; Koh, J.; Bae, Y.H.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Ahn, M.J. The CDK4/6 inhibitor LY2835219 has potent activity in combination with mTOR inhibitor in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 14803–14813. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Demartines, N.; Dormond, O. Resistance to mTORC1 inhibitors in cancer therapy: From kinase mutations to intratumoral heterogeneity of kinase activity. Oxid. Med. Cell. Longev. 2017, 2017, 1726078. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Fala, F.; Tazzari, P.L.; Ricci, F.; Astolfi, A.; Pession, A.; Pagliaro, P.; McCubrey, J.A.; Martelli, A.M. Dual inhibition of class IA phosphatidylinositol 3-kinase and mammalian target of rapamycin as a new therapeutic option for T-cell acute lymphoblastic leukemia. Cancer Res. 2009, 69, 3520–3528. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, F.; Grimaldi, C.; Ricci, F.; Tazzari, P.L.; Evangelisti, C.; Ognibene, A.; Battistelli, M.; Falcieri, E.; Melchionda, F.; Pession, A.; et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res. 2010, 70, 8097–8107. [Google Scholar] [CrossRef] [PubMed]
- Gazi, M.; Moharram, S.A.; Marhall, A.; Kazi, J.U. The dual specificity PI3K/mTOR inhibitor PKI-587 displays efficacy against T-cell acute lymphoblastic leukemia (T-ALL). Cancer Lett. 2017, 392, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.; Banerjee, L.; Cheung, C.W.; Mansour, M.R.; Jenkinson, S.; Gale, R.E.; Khwaja, A. PI3K/mTOR inhibition upregulates NOTCH-MYC signalling leading to an impaired cytotoxic response. Leukemia 2013, 27, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Schult, C.; Dahlhaus, M.; Glass, A.; Fischer, K.; Lange, S.; Freund, M.; Junghanss, C. The dual kinase inhibitor NVP-BEZ235 in combination with cytotoxic drugs exerts anti-proliferative activity towards acute lymphoblastic leukemia cells. Anticancer Res. 2012, 32, 463–474. [Google Scholar] [PubMed]
- Hall, C.P.; Reynolds, C.P.; Kang, M.H. Modulation of glucocorticoid resistance in pediatric T-cell acute lymphoblastic leukemia by increasing BIM expression with the PI3K/mTOR inhibitor BEZ235. Clin. Cancer Res. 2016, 22, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Tosello, V.; Saccomani, V.; Yu, J.; Bordin, F.; Amadori, A.; Piovan, E. Calcineurin complex isolated from T-cell acute lymphoblastic leukemia (T-ALL) cells identifies new signaling pathways including mTOR/AKT/S6K whose inhibition synergize with calcineurin inhibition to promote T-ALL cell death. Oncotarget 2016, 7, 45715–45729. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G. Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 2017, 63, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Passaro, D.; Irigoyen, M.; Catherinet, C.; Gachet, S.; Da Costa De Jesus, C.; Lasgi, C.; Tran Quang, C.; Ghysdael, J. CXCR4 is required for leukemia-initiating cell activity in T cell acute lymphoblastic leukemia. Cancer Cell 2015, 27, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Gachet, S.; Genesca, E.; Passaro, D.; Irigoyen, M.; Alcalde, H.; Clemenson, C.; Poglio, S.; Pflumio, F.; Janin, A.; Lasgi, C.; et al. Leukemia-initiating cell activity requires calcineurin in T-cell acute lymphoblastic leukemia. Leukemia 2013, 27, 2289–2300. [Google Scholar] [CrossRef] [PubMed]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef] [PubMed]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, C.; Ricci, F.; Tazzari, P.; Tabellini, G.; Battistelli, M.; Falcieri, E.; Chiarini, F.; Bortul, R.; Melchionda, F.; Pagliaro, P.; et al. Targeted inhibition of mTORC1 and mTORC2 by active-site mTOR inhibitors has cytotoxic effects in T-cell acute lymphoblastic leukemia. Leukemia 2011, 25, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Sim, H.; Lee, K. Rapamycin-resistant and torin-sensitive mTOR signaling promotes the survival and proliferation of leukemic cells. BMB Rep. 2016, 49, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Li, J.Y.; Xu, W. A review of the role of Puma, Noxa and Bim in the tumorigenesis, therapy and drug resistance of chronic lymphocytic leukemia. Cancer Gene Ther. 2013, 20, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Banerjee, S. p27 and leukemia: Cell cycle and beyond. J. Cell. Physiol. 2015, 230, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; Vincelette, N.D.; Knorr, K.L.; Almada, L.L.; Schneider, P.A.; Peterson, K.L.; Flatten, K.S.; Dai, H.; Pratz, K.W.; Hess, A.D.; et al. 4EBP1/c-MYC/PUMA and NF-κB/EGR1/BIM pathways underlie cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells. Blood 2016, 127, 2711–2722. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; O’Brien, S.; Smith, T.L.; Cortes, J.; Giles, F.J.; Beran, M.; Pierce, S.; Huh, Y.; Andreeff, M.; Koller, C.; et al. Results of treatment with hyper-CVAD, a dose-intensive regimen, in adult acute lymphocytic leukemia. J. Clin. Oncol. 2000, 18, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Boumber, Y.; Kantarjian, H.; Ravandi, F.; Cortes, J.; Rytting, M.E.; Kawedia, J.D.; Basnett, J.; Culotta, K.S.; Zeng, Z.; et al. A phase I/II study of the mTOR inhibitor everolimus in combination with HyperCVAD chemotherapy in patients with relapsed/refractory acute lymphoblastic leukemia. Clin. Cancer Res. 2015, 21, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Rheingold, S.R.; Tasian, S.K.; Whitlock, J.A.; Teachey, D.T.; Borowitz, M.J.; Liu, X.; Minard, C.G.; Fox, E.; Weigel, B.J.; Blaney, S.M. A phase 1 trial of temsirolimus and intensive re-induction chemotherapy for 2nd or greater relapse of acute lymphoblastic leukaemia: A Children’s Oncology Group study (ADVL1114). Br. J. Haematol. 2017, 177, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Place, A.E.; Pikman, Y.; Stevenson, K.E.; Harris, M.H.; Pauly, M.; Sulis, M.L.; Hijiya, N.; Gore, L.; Cooper, T.M.; Loh, M.L.; et al. Phase I trial of the mTOR inhibitor everolimus in combination with multi-agent chemotherapy in relapsed childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2018, 65, e27062. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, A.A.; Lopez-Otin, C. Clonal evolution in leukemia. Nat. Med. 2017, 23, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Duval, A.P.; Planche, A.; Uldry, E.; Santoro, T.; Pythoud, C.; Stehle, J.C.; Horlbeck, J.; Letovanec, I.; Riggi, N.; et al. Acidic tumor microenvironment abrogates the efficacy of mTORC1 inhibitors. Mol. Cancer 2016, 15, 78. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; O’Regan, R.; Ozguroglu, M.; Toi, M.; Xu, B.; Jerusalem, G.; Masuda, N.; Wilks, S.; Arena, F.; Isaacs, C.; et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [Google Scholar] [CrossRef]
- Moscetti, L.; Vici, P.; Gamucci, T.; Natoli, C.; Cortesi, E.; Marchetti, P.; Santini, D.; Giuliani, R.; Sperduti, I.; Mauri, M.; et al. Safety analysis, association with response and previous treatments of everolimus and exemestane in 181 metastatic breast cancer patients: A multicenter Italian experience. Breast 2016, 29, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Gatzka, M.V. Targeted tumor therapy remixed-an update on the use of small-molecule drugs in combination therapies. Cancers 2018, 10, 155. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortes, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Davies, M.; Saxena, A.; Kingswood, J.C. Management of everolimus-associated adverse events in patients with tuberous sclerosis complex: A practical guide. Orphanet J. Rare Dis. 2017, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Morviducci, L.; Rota, F.; Rizza, L.; Di Giacinto, P.; Ramponi, S.; Nardone, M.R.; Tubili, C.; Lenzi, A.; Zuppi, P.; Baldelli, R. Everolimus is a new anti-cancer molecule: Metabolic side effects as lipid disorders and hyperglycemia. Diabetes Res. Clin. Pract. 2018. [Google Scholar] [CrossRef] [PubMed]
- Eiden, A.M.; Zhang, S.; Gary, J.M.; Simmons, J.K.; Mock, B.A. Molecular pathways: Increased susceptibility to infection is a complication of mTOR inhibitor use in cancer therapy. Clin. Cancer Res. 2016, 22, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Curwen, J.; Bihani, T.; D’Cruz, C.M.; Yates, J.W.; Grondine, M.; Howard, Z.; Davies, B.R.; Bigley, G.; Klinowska, T.; et al. AZD2014, an inhibitor of mTORC1 and mTORC2, is highly effective in ER+ breast cancer when administered using intermittent or continuous schedules. Mol. Cancer Ther. 2015, 14, 2508–2518. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; LoRusso, P.; Arzt, J.; Busman, T.A.; Lian, G.; Rudersdorf, N.S.; Vanderwal, C.A.; Waring, J.F.; Yang, J.; Holen, K.D.; et al. Safety, efficacy, and pharmacokinetics of navitoclax (ABT-263) in combination with irinotecan: Results of an open-label, phase 1 study. Cancer Chemother. Pharmacol. 2015, 76, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.W.; Dudley, P.; Cheng, J.; D’Cruz, C.; Davies, B.R. Validation of a predictive modeling approach to demonstrate the relative efficacy of three different schedules of the AKT inhibitor AZD5363. Cancer Chemother. Pharmacol. 2015, 76, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Yi, Z.; Ma, F. Biomarkers of everolimus sensitivity in hormone receptor-positive breast cancer. J. Breast Cancer 2017, 20, 321–326. [Google Scholar] [CrossRef]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013, 6, rs6. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xing, X.; Dowlut, D.; Zeng, Y.; Liu, J.; Liu, X. Integrating phosphoproteomics into kinase-targeted cancer therapies in precision medicine. J. Proteom. 2018. [Google Scholar] [CrossRef] [PubMed]
- Van der Sligte, N.E.; Scherpen, F.J.; Meeuwsen-de Boer, T.G.; Lourens, H.J.; Ter Elst, A.; Diks, S.H.; Guryev, V.; Peppelenbosch, M.P.; van Leeuwen, F.N.; de Bont, E.S. Kinase activity profiling reveals active signal transduction pathways in pediatric acute lymphoblastic leukemia: A new approach for target discovery. Proteomics 2015, 15, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Montano, A.; Forero-Castro, M.; Marchena-Mendoza, D.; Benito, R.; Hernandez-Rivas, J.M. New challenges in targeting signaling pathways in acute lymphoblastic leukemia by NGS approaches: An update. Cancers 2018, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Wilkes, E.H.; Miraki-Moud, F.; Hadi, M.M.; Rio-Machin, A.; Rajeeve, V.; Pike, R.; Iqbal, S.; Marfa, S.; Lea, N.; et al. Proteomic and genomic integration identifies kinase and differentiation determinants of kinase inhibitor sensitivity in leukemia cells. Leukemia 2018. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; van der Windt, G.J.; Kishton, R.J.; Cohen, S.; Eisner, W.; MacIver, N.J.; Kater, A.P.; Weinberg, J.B.; Rathmell, J.C. Suppression of Glut1 and glucose metabolism by decreased Akt/mTORC1 signaling drives T cell impairment in B cell leukemia. J. Immunol. 2016, 197, 2532–2540. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Wu, J.; Ye, A.; Wu, J.; Yu, K.; Zhang, S.; Han, Y. Increased Th17 cells and IL-17A exist in patients with B cell acute lymphoblastic leukemia and promote proliferation and resistance to daunorubicin through activation of Akt signaling. J. Transl. Med. 2016, 14, 132. [Google Scholar] [CrossRef] [PubMed]
- Brinda, B.; Khan, I.; Parkin, B.; Konig, H. The rocky road to personalized medicine in acute myeloid leukaemia. J. Cell. Mol. Med. 2018, 22, 1411–1427. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.E.R. Precision medicine for breast cancer: The paths to truly individualized diagnosis and treatment. Int. J. Breast Cancer 2018, 2018, 4809183. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Freeman, M.R.; Kyprianou, N. Personalization of prostate cancer therapy through phosphoproteomics. Nat. Rev. Urol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Yuan, K.; Ding, W.; Lin, M. Notch signaling: A potential therapeutic target for hematologic malignancies. Crit. Rev. Eukaryot. Gene Expr. 2016, 26, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Knoechel, B.; Bhatt, A.; Pan, L.; Pedamallu, C.S.; Severson, E.; Gutierrez, A.; Dorfman, D.M.; Kuo, F.C.; Kluk, M.; Kung, A.L.; et al. Complete hematologic response of early T-cell progenitor acute lymphoblastic leukemia to the γ-secretase inhibitor BMS-906024: Genetic and epigenetic findings in an outlier case. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000539. [Google Scholar] [CrossRef] [PubMed]
- Papayannidis, C.; DeAngelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evangelisti, C.; Chiarini, F.; McCubrey, J.A.; Martelli, A.M. Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update. Int. J. Mol. Sci. 2018, 19, 1878. https://doi.org/10.3390/ijms19071878
Evangelisti C, Chiarini F, McCubrey JA, Martelli AM. Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update. International Journal of Molecular Sciences. 2018; 19(7):1878. https://doi.org/10.3390/ijms19071878
Chicago/Turabian StyleEvangelisti, Camilla, Francesca Chiarini, James A. McCubrey, and Alberto M. Martelli. 2018. "Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update" International Journal of Molecular Sciences 19, no. 7: 1878. https://doi.org/10.3390/ijms19071878
APA StyleEvangelisti, C., Chiarini, F., McCubrey, J. A., & Martelli, A. M. (2018). Therapeutic Targeting of mTOR in T-Cell Acute Lymphoblastic Leukemia: An Update. International Journal of Molecular Sciences, 19(7), 1878. https://doi.org/10.3390/ijms19071878