Development of a Fingerprint-Based Scoring Function for the Prediction of the Binding Mode of Carbonic Anhydrase II Inhibitors


 , ,
, ,  and
and 
Abstract
1. Introduction
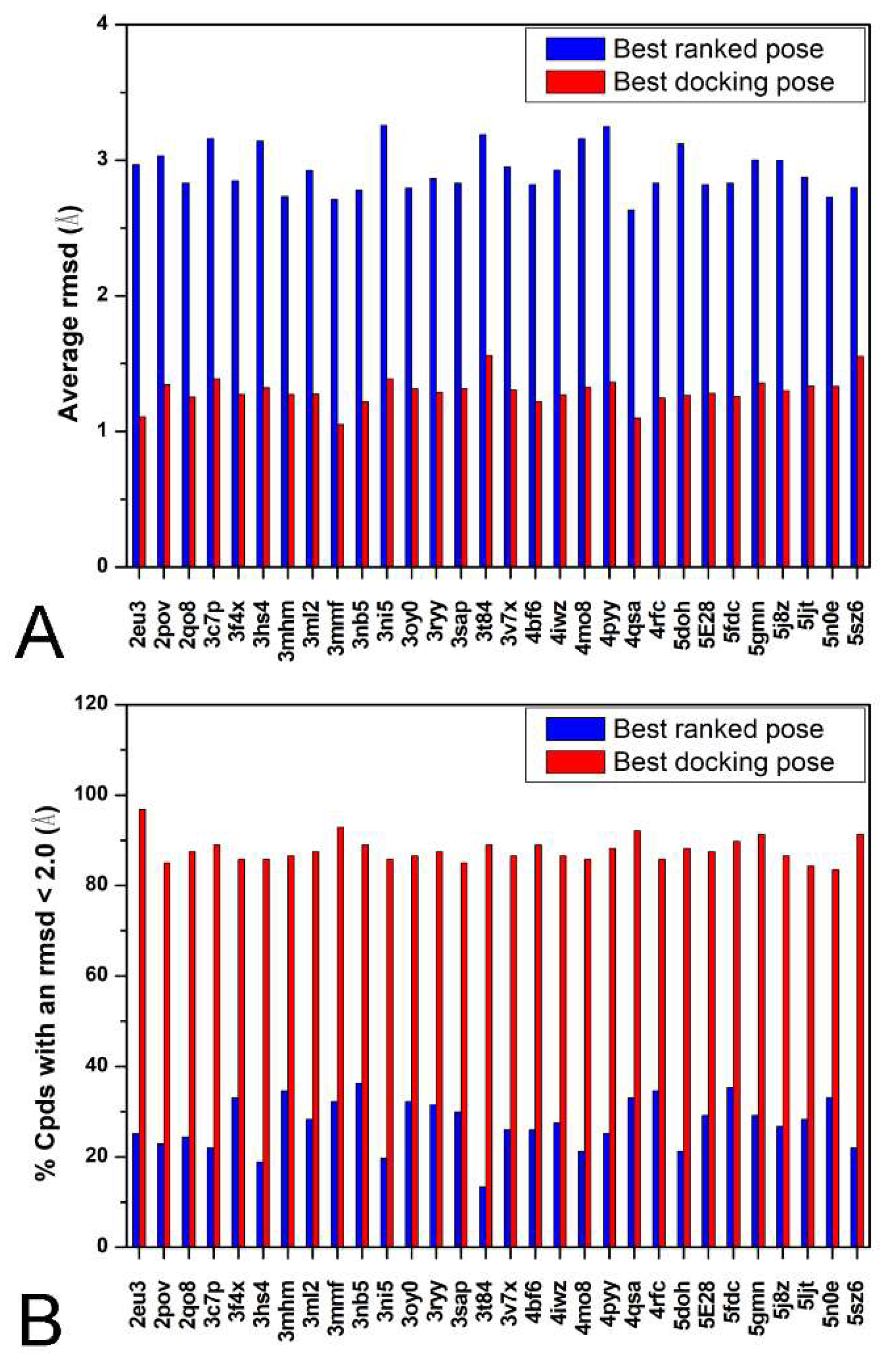
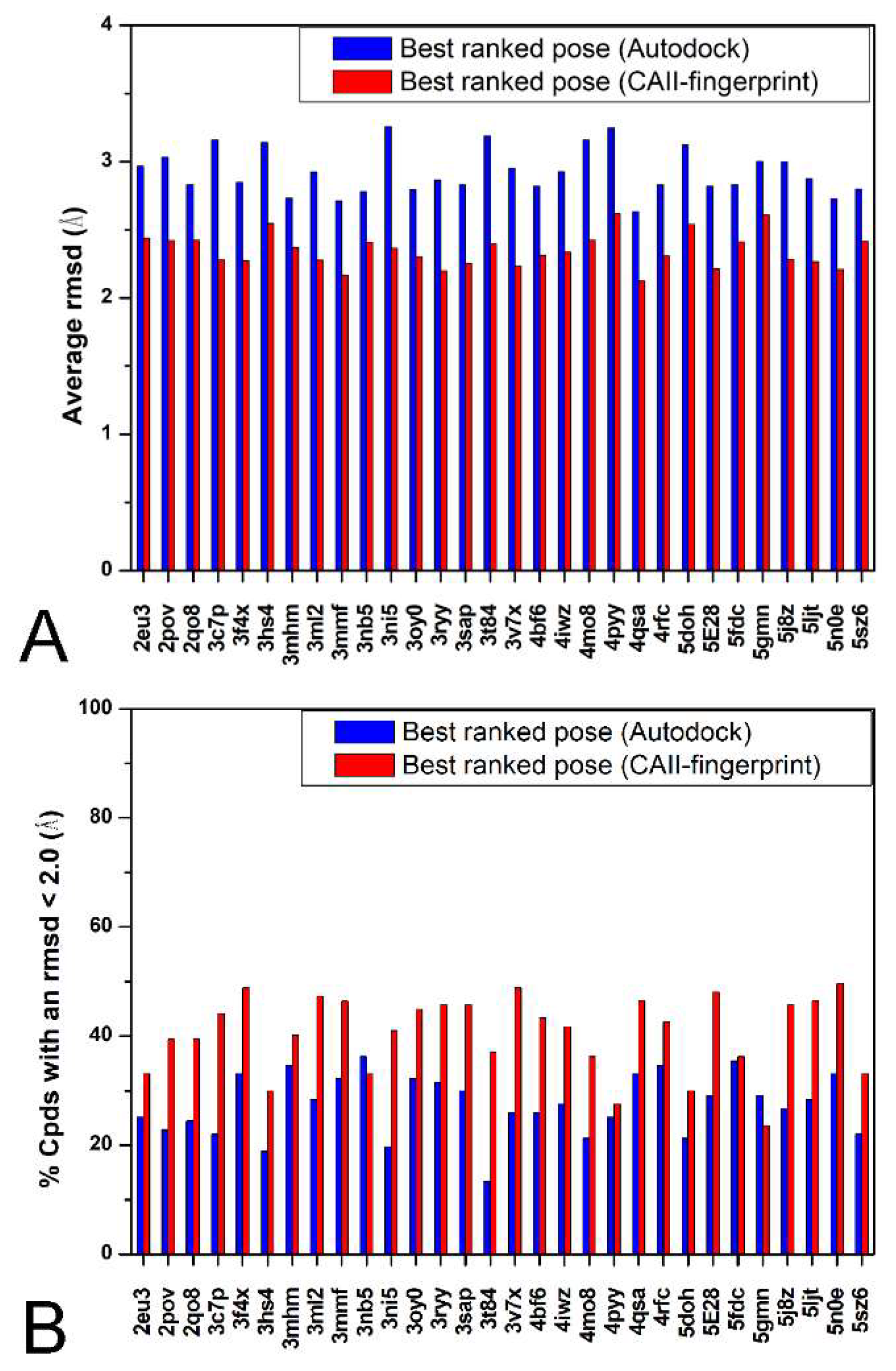
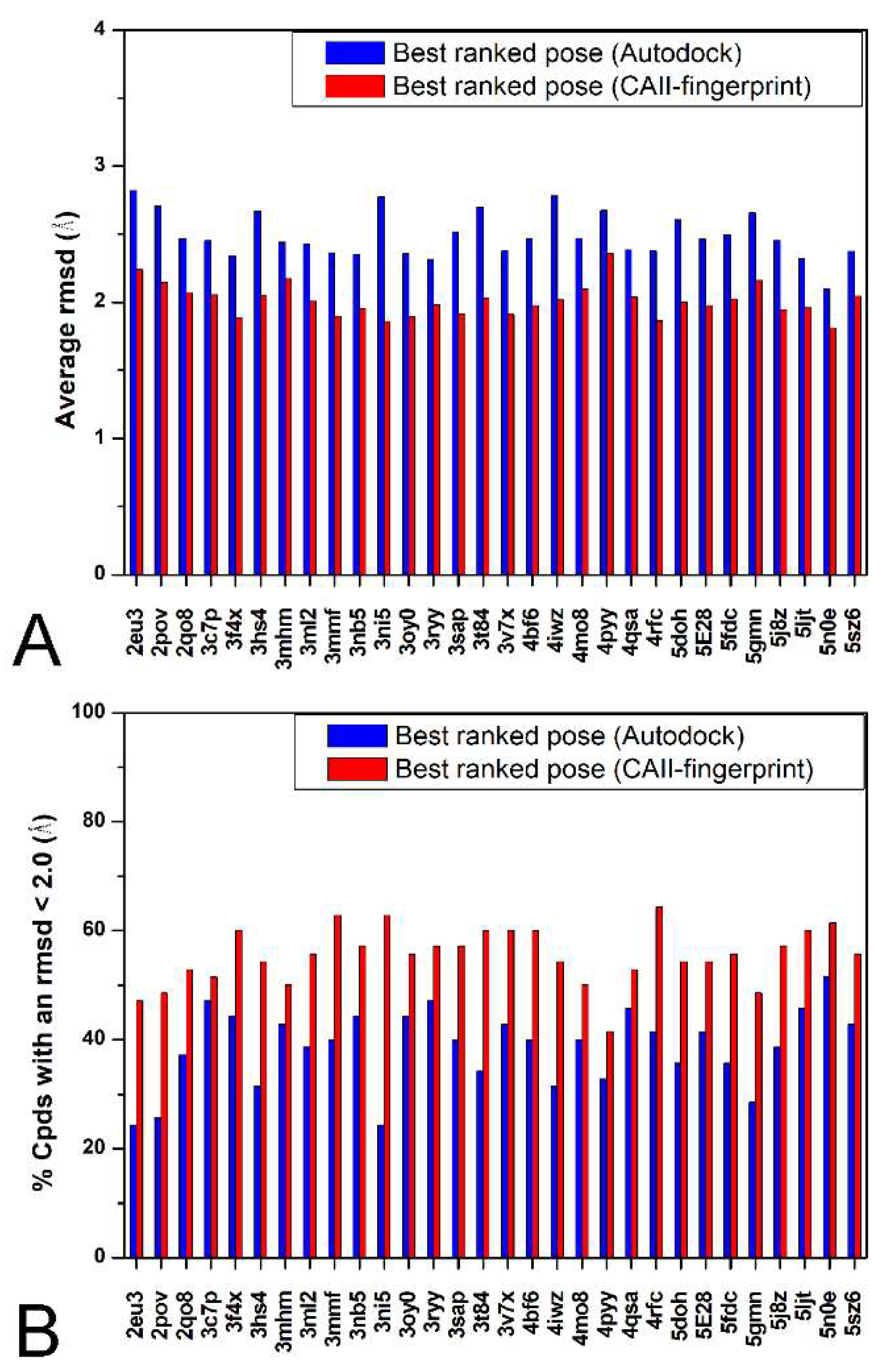
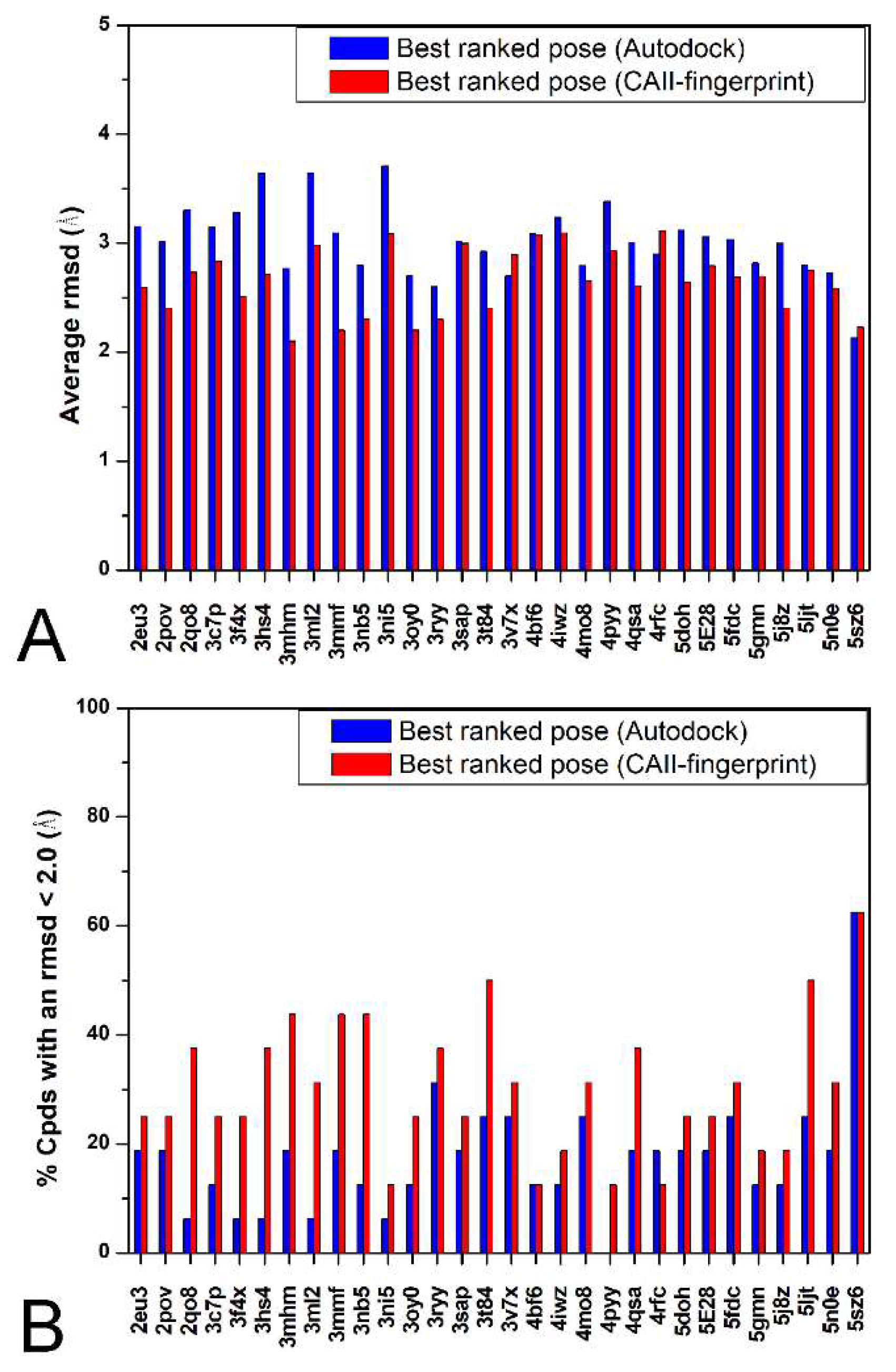
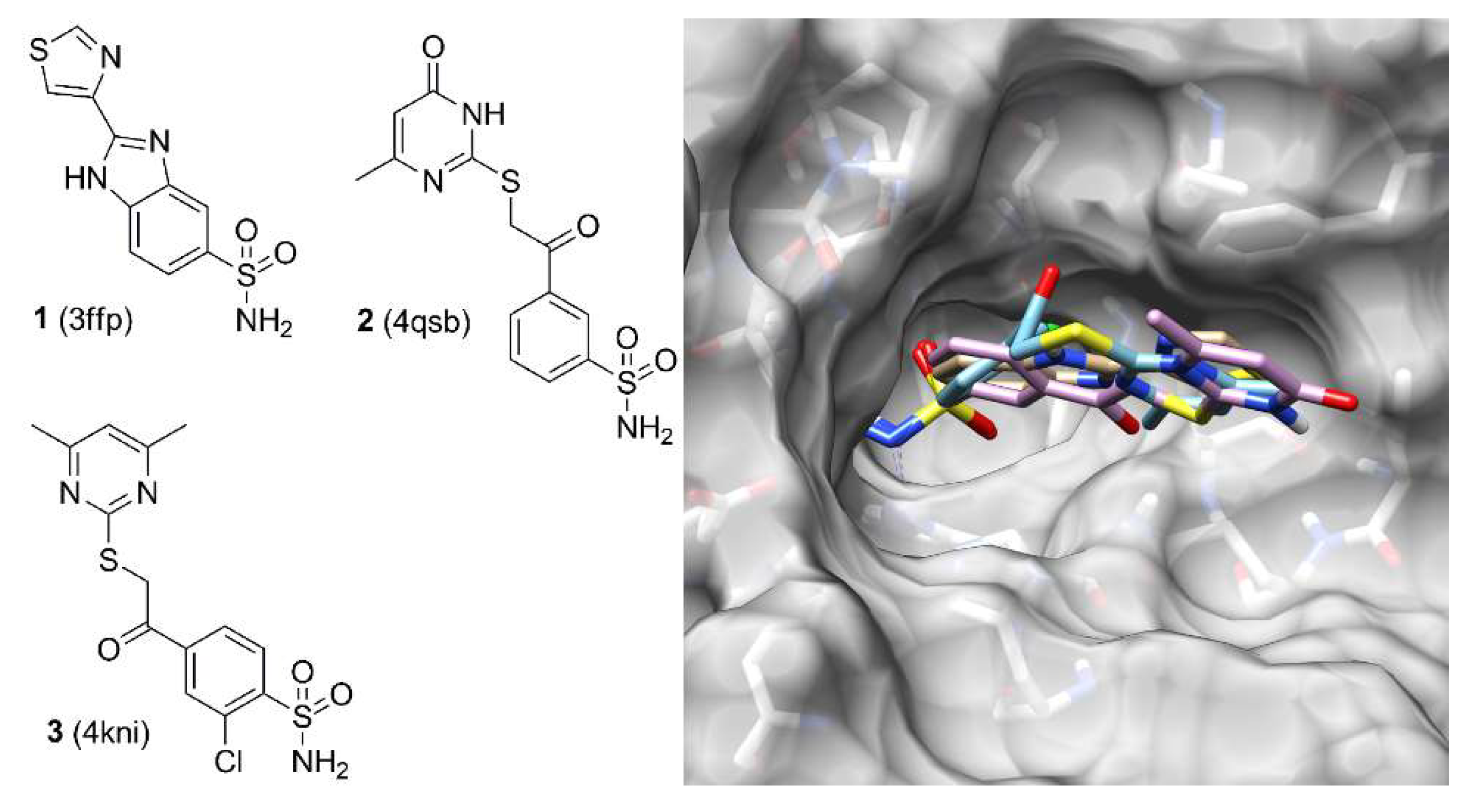
2. Results and Discussion
3. Materials and Methods
3.1. Protein-Ligand Complex Structures
3.2. Docking Procedures
3.3. Cross-Docking Analysis
3.4. CAII-rIFP Generation
3.5. Tc-IFP Calculation
3.6. Clustering of the CAII Inhibitors
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Żołnowska, B.; Sławiński, J.; Szafrański, K.; Angeli, A.; Supuran, C.T.; Kawiak, A.; Wieczór, M.; Zielińska, J.; Bączek, T.; Bartoszewska, S. Novel 2-(2-arylmethylthio-4-chloro-5-methylbenzenesulfonyl)-1-(1,3,5-triazin-2-ylamino)guanidine derivatives: Inhibition of human carbonic anhydrase cytosolic isozymes I and II and the transmembrane tumor-associated isozymes IX and XII, anticancer activit. Eur. J. Med. Chem. 2018, 143, 1931–1941. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Vats, L.; Sharma, V.; Angeli, A.; Kumar, R.; Supuran, C.T.; Sharma, P.K. Synthesis of novel 4-functionalized 1,5-diaryl-1,2,3-triazoles containing benzenesulfonamide moiety as carbonic anhydrase I, II, IV and IX inhibitors. Eur. J. Med. Chem. 2018, 150, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Perfetto, R.; del Prete, S.; Vullo, D.; Sansone, G.; Barone, C.; Rossi, M.; Supuran, C.T.; Capasso, C. Biochemical characterization of the native α-carbonic anhydrase purified from the mantle of the Mediterranean mussel, Mytilus galloprovincialis. J. Enzyme Inhib. Med. Chem. 2017, 32, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: From biomedical applications of the inhibitors and activators to biotechnological use for CO2capture. J. Enzyme Inhib. Med. Chem. 2013, 28, 229–230. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Alterio, V.; Supuran, C.T. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin. Drug Discov. 2013, 8, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, J.; Nordfors, K.; Järvelä, S.; Bragge, H.; Rantala, I.; Parkkila, A.-K.; Haapasalo, H.; Parkkila, S. Carbonic anhydrase II in the endothelium of glial tumors: A potential target for therapy. Neuro. Oncol. 2007, 9, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Korsakov, M.; Dorogov, M.; Tuccinardi, T.; Dedeoglu, N.; Supuran, C.T. Probing the “bipolar” nature of the carbonic anhydrase active site: Aromatic sulfonamides containing 1,3-oxazol-5-yl moiety as picomolar inhibitors of cytosolic CA I and CA II isoforms. Eur. J. Med. Chem. 2015, 101, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T. Docking-Based Virtual Screening: Recent Developments. Comb. Chem. High Throughput Screen. 2009, 12, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Da, C.; Kireev, D. Structural protein-ligand interaction fingerprints (SPLIF) for structure-based virtual screening: Method and benchmark study. J. Chem. Inf. Model. 2014, 54, 2555–2561. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Su, M.; Liu, Z.; Li, J.; Li, Y.; Wang, R. Enhance the performance of current scoring functions with the aid of 3D protein-ligand interaction fingerprints. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Marcou, G.; Rognan, D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. BINANA: A novel algorithm for ligand-binding characterization. J. Mol. Graph. Model. 2011, 29, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Van Linden, O.P.J.; Kooistra, A.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.; Kooistra, A.J.; Kanev, G.K.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. PDEStrIAn: A Phosphodiesterase Structure and Ligand Interaction Annotated Database As a Tool for Structure-Based Drug Design. J. Med. Chem. 2016, 59, 7029–7065. [Google Scholar] [CrossRef] [PubMed]
- Maestro, Version 10.6. Portland (OR): Schrödinger Inc 2016. Available online: https://www.schrodinger.com/ (accessed on 2 May 2018).
- Rostkowski, M.; Olsson, M.H.; Søndergaard, C.R.; Jensen, J.H. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct. Biol. 2011, 11. [Google Scholar] [CrossRef] [PubMed]
- Macromodel, Version 9.7. Portland (OR): Schrödinger Inc 2009. Available online: https://www.schrodinger.com/ (accessed on 2 May 2018).
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Nuti, E.; Ortore, G.; Supuran, C.T.; Rossello, A.; Martinelli, A. Analysis of human carbonic anhydrase II: Docking reliability and receptor-based 3D-QSAR study. J. Chem. Inf. Model. 2007, 47, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Giuntini, N.; Martinelli, A.; Tuccinardi, T. Application of a FLAP-consensus docking mixed strategy for the identification of new fatty acid amide hydrolase inhibitors. J. Chem. Inf. Model. 2015, 55, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Granchi, C.; Rizzolio, F.; Caligiuri, I.; Battistello, V.; Toffoli, G.; Minutolo, F.; Macchia, M.; Martinelli, A. Identification and characterization of a new reversible MAGL inhibitor. Bioorganic Med. Chem. 2014, 22, 3285–3291. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Gelain, A.; Porta, F.; Asai, A.; Martinelli, A.; Tuccinardi, T. Identification of a new STAT3 dimerization inhibitor through a pharmacophore-based virtual screening approach. J. Enzyme Inhib. Med. Chem. 2016, 31, 1011–1017. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anum | Name and Number of the Residue (for Example R92) |
|---|---|
| 0 or 1 | H-bonds (acceptor) |
| 0 or 1 | H-bonds (donor) |
| 0 or 1 | Hydrophobic contacts |
| 0 or 1 | π—π stacking interaction |
| 0 or 1 | T-stacking interaction |
| 0 or 1 | Cation-π interaction |
| 0 or 1 | Salt bridge |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poli, G.; Jha, V.; Martinelli, A.; Supuran, C.T.; Tuccinardi, T. Development of a Fingerprint-Based Scoring Function for the Prediction of the Binding Mode of Carbonic Anhydrase II Inhibitors. Int. J. Mol. Sci. 2018, 19, 1851. https://doi.org/10.3390/ijms19071851
Poli G, Jha V, Martinelli A, Supuran CT, Tuccinardi T. Development of a Fingerprint-Based Scoring Function for the Prediction of the Binding Mode of Carbonic Anhydrase II Inhibitors. International Journal of Molecular Sciences. 2018; 19(7):1851. https://doi.org/10.3390/ijms19071851
Chicago/Turabian StylePoli, Giulio, Vibhu Jha, Adriano Martinelli, Claudiu T. Supuran, and Tiziano Tuccinardi. 2018. "Development of a Fingerprint-Based Scoring Function for the Prediction of the Binding Mode of Carbonic Anhydrase II Inhibitors" International Journal of Molecular Sciences 19, no. 7: 1851. https://doi.org/10.3390/ijms19071851
APA StylePoli, G., Jha, V., Martinelli, A., Supuran, C. T., & Tuccinardi, T. (2018). Development of a Fingerprint-Based Scoring Function for the Prediction of the Binding Mode of Carbonic Anhydrase II Inhibitors. International Journal of Molecular Sciences, 19(7), 1851. https://doi.org/10.3390/ijms19071851