1. Introduction
Pannexins (Panx1, Panx2, and Panx3) are membrane-spanning glycoproteins capable of forming large pore channels that allow the passage of ions and macromolecules involved in paracrine and autocrine signaling [
1,
2]. Panx1 is the most widely expressed pannexin and the most studied, with evidence supporting that its channels act as ATP and Ca
2+ conduits [
3,
4,
5,
6] and are implicated in critical cellular processes such as cell death after brain ischemia [
7] and inflammation [
8]. On the other hand, Panx2 is the largest member of the family and its expression was thought to be restricted to the central nervous system (CNS). Recently, it has been reported that Panx2 can also be expressed in other tissues such as skin, kidney and liver [
9,
10], while Panx3 is predominantly expressed in skin, cartilage and bone [
4,
5,
11,
12,
13]. In contrast to the hexameric type of channels formed by Panx1, it has been suggested that Panx2 can form octameric or heptameric channels [
14] and it is still unclear whether its channel function would be similar to the other family members. Interestingly, Panx1 and Panx2 expression have been found to overlap in adult rodent brains although they are inversely regulated throughout development, with Panx1 being more abundant in neonatal and young tissues, whereas Panx2 is more abundant in the adult [
15,
16,
17,
18]. Under ischemic conditions, both Panx1 and Panx2 are expressed in the brain and their overlapping channel functions contribute to neurodegeneration. In fact, the deletion of Panx1 and Panx2 in a double knockout mouse model was necessary to observe a reduction in cell death after ischemia, perhaps due to their redundant and/or complementary functions [
19].
N-glycosylation is a posttranslational modification that occurs in the endoplasmic reticulum (ER) and is recognized to have profound effects on protein folding and trafficking of membrane-bound proteins [
20]. The prediction of putative N-linked glycosylation sites for pannexins has been done in the past, and it has been demonstrated that Panx1 and Panx3 have sites for N-linked glycosylation at Asn (N) 254 and N71, respectively. These studies comprised further characterization using enzymatic digestion with endoglycosidases that confirmed that all three members of the pannexin family are differentially N-glycosylated but not O-glycosylated [
11,
21]. However, for Panx2 the predicted site of N-glycosylation at residue N86 remains to be validated [
22].
Unlike other pannexins, Panx2 is only modified to a high-mannose glycosylation species (termed as Gly-1) and presents a predominantly intracellular localization that has been associated with this lower level of N-glycosylation [
22,
23,
24,
25,
26]. Previous evidence supports the concept that complex glycoprotein species (Gly-2) (further processed at Golgi) present in Panx1 and Panx3 traffic readily to the cell surface [
22]. However, our group and others have stated that under certain circumstances Panx2 can also translocate to the plasma membrane [
14,
17,
22], but it is still unclear whether glycosylation plays a role in regulating Panx2 trafficking.
Panx2 has been shown to interact with Panx1, and when co-expressed together they can form heteromeric channels with reduced channel properties compared to homomeric ones [
22,
27]. Although this has only been tested in ectopic expression systems it has been suggested that it might function as a mode of regulation in cells that endogenously express both proteins. Interestingly, the Panx1/Panx2 interaction only occurs with the Gly-0 and Gly-1 species of Panx1 [
22] and it is unknown whether Panx2 glycosylation has any impact on the formation of these intermixed channels. Since previous studies have shown that Panx1 and Panx2 are often co-expressed in the same cells under normal conditions [
15,
18,
27,
28,
29,
30], and that their co-expression modulates important processes such as ischemia-induced neurodegeneration and brain damage in vivo [
31], it is important to understand how these channels may be regulated by post-translational modifications (PTMs) such as N-glycosylation, and how this PTM may regulate their interaction.
The present study aimed to validate the predicted N-linked-glycosylation site of Panx2 and determine its role in the regulation of the subcellular localization and intermixing of Panx2 with Panx1. We generated a Panx2 mutant protein completely devoid of N-glycosylation that, when overexpressed, exhibits a high level of intracellular aggregation with decreased traffic to the plasma membrane compared with wild-type Panx2. We found that N-glycosylation of Panx2 is not required for Panx1/Panx2 intermixing but facilitates Panx2 trafficking and localization at the plasma membrane when co-expressed with Panx1. The intracellular localization of un-glycosylated Panx2 reduces its co-localization with Panx1 at the cell surface and may impact their channel function in cells that co-express both glycoproteins, such as neurons. Collectively, we propose that N-glycosylation may be necessary for proper processing of Panx2 at the endoplasmic reticulum, regulating its intracellular distribution, but it is not required for interacting with Panx1.
3. Discussion
Pannexins are a family of channel proteins implicated in important physiological and pathological functions and most of the current research has been conducted to analyze their level of expression and distribution within mammalian tissues and their role in diverse diseases [
34]. However, there is still a need to understand the biophysical properties of these channels and the different ways of regulation that prevent the detrimental effects of their exacerbated channel activity. Pannexins are N-glycosylated and as integral membrane proteins, this modification seems to be essential in regulating their trafficking, as was demonstrated formerly for Panx1 and Panx3 [
11,
22]. Unlike the other pannexins, Panx2 is modified only to a high-mannose glycosylation (Gly-1) which is known to be an early post-translational modification occurring in the ER lumen. For many other glycoproteins, this step is generally followed by further oligosaccharide editing in the Golgi (complex glycosylation). To date, there is no evidence showing further processing of Panx2 in Golgi and only studies in Panx1 and Panx3 showed that trafficking of these two pannexins to the plasma membrane is mediated by Sar1-dependent COPII vesicles [
35] with N-glycosylation affecting their final delivery [
11,
36,
37]. This suggests that N-glycosylation regulates the route of pannexin trafficking and modifies their localization and channel formation in immortalized culture cells. A previous report of Panx2 localization pointed to a predominantly intracellular distribution that can also be modified by other PTMs like palmytoilation, which can determine its subcellular localization in neurons [
17].
Panx2 seems to be mostly intracellularly localized, but in some instances, it can also translocate to the plasma membrane [
14,
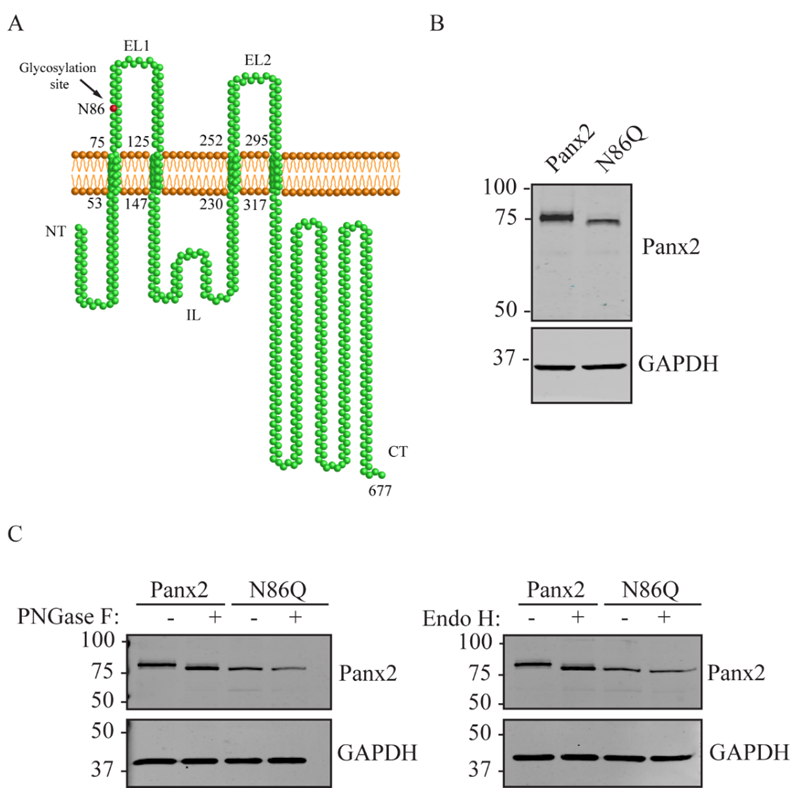
17]. However, the exact site of N-glycosylation and whether this modification affects Panx2 trafficking was unknown. In this study, we sought to determine the N-glycosylation site by generating a glycosylation-deficient mutant (N86Q) based on the predicted N-linked glycosylation site reported by Penuela et al. [
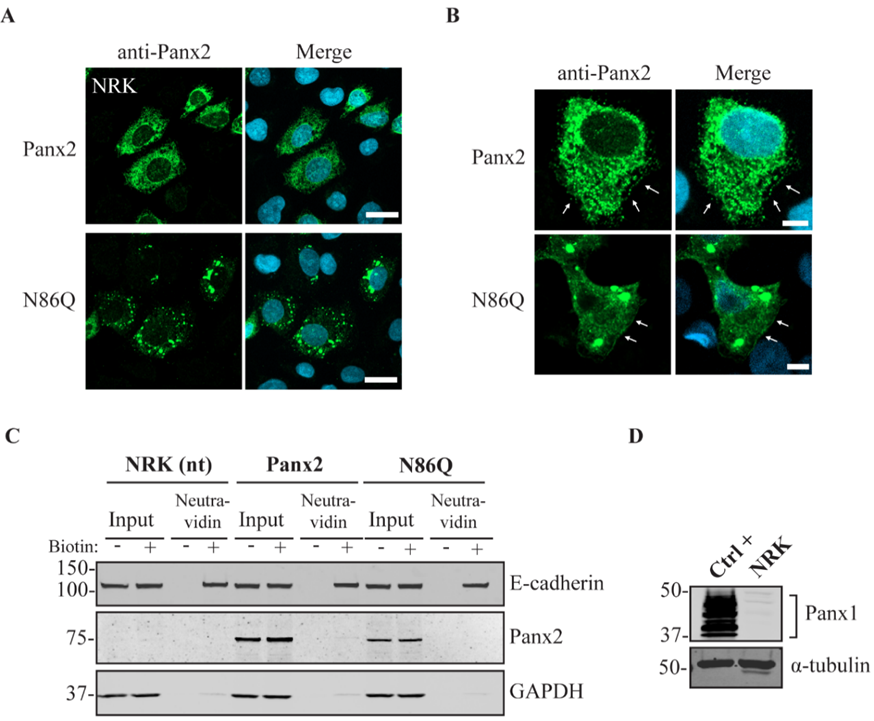
22]. For our experiments we transfected constructs encoding mouse Panx2 and an N86Q mutant into HEK293T cells. Our results showed that N86Q substitution generated a Panx2 mutant with a faster-migrating electrophoretic band compared to Panx2 WT, that does not shift after specific N-glycosidase digestion with endoglycosidases, thus confirming that N86 is the only N-glycosylation site for Panx2. In the other cell lines assayed (NRK and AD293) we also observed that overexpression of the same Panx2 WT and N86Q constructs exhibited the same electrophoretic properties seen with HEK293T (
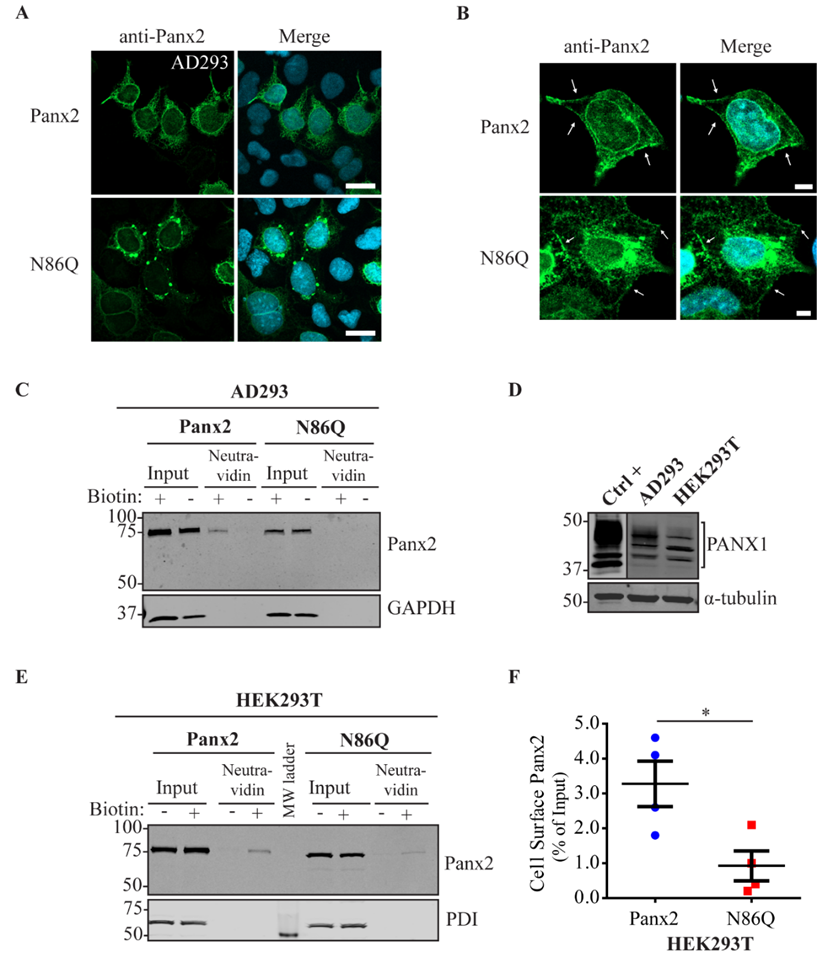
Figure 2C and
Figure 3C).
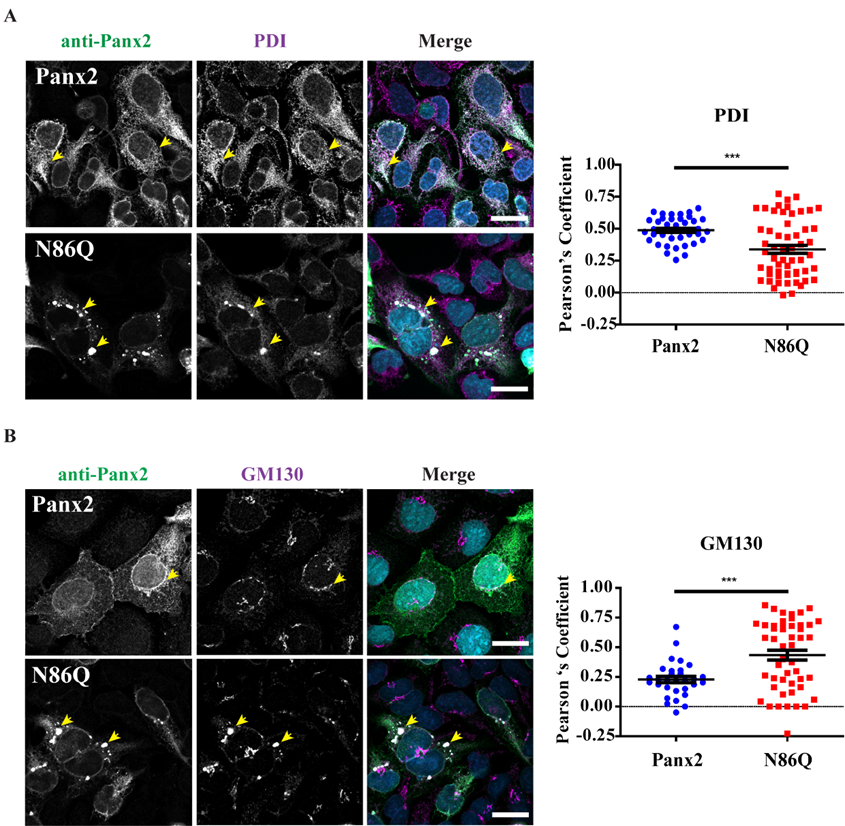
Independently of the cell-type used for ectopic expression, the N86Q mutant appeared to form punctate aggregates that were localized to intracellular compartments along with the ER-chaperone PDI, and the cis-Golgi matrix marker GM130. Compared to the N-glycosylation-deficient mutants of Panx1 and Panx3 [
11], un-glycosylated Panx2 exhibited an exacerbated abnormal intracellular aggregation. This raises the possibility that the lack of N-glycosylation may have affected proper protein folding of Panx2, which is the largest member of the pannexin family. We cannot rule out that the intracellular accumulation of N86Q could be an artifact of overexpression, that concomitant with the lack of glycosylation of the Panx2 mutant, might have induced misfolding and ER-stress [
38].
A previous study in murine postnatal hippocampal neural progenitor cells (NPC)s [
17] showed that treatment with glycosidases had no effect on endogenous Panx2 electrophoretic mobility, suggesting that they were un-glycosylated. That study relied on antibody detection of endogenous Panx2, and the protein bands identified were of lower molecular weight than the predicted full-length Panx2 used in this study (677 aa, [
39]). Further studies are needed to determine first, if the expression of certain un-glycosylated endogenous isoforms of Panx2 is cell-type specific, and second, whether glycosylation has measurable effects on the Panx2 cellular function. To date, there are no reports of mutations in the
Panx2 gene, but our results would predict significant changes in Panx2 behavior if its glycosylation is affected. It is also possible that the un-glycosylated form of Panx2 may be preferentially expressed in some cells and tissues determining the primary function of the Panx2 channel and its subcellular localization.
In our study, we detected large intracellular subpopulations of Panx2 likely localized in the ER, and partially colocalized with cis-Golgi marker. Interestingly, we found that overexpression of the N86Q mutant changed the distribution of PDI and GM130 compared to Panx2 WT, suggesting that the formation of aggregates may disrupt the morphology of ER and cis-Golgi. GM-130 is a peripheral membrane protein in
cis-Golgi matrix that is important for maintenance of Golgi structure [
40], and the regulation of ER-to-Golgi transport of proteins and glycosylation [
41]. It is possible that the aggregation of N86Q may interfere with the mutant protein transport from the ER causing accumulation of GM130.
As reviewed by Boyce et al. [
42], pannexins contain putative recognition sequences for endocytic and endo-lysosomal targeting which could account for the control of pannexin trafficking. Interestingly, recently published work by Boassa et al., described the localization of a recombinant Panx2 fused to mini-SOG tag that was transiently expressed in HeLa cells [
26]. These authors used correlated light and electron microscopy imaging to detect Panx2 localization at cytoplasmic protrusions. Also, immuno-colocalization in HEK293T cells using assorted vesicular markers displayed Panx2 WT (untagged) localized to early or recycling endosomes rather than ER. Although, they mentioned in the manuscript that when the ER marker calnexin was used they detected colocalization with overexpressed Panx2 in HEK293T cells. Our findings are consistent with these reports [
24], since we found Panx2 primarily in the ER (
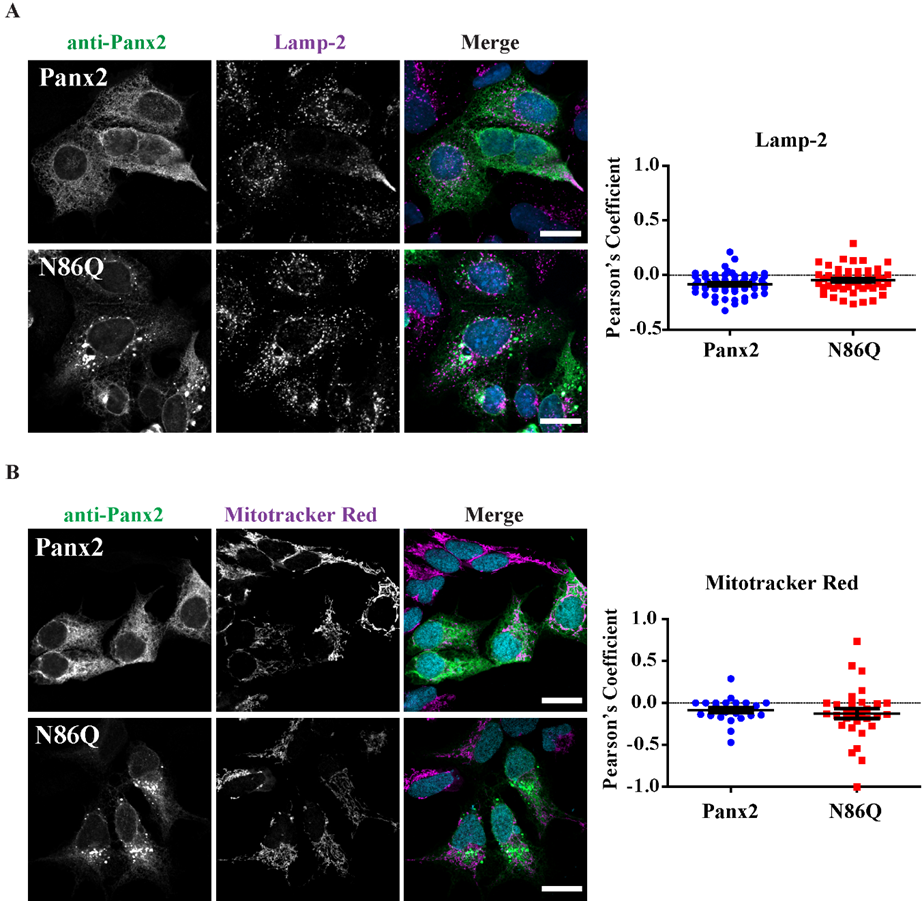
Figure 4A). However, we did not find conclusive evidence of Panx2 in endo-lysosome compartments (
Figure 5A) as others have reported [
25,
26]. Based on our results, it is possible that Panx2 may have an intracellular channel function in the ER, similar to the proposed calcium-leak channels formed by Panx1 and Panx3 [
6].
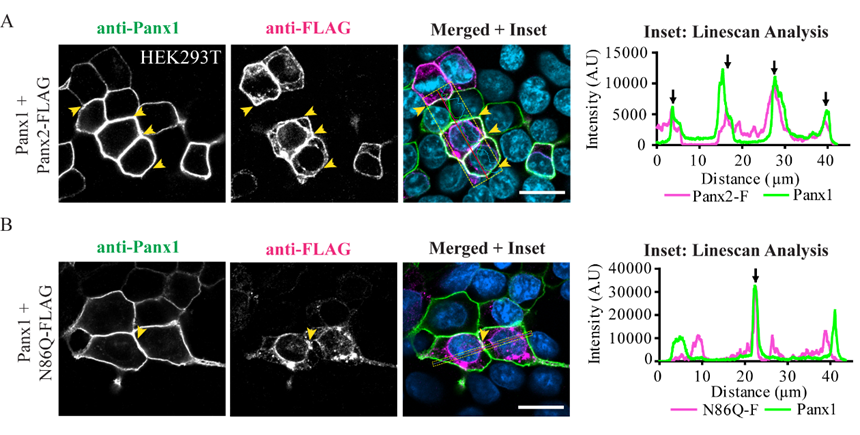
In some instances, Panx2 distribution exhibited limited cell surface localization that was more apparent when higher endogenous or ectopic Panx1 protein was expressed in the studied cell lines. This is consistent with our previous observation of increased Panx2 at the cell surface when co-expressed with Panx1 under overexpression conditions [
22]. Here, we used three different cells lines with varying levels of endogenous Panx1 and a different capacity of protein production. We noticed that in cells with low endogenous Panx1 (e.g., NRKs), there was barely any Panx2 at the cell surface based on immunolabeling and cell surface biotinylation pull-downs. In the case of AD293 and HEK293T cells, it was possible to detect low levels of overexpressed Panx2 at the cell surface, while the mutant Panx2 (N86Q) had a detectable but significantly decreased presence only at the cell membrane of HEK293T cells. This result could be attributed to the increased protein expression in HEK293T cells that contains the SV40 T-antigen and have high transfection efficiency. This feature might have allowed the overexpressed Panx2 to bypass mechanisms of protein quality control resulting in more Panx2 trafficking to the plasma membrane [
43].
Further research is needed to evaluate endogenous Panx2 in terms of N-glycosylation and subcellular localization of its isoforms, and to examine if Panx2 can form channels at the plasma membrane under physiological conditions. To date, limited studies have attempted to evaluate the Panx2 channel function [
14,
15] and several factors make it difficult to test Panx2 channel activity, such as its intracellular localization, the lack of evidence of in vivo functional channel formation [
27] and the unknown mechanisms of activation [
14].
Work done by Bruzzone et al. [
15], showed that Panx1 and Panx2 were abundantly expressed in the CNS, and co-injection of both pannexin RNAs in paired
Xenopus oocytes resulted in the formation of heteromeric channels with functional characteristics different from those formed by Panx1 monomers but with similar pharmacological sensitivity [
27]. Ambrosi et al. [
14] suggested that Panx1/Panx2 heteromeric channels tend to be unstable and they attributed that to differences in monomers size and oligomeric symmetry between these two pannexins. We have previously shown that Panx1 and Panx2 do form a complex as determined by co-IP experiments. Interestingly, when both pannexins are co-expressed, the level of interaction between Panx2 and glycosylated-species of Panx1 is dependent of the glycosylation of the latter [
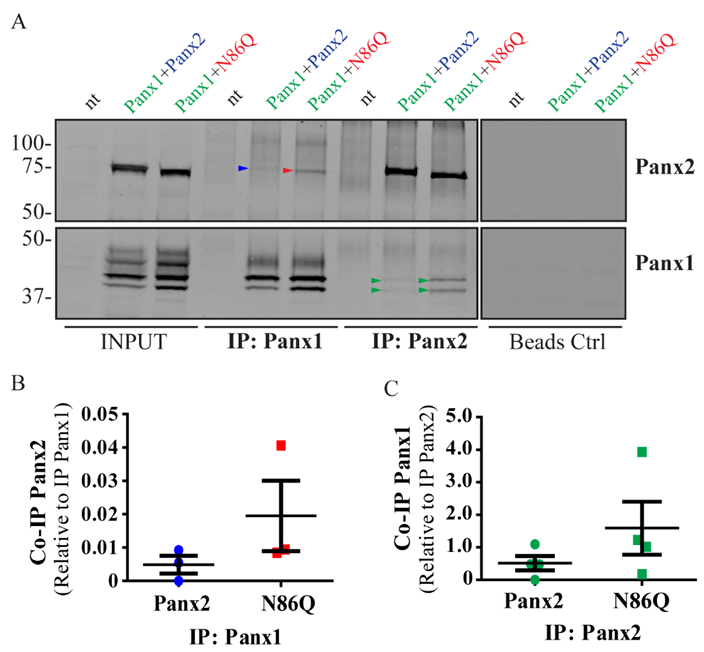
22]. Here, we showed in vitro, that the Panx2 glycosylation-deficient mutant can readily form complexes with Panx1, thus Panx2 glycosylation is not required for intermixing of the two pannexins. In fact, although it was not statistically significant, N86Q seemed to pull-down Panx1 more efficiently than the WT Panx2. However, in confocal images the N86Q aggregates did not show higher overlap with Panx1-immunolabeling than Panx2 WT. Consistent with our previous report [
22], complex N-glycosylation of Panx1 hinders their interaction, since both Panx2 and N86Q interacted only with the Gly-0 and Gly-1 forms of Panx1. Taken together, these results suggest that in an ectopic expression system, glycosylation of Panx2 is not required for Panx1/Panx2 intermixing, but it does help with the transport of Panx2 to the cell surface, which is also increased by the presence of Panx1.
Finally, we propose that N-glycosylation of pannexins is an important post-translational modification that partially regulates their subcellular localization. Whether N-glycosylation represents a post-translational mechanism that regulates trafficking and Panx1/Panx2 interactions in vivo would be important questions to address in future studies. In cells where both pannexins are co-expressed, glycosylation may act as a form of regulation defining whether these channels will serve as intracellular or plasma membrane channels with different physiological and pathological functions.
4. Materials and Methods
4.1. Cell Lines, Constructs and Transient Transfections
Media, supplements and reagents were obtained from GIBCO
® and Invitrogen™ (Carlsbad, CA, USA). Normal rat kidney (NRK) (ATCC
® CRL-6509™) and human embryonic kidney cells (HEK293T) (ATCC
® CRL-3216™) were obtained from ATCC (Manassas, VA, USA). Adherent HEK293 cells (AD293, Cat# 240085) were obtained from Agilent Technologies, Inc. (Santa Clara, CA, USA). Cell cultures were grown in high-glucose DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 µg/mL streptomycin and 2mM L-Glutamine. At ~50% of confluency, cells were transfected adding Lipofectamine 3000 (Invitrogen™) following manufacturer directions. 2 µg of pcDNA3.1 (Invitrogen™) plasmids encoding mouse Panx2 [
22] or Panx2
N86Q or their respective FLAG-tagged versions were used for transfections in 35 mm culture plates. After 48 h for single transfections and 72 h for co-transfections, proteins were extracted, and expression levels were determined by Western blot. For co-transfections experiments with mPanx1 plasmid [
11], levels were reduced to 0.5 µg of DNA.
4.2. Mutagenesis and Cloning of New FLAG-Tagged Panx2 Constructs
As described previously [
11], Panx2 has a predicted N-glycosylation consensus site located at asparagine (N) 86 on the first extracellular loop. Site-directed mutagenesis service (NorClone Biotech Labs, London, ON, Canada) was used to generate a new expression Panx2 construct encoding a replacement of asparagine by glutamine at position 86, referred to as Panx2 N86Q. FLAG-tagged Panx2 was obtained by inserting a single FLAG sequence with In-Fusion HD Cloning Kit (Clontech Laboratories, Inc., Takara Bio, Inc., Mountain View, CA, USA) at the end of the coding region of the Panx2 C-termini. Primers used for FLAG insertion were Forward: 5′-GTTTAAACTTAAGCTTCATGCACCACCTCCTGGAG-3′ and Reverse: 5′-GCCCTCTAGACTCGAGCTCACTTGTCATCGTCGTCCTTGTAATCAAACTCCACAGTACT-3′. All the constructs were verified by sequencing.
4.3. Protein Extractions and Western Blots
For co-immunoprecipitation (Co-IP) assays, cell lysates were obtained using a Triton-based extraction buffer (IP buffer) (1% Triton X-100, 150 mM NaCl, 10 mM Tris, 1 mM EDTA, 1 mM EGTA, 0.5% NP-40). The rest of the protein extractions were performed with SDS-based buffer (RIPA buffer) (0.1% SDS, 50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40 and 0.5% Sodium Deoxycholate). In each case, lysis buffers were complemented with a final concentration of 1 mM NaF, 1 mM Na
3VO
4, and one tablet of cOmplete™-mini, EDTA-free Protease Inhibitor Cocktail (Roche, Mannheim, Germany). Total protein concentrations were quantitated with Pierce™ BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA). For Western Blots, 50 µg of total protein were resolved in 8% SDS-PAGE and transferred onto nitrocellulose membranes using an iBlot™ Blotting System (Invitrogen, Carlsbad, CA, USA). Membranes were blocked with 3% bovine serum albumin (BSA, Burlington, ON, Canada) and 0.05% Tween 20-Phosphate Buffer Saline (T-PBS) for 45 min at room temperature and probed overnight with a 1:1000 dilution of the rabbit affinity-purified antibodies anti-Panx2-CT-523 [
22]. Mouse monoclonal anti-FLAG
® M2 (Sigma, St. Louis, MO, USA, Cat# F3165), monoclonal mouse anti-GAPDH (Millipore, Burlington, MA, USA, Cat# MAB374, RRID: AB_2107445), and anti-α-Tubulin (Millipore-Sigma Cat# 05-829, clone DM1A) antibodies were used at 1:2000, 1:1000 and 1:5000 dilutions, respectively. For detection, IRDye-800 and -680RD (Life Technologies™, Carlsbad, CA, USA) were used as secondary antibodies at 1:10,000 dilution and the membranes were scanned on a Li-Cor Odyssey infrared system (Li-Cor, Lincoln, NL, USA). In most cases, GAPDH was used as a loading control.
4.4. Immunofluorescence, Confocal Imaging, Linescans and Colocalization Analysis
Cells were grown on coverslips at ~70% of confluency and were transfected as described previously in
Section 4.1. After 48 h of transfection, coverslips were washed with D-PBS (Gibco
®) and fixed with ice-cold 80% methanol and 20% acetone for 15 min at 4 °C. Coverslips were blocked with 2% BSA-PBS for 1h and primary antibodies were used diluted in blocking buffer as follows: polyclonal anti-Panx2-CT (2 mg/mL, 1:100 dilution), polyclonal anti-Panx1-CT (1 mg/mL, 1:100 dilution), anti-PDI monoclonal antibody (1 mg/mL, 1:400 dilution) (1D3, Enzo
® Life Sciences, Burlington, ON, Canada, ADI-SPA-891-D), cis-Golgi marker anti-GM130 (1 mg/mL, 1:300) (Abcam, Toronto, ON, Canada, Prod#: ab169276), anti-FLAG
® M2 (1 mg/mL, 1:500) (F3165, Sigma, St. Louis, MO, USA), mouse monoclonal anti-Lamp-2 (1 mg/mL, 1:300) (DHSB, clone H4B4). Coverslips were incubated with primary antibodies for 1 h at room temperature, then washed with PBS and incubated with the secondary antibodies: Alexa Fluor 488 goat anti-rabbit IgG (2 mg/mL, 1:700) or goat anti-mouse antibody Alexa Fluor 647 (2 mg/mL, 1:400) (Life Technologies), that were selected to avoid bleed-through between dyes. Mitochondrial labeling was performed by using MitoTracker™ Red CMXRos (M7512, Thermofisher, Life Technologies, Eugene, OR, USA) as per manufacturer directions, and cells were fixed with freshly prepared paraformaldehyde (4%) and then permeabilized with 0.1% Triton X-100 before the blocking step. Coverslips were rinsed with PBS followed by water once and counterstained with Hoechst 33342 (H3570, Life Technologies™, Eugene, OR, USA) (1:1000, in water) for 5 min to stain nuclei and then were mounted with the custom-made Airvol mounting medium. Imaging was performed with an LSM 800 Confocal Microscope (Carl Zeiss, Oberkochen, Germany) using a Plan-Apochromat 63x/1.40 Oil DIC objective (Carl Zeiss, Oberkochen, Germany). Image acquisition for colocalization was performed with sequential laser scanning and with the multitracking feature of the Zeiss software with settings to avoid wrong excitation-crosstalk and bleed-through of the channels. Colocalization was quantitated with the colocalization plug-in of the Zeiss software (ZEN, version 2.3, blue edition). Regions of interest (ROI) were drawn in dual-labeled cells selecting individual cells expressing Panx2 or the mutant and co-stained with organelle markers. Controls of single-labeled cells were used to determine thresholds of intensities for each single channel and a manual thresholding was used to determine the region of pixels colocalized in the intensities scatterplots. Pearson correlation coefficient was determined for each cell as a measure of colocalization and was expressed as means ± S.E.M., representative of at least three independent transfections. Linescans using Zeiss software tool were used to detect overlaps of fluorescence peaks in cells co-transfected with Panx1.
4.5. Cell Surface Biotinylation Assays
Cell surface biotinylation assays were performed as described in Reference [
22]. Briefly, cells were grown in 60 mm plates and used for biotinylation 72 h following transfection with Panx2 or N86Q constructs. After, culture media was aspirated, the cell monolayer was rinsed twice with ice-cold D-PBS supplemented with Ca
2+ and Mg
2+ (Gibco
®). Then, cells were incubated only with D-PBS (non-labeling as a negative control) or with a solution of 1.5 mg/mL EZ-Link™ Sulfo-NHS-SS-Biotin (Thermo Scientific, Rockford, IL, USA) in D-PBS for 30 min on ice and covered from light. Plates were washed once again with D-PBS and then incubated with 100 mM glycine dissolved in D-PBS for 30 min to quench the remaining labeling biotin washed once more with D-PBS. Lysates were prepared using RIPA buffer as described before. For pull-down of cell surface biotinylated proteins, 250 µg of total protein was incubated overnight with 50% slurry of 50 µL NeutrAvidin agarose beads (Thermo Scientific, Rockford, IL, USA). Samples from lysates and initial flow-through wash were collected, the beads were spun down (at 500×
g, 4 °C) and then washed three times with RIPA buffer. The samples and the beads were then mixed with 2X Laemmli buffer and 10% β-mercaptoethanol and placed at 95 °C in heat block for 5 min. 50 µg of total protein from lysates and the beads supernatant were resolved in parallel with 8% SDS-PAGE gel and then transferred to nitrocellulose membranes as previously described. PDI or GAPDH were used as controls of non-specific biotinylation of intracellular/cytoplasmic proteins and E-cadherin was used as positive control of cell surface protein.
4.6. Co-Immunoprecipitation (Co-IP) Assays
Co-immunoprecipitation (Co-IP) of protein complexes was performed at 4 °C by incubating overnight 1 mg of total protein from pre-cleared (with Protein A/G beads alone) lysates with 5 µg/mL of rabbit polyclonal anti-Panx2-CT or anti-Panx1-CT affinity-purified antibody crosslinked to Pierce Protein A/G-Agarose beads (Thermo Scientific); the same amount of beads (~30 µL) were used for IP in each case. Control experiments to evaluate unspecific binding to the beads were performed in parallel using beads with no antibody. To remove un-bound proteins, four washes with 500 µL of ice-cold IP buffer were performed. Then, beads were dried by aspiration and re-suspended in 2X Laemmli buffer (10% (v:v) β-mercaptoethanol), boiled for 5 min, spun down and the supernatants (IP samples) were used for WB. For WB analysis, 50 µg of protein of each lysate was loaded into the INPUT lanes and ran along with the IP samples. The intensities of the bands in each lane were obtained by densitometry and were used for quantitation.
4.7. De-Glycosylation Assays
Lysates from HEK293T cells ectopically expressing mouse Panx2 and the N86Q were used for validation of N-glycosylation site of the mouse Panx2 construct. Enzymatic de-glycosylation with Peptide-N-glycosidase F (PNGase F) and Endoglycosidase H (EndoH) were used to detect the presence of all complex forms of N-glycosylation and high-mannose modification, respectively. PNGase F (Roche, Indianapolis, IN, USA) and EndoH (New England Biolabs Ipswich, MA, USA) digestions were performed according to their manufacturer’s instructions. Briefly, at least 35 µg of total protein was denatured at 100 °C for 5 min in denaturing buffer (0.1% (v/v) SDS, 0.05 M 2-mercaptoethanol, 50 mM phosphate buffer, pH 7.5) and subsequently incubated for 1h at 37 °C with 10 units of the PNGase F, 0.7% (v/v) of Triton X-100 or 2500 U of Endo H in supplier’s digestion buffer. In the parallel control, samples were assayed without endoglycosidases. Protein samples were separated on an 8% SDS-polyacrylamide gel electrophoresis gel (PAGE) and transferred to nitrocellulose membranes for WB.
4.8. Densitometric Analysis of Western Blots
Densitometry analysis was performed in the Odyssey Application Software Version 3.0.16 (LI-COR Biosciences, Lincoln, NL, USA) as follows: For cell surface biotinylation experiments, the fraction of biotinylated-protein detected at the cell surface was calculated using the integrated intensity (I.I.) of protein bands detected in the Neutravidin lanes divided by the I.I. of protein bands detected in their corresponding input-lysate lanes. For Co-IP experiments, quantitative analysis was performed by calculating the ratio of the I.I detected for Co-IP protein divided by the I.I of the IP-target protein. Quantitation results were expressed as means ± S.E.M. representative of at least three independent experiments.
4.9. Statistics
Statistical analysis was performed using the statistical package of GraphPad Prism® Ver. 5.03 (GraphPad Software, Inc., San Diego, CA, USA). Cell surface biotinylation data were analyzed with non-parametric Mann-Whitney U test for unpaired data. Data derived from quantitation of Co-IP assays and colocalization were analyzed with a two-tailed unpaired t test with Welch’s correction. A probability of p < 0.05 was considered as statistically significant.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






