Role of the DNA Damage Response in Human Papillomavirus RNA Splicing and Polyadenylation


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The Life Cycle of HPV
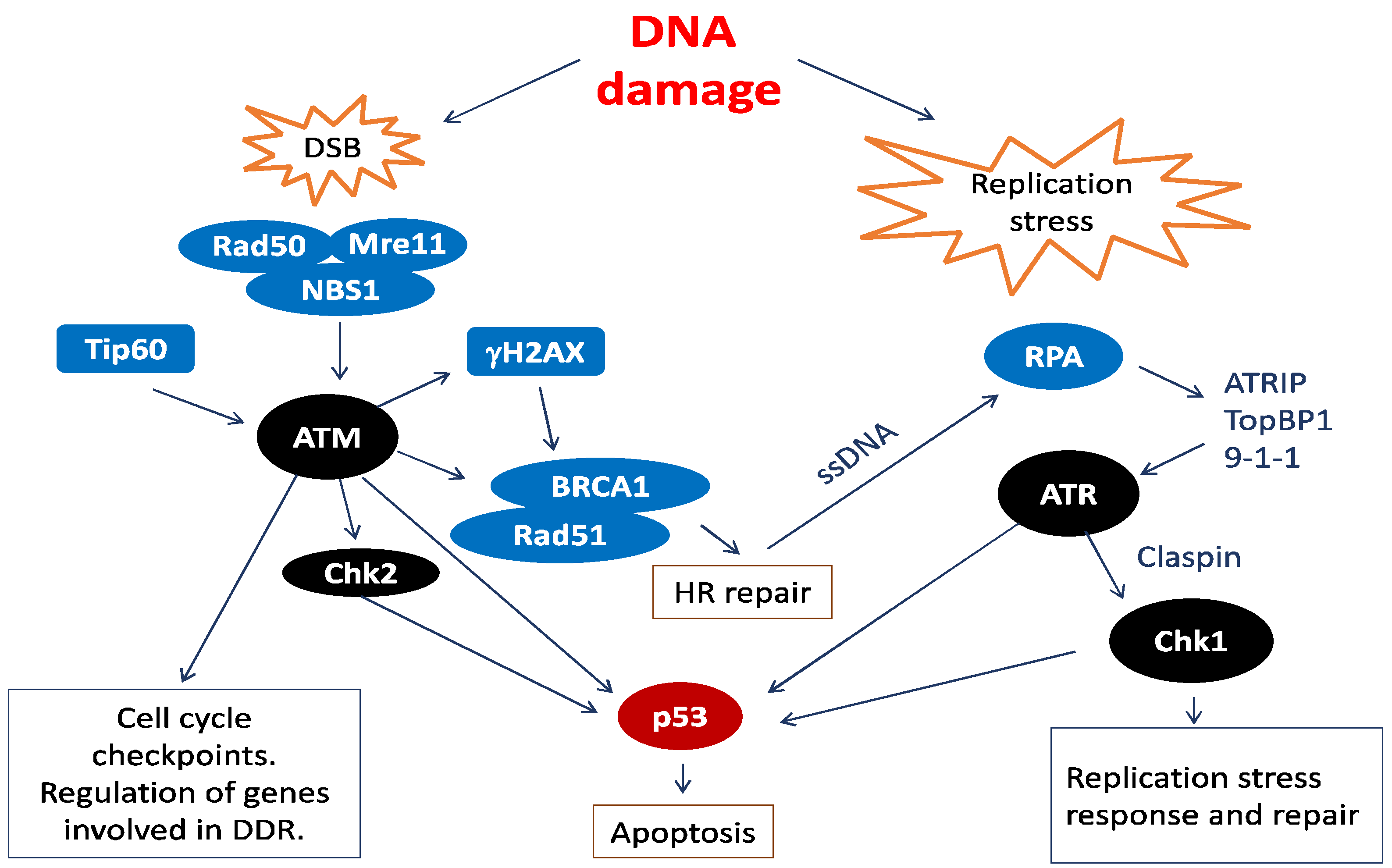
2. Human Papillomavirus (HPV) and the Cellular DNA Damage Response (DDR)
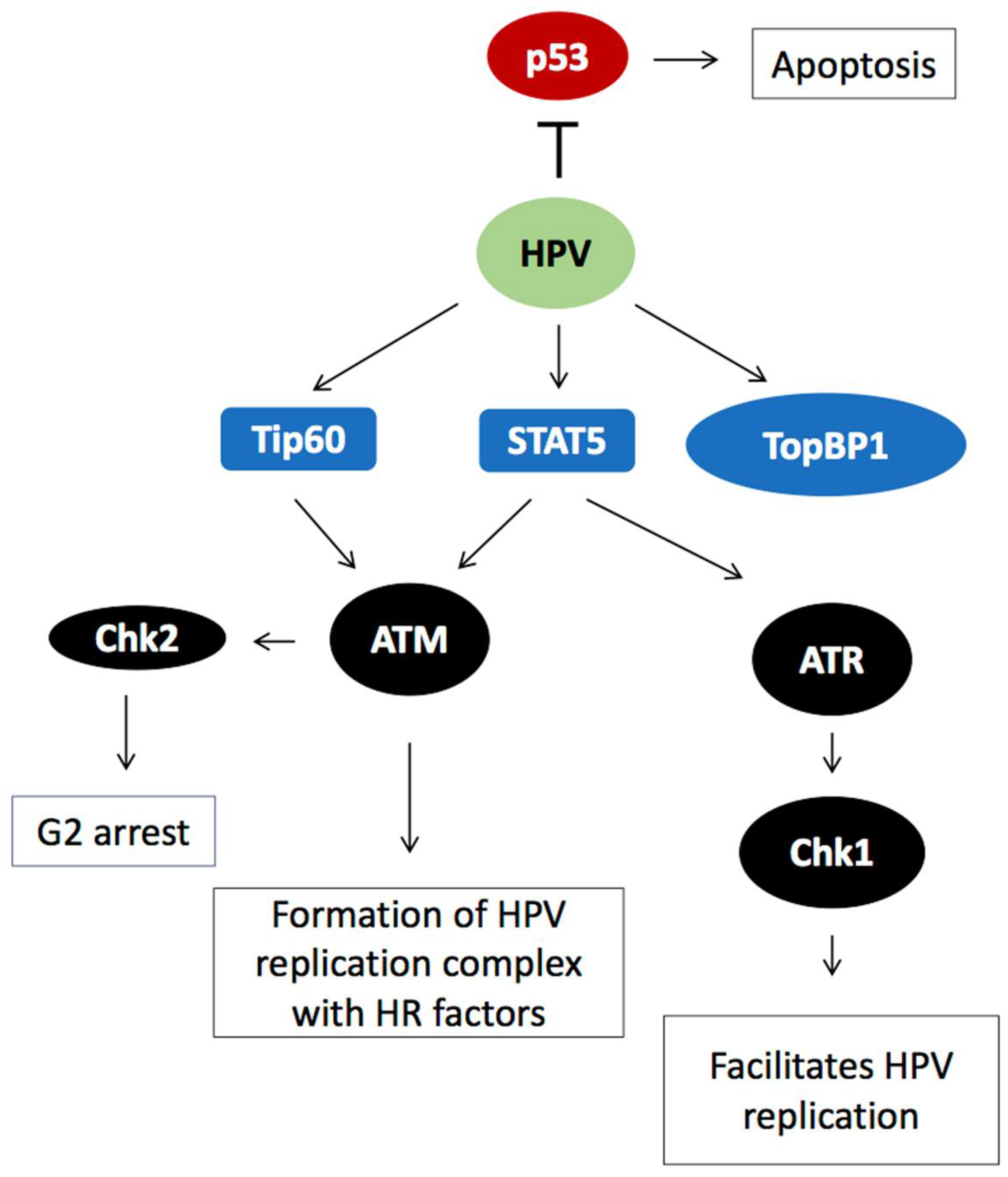
2.1. HPV Employs the Cellular DNA Damage Response for Genome Amplification
2.2. HPV Proteins Perturb Cell Differentiation to Allow for Replication of HPV DNA
2.3. DDR Factors Contribute to HPV DNA Replication
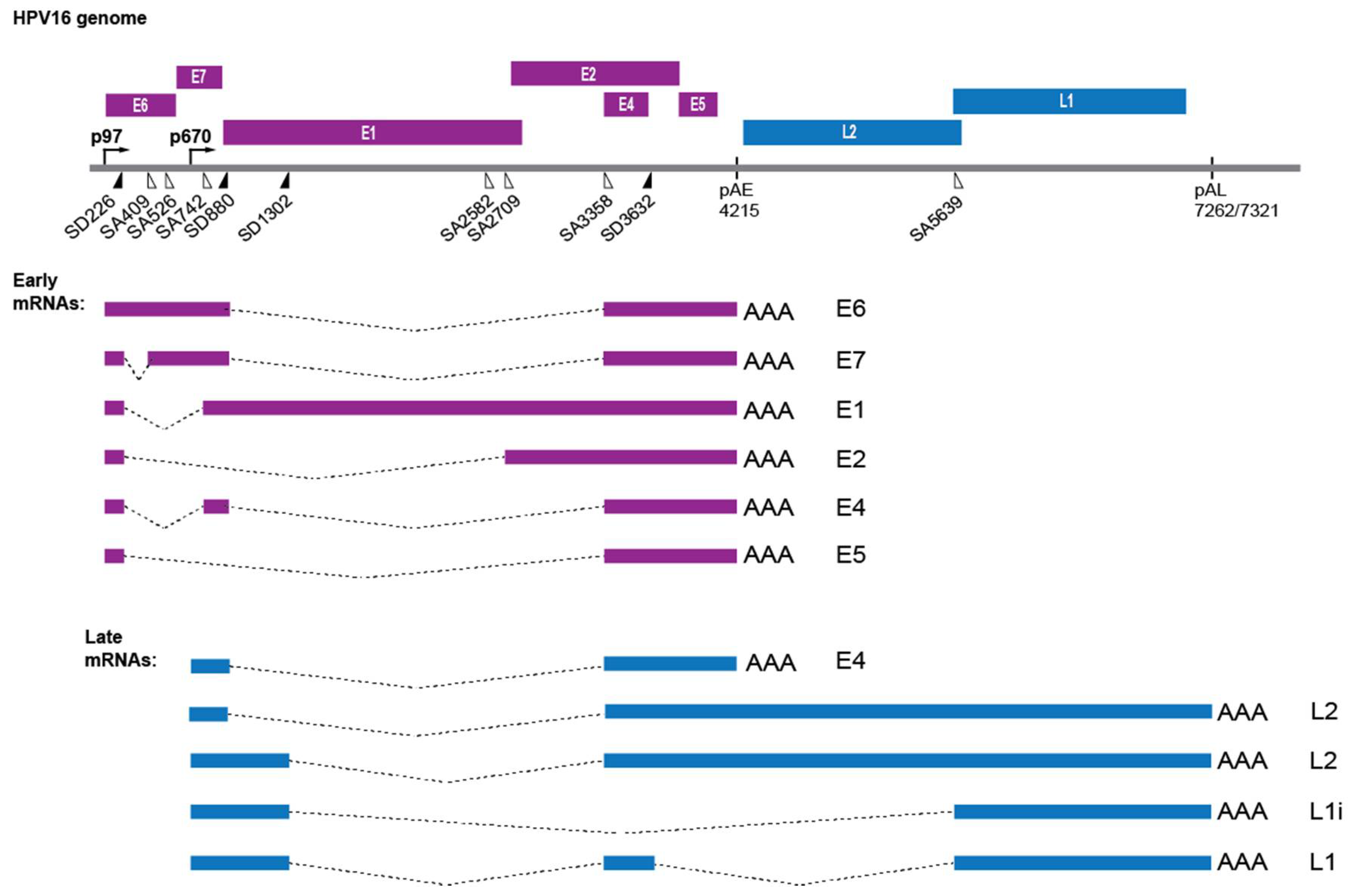
2.4. HPV Gene Regulation
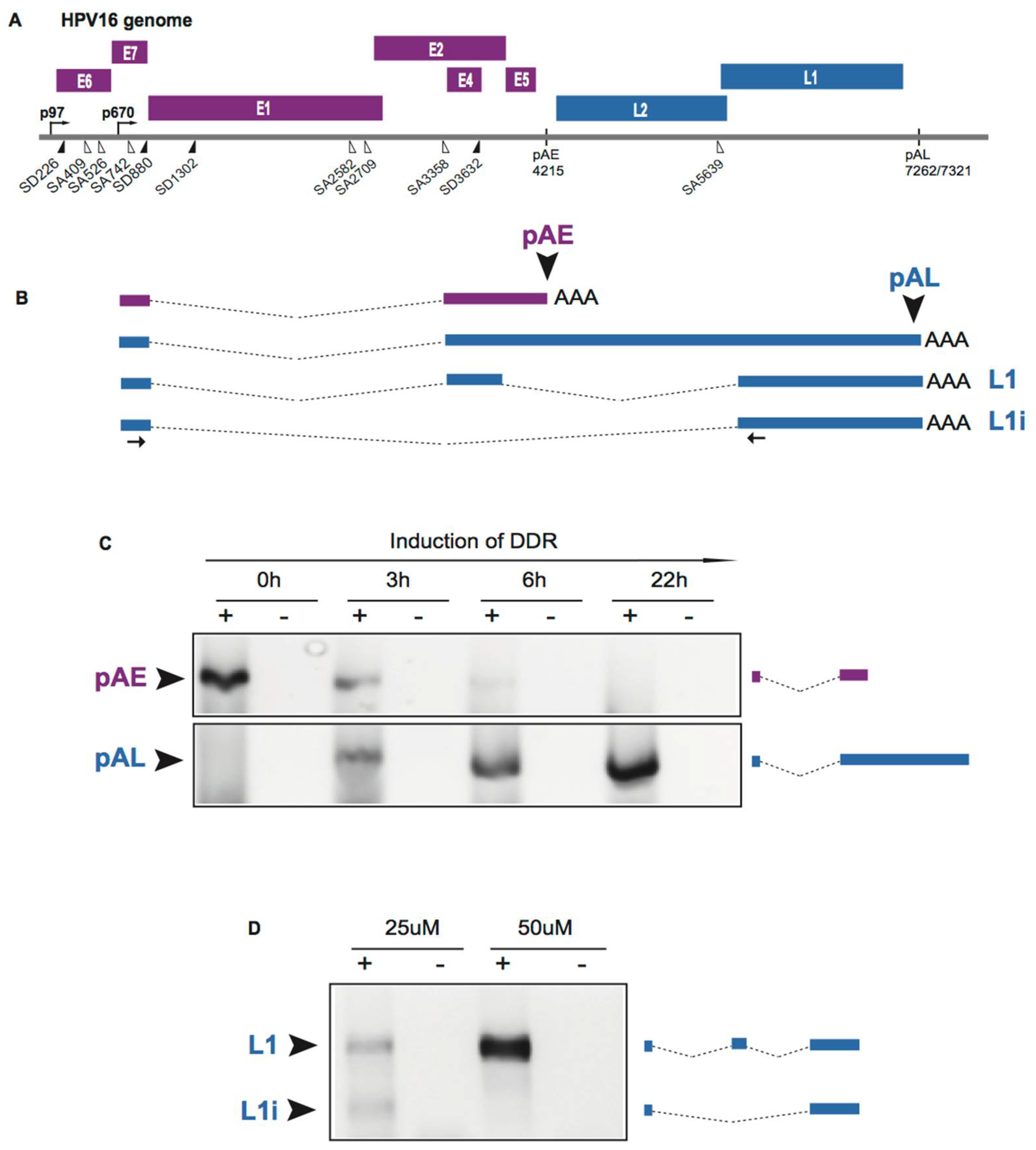
2.5. Induction of HPV Late Gene Expression by the DNA Damage Response
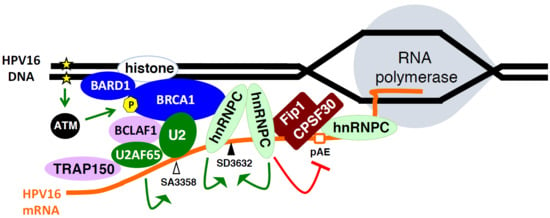
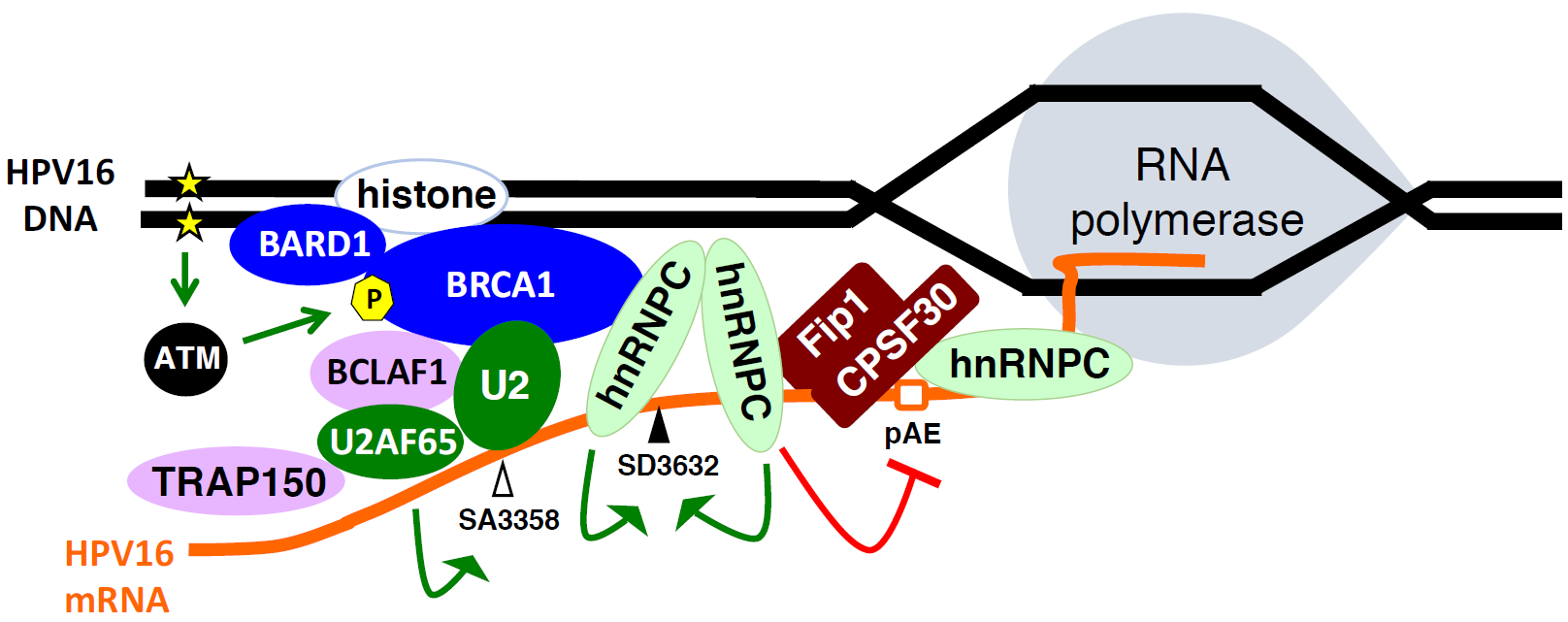
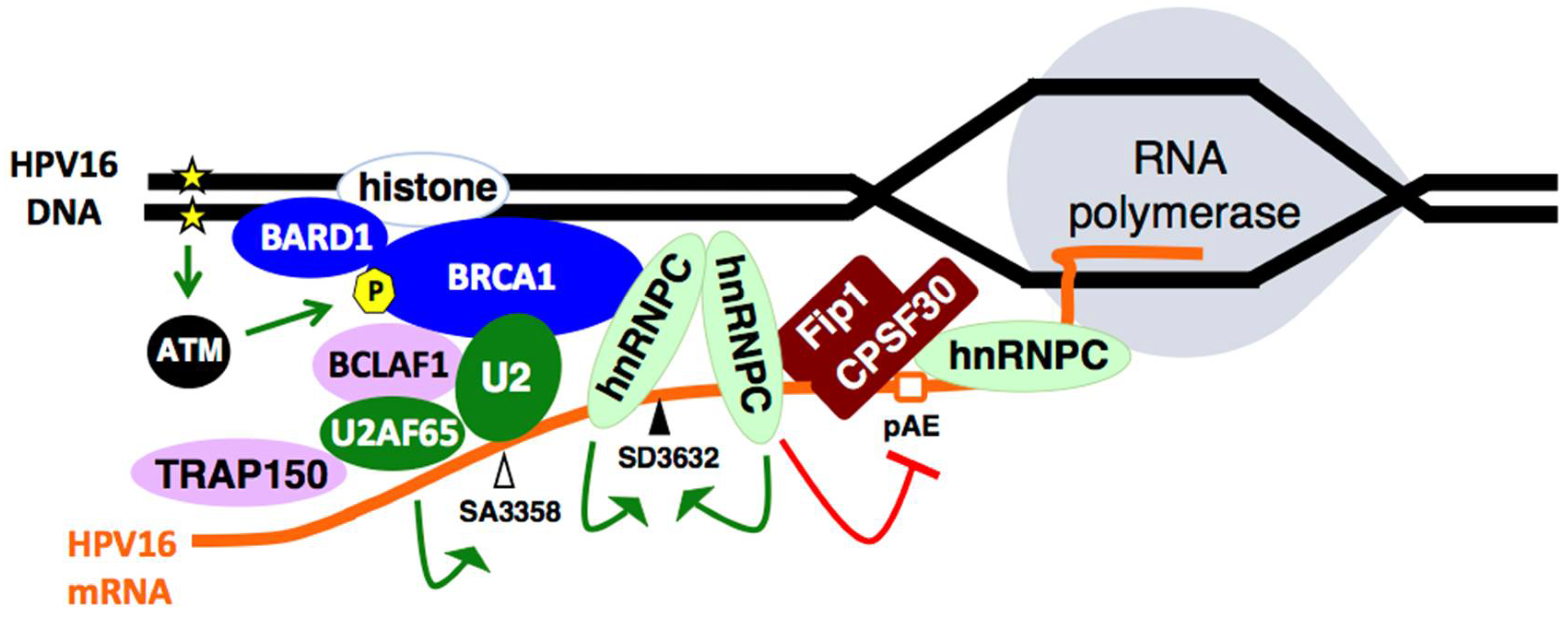
2.6. Cellular DNA Damage Response Factors Associate with HPV16 DNA and Recruit Cellular RNA Processing Factors
2.7. Increased Association between HPV16 mRNA-Binding Proteins and Cellular Polyadenylation Factors in Response to DNA Damage
2.8. DNA Damage Response Factors Recruit Splicing Factors to HPV16 DNA That Alter Splicing of HPV16 mRNAs
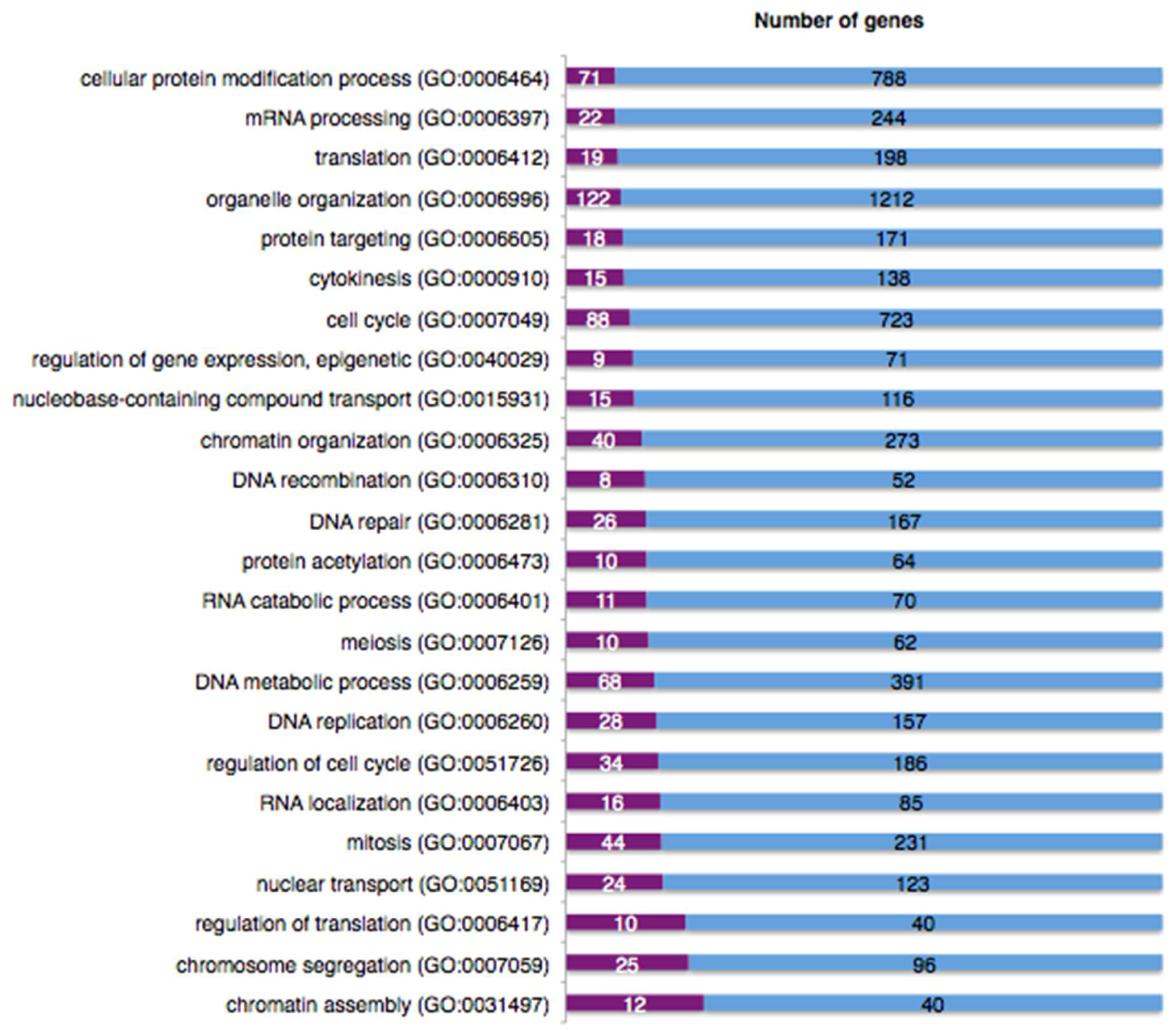
2.9. The DNA Damage Response Affects Alternative Splicing of Cellular mRNAs
3. Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Lowy, D.R. Papillomaviridae. In Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott/The Williams & Wilkins Co.: Philadelphia, PA, USA, 2006; Volume 2, pp. 2299–2354. [Google Scholar]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Kadaja, M.; Silla, T.; Ustav, E.; Ustav, M. Papillomavirus DNA replication—From initiation to genomic instability. Virology 2009, 384, 360–368. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Breitburd, F.; Croissant, O.; Orth, G. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J. Virol. 1977, 21, 1205–1209. [Google Scholar] [PubMed]
- Chow, L.T.; Broker, T.R.; Steinberg, B.M. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 2010, 118, 422–449. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U. Regulatory elements in the viral genome. Virology 2013, 445, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Thierry, F. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 2009, 384, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Schwartz, S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat. Rev. Microbiol. 2013, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Zheng, Z.M. Regulation of bovine papillomavirus type 1 gene expression by RNA processing. Front. Biosci. 2009, 14, 1270–1282. [Google Scholar] [CrossRef]
- Graham, S.V.; Faizo, A.A. Control of human papillomavirus gene expression by alternative splicing. Virus Res. 2017, 231, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Kajitani, N.; Schwartz, S. Splicing and Polyadenylation of Human Papillomavirus Type 16 mRNAs. Int. J. Mol. Sci. 2017, 18, 366. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Schwartz, S. RNA Binding Proteins that Control Human Papillomavirus Gene Expression. Biomolecules 2015, 5, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Bodaghi, S.; Jia, R.; Zheng, Z.M. Human papillomavirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNAs with suboptimal splice sites. Virology 2009, 386, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, 012716. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Smolka, M.B.; Albuquerque, C.P.; Chen, S.H.; Zhou, H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 10364–10369. [Google Scholar] [CrossRef] [PubMed]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Peh, W.L.; Doorbar, J.; Lee, D.; Lambert, P.F. Human papillomavirus type 16 E1E4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 2005, 79, 13150–13165. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Taylor, W.R. Control of the G2/M transition. Mol. Biotechnol. 2006, 32, 227–248. [Google Scholar] [CrossRef]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 Regulates Transcription of the Topoisomerase IIbeta-Binding Protein 1 (TopBP1) Gene To Activate the ATR Pathway and Promote Human Papillomavirus Replication. MBio 2015, 6, e02006–e02015. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Aloor, H.L.; Shepard, C.N.; Lenzi, G.M.; Johnson, B.A.; Kim, B.; Moody, C.A. HPV31 utilizes the ATR-Chk1 pathway to maintain elevated RRM2 levels and a replication-competent environment in differentiating Keratinocytes. Virology 2016, 499, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed]
- Moody, C. Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses 2017, 9, 261. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y. DNA damage response is hijacked by human papillomaviruses to complete their life cycle. J. Zhejiang Univ. Sci. B 2017, 18, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Moody, C.A. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017, 231, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J. Virol. 2015, 89, 4980–4991. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Aloor, H.L.; Moody, C.A. The Rb binding domain of HPV31 E7 is required to maintain high levels of DNA repair factors in infected cells. Virology 2017, 500, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce HPV 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dutta, A.; Laimins, L.A. The acetyltransferase Tip60 is a critical regulator of the differentiation-dependent amplification of human papillomaviruses. J. Virol. 2015, 89, 4668–4675. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S. Papillomavirus transcripts and posttranscriptional regulation. Virology 2013, 445, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Papillomavirus 3’UTR regulatory elements. Front. Biosci. 2008, 13, 5646–5663. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Johansson, C.; Glahder, J.; Mossberg, A.K.; Schwartz, S. Suppression of HPV-16 late L1 5’-splice site SD3632 by binding of hnRNP D proteins and hnRNP A2/B1 to upstream AUAGUA RNA motifs. Nucleic Acids Res. 2013, 22, 10488–10508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rush, M.; Schwartz, S. Identification of an hnRNP A1 dependent splicing silencer in the HPV-16 L1 coding region that prevents premature expression of the late L1 gene. J. Virol. 2004, 78, 10888–10905. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Somberg, M.; Li, X.; Backström Winquist, E.; Fay, J.; Ryan, F.; Pim, D.; Banks, L.; Schwartz, S. HPV-16 E2 contributes to induction of HPV-16 late gene expression by inhibiting early polyadenylation. EMBO J. 2012, 13, 3212–3227. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Anantha, R.W.; Alcivar, A.L.; Ma, J.; Cai, H.; Simhadri, S.; Ule, J.; Konig, J.; Xia, B. Requirement of heterogeneous nuclear ribonucleoprotein C for BRCA gene expression and homologous recombination. PLoS ONE 2013, 8, e61368. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Li, J.M.; Ji, X.Y.; Wu, C.S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jimenez, A.E.; Jin, J.; Zou, L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Kai, M. Roles of RNA-Binding Proteins in DNA Damage Response. Int. J. Mol. Sci. 2016, 17, 310. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.H.; Choi, H.K.; Jung, S.Y.; Hyle, J.; Kim, B.J.; Yoon, K.; Cho, E.J.; Youn, H.D.; Lahti, J.M.; Qin, J.; et al. CHK2 kinase promotes pre-mRNA splicing via phosphorylating CDK11(p110). Oncogene 2014, 33, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Shkreta, L.; Chabot, B. The RNA Splicing Response to DNA Damage. Biomolecules 2015, 5, 2935–2977. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Vagner, S. DNA-Damage Response RNA-Binding Proteins (DDRBPs): Perspectives from a New Class of Proteins and Their RNA Targets. J. Mol. Biol. 2017, 429, 3139–3145. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Jamal Fattah, T.; Yu, H.; Nygren, J.; Mossberg, A.K.; Schwartz, S. Acetylation of intragenic histones on HPV16 correlates with enhanced HPV16 gene expression. Virology 2015, 482, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, K.; Wu, C.; Kajitani, N.; Yu, H.; Tsimtsirakis, E.; Gong, L.; Winquist, E.B.; Glahder, J.; Ekblad, L.; Wennerberg, J.; et al. The DNA damage response activates HPV16 late gene expression at the level of RNA processing. Nucleic Acids Res. 2018, 46, 5029–5049. [Google Scholar] [CrossRef] [PubMed]
- Sarras, H.; Alizadeh Azami, S.; McPherson, J.P. In search of a function for BCLAF1. Sci. World J. 2010, 10, 1450–1461. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Hsu Ia, W.; Tarn, W.Y. TRAP150 activates pre-mRNA splicing and promotes nuclear mRNA degradation. Nucleic Acids Res. 2010, 38, 3340–3350. [Google Scholar] [CrossRef] [PubMed]
- Yarosh, C.A.; Tapescu, I.; Thompson, M.G.; Qiu, J.; Mallory, M.J.; Fu, X.D.; Lynch, K.W. TRAP150 interacts with the RNA-binding domain of PSF and antagonizes splicing of numerous PSF-target genes in T cells. Nucleic Acids Res. 2015, 43, 9006–9016. [Google Scholar] [CrossRef] [PubMed]
- Shepard, P.J.; Hertel, K.J. The SR protein family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Long, J.C.; Caceres, J.F. The SR protein family of splicing factors: Master regulators of gene expression. Biochem. J. 2009, 417, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Dhanjal, S.; Kajitani, N.; Glahder, J.; Mossberg, A.K.; Johansson, C.; Schwartz, S. Heterogeneous Nuclear Ribonucleoprotein C Proteins Interact with the Human Papillomavirus Type 16 (HPV16) Early 3’-Untranslated Region and Alleviate Suppression of HPV16 Late L1 mRNA Splicing. J. Biol. Chem. 2015, 290, 13354–13371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Öberg, D.; Rush, M.; Fay, J.; Lambkin, H.; Schwartz, S. A 57 nucleotide upstream early polyadenylation element in human papillomavirus type 16 interacts with hFip1, CstF-64, hnRNP C1/C2 and PTB. J. Virol. 2005, 79, 4270–4288. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; Chuen-Im, T.; Zhang, J.; Graham, S.V. The RNA stability regulator HuR regulates L1 protein expression in vivo in differentiating cervical epithelial cells. Virology 2009, 383, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.J.; Schmidt, R.; Gruber, A.R.; Martin, G.; Ghosh, S.; Belmadani, M.; Keller, W.; Zavolan, M. A comprehensive analysis of 3’ end sequencing data sets reveals novel polyadenylation signals and the repressive role of heterogeneous ribonucleoprotein C on cleavage and polyadenylation. Genome Res. 2016, 26, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Gong, L.; Wu, C.; Nilsson, K.; Li-Wang, X.; Schwartz, S. hnRNP G prevents inclusion on the HPV16 L1 mRNAs of the central exon between splice sites SA3358 and SD3632. J. Gen. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Liu, X.; Tao, M.; Kruhlak, M.; Guo, M.; Meyers, C.; Baker, C.C.; Zheng, Z.M. Control of the papillomavirus early-to-late switch by differentially expressed SRp20. J. Virol. 2009, 83, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Somberg, M.; Li, X.; Johansson, C.; Orru, B.; Chang, R.; Rush, M.; Fay, J.; Ryan, F.; Schwartz, S. SRp30c activates human papillomavirus type 16 L1 mRNA expression via a bimodal mechanism. J. Gen. Virol. 2011, 92, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Somberg, M.; Schwartz, S. Multiple ASF/SF2 sites in the HPV-16 E4-coding region promote splicing to the most commonly used 3’-splice site on the HPV-16 genome. J. Virol. 2010, 84, 8219–8230. [Google Scholar] [CrossRef] [PubMed]
- Rush, M.; Zhao, X.; Schwartz, S. A splicing enhancer in the E4 coding region of human papillomavirus type 16 is required for early mRNA splicing and polyadenylation as well as inhibition of premature late gene expression. J. Virol. 2005, 79, 12002–12015. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Johansson, C.; Cardoso-Palacios, C.; Mossberg, A.; Dhanjal, S.; Bergvall, M.; Schwartz, S. Eight nucleotide substitutions inhibit splicing to HPV-16 3’-splice site SA3358 and reduce the efficiency by which HPV-16 increases the life span of primary human keratinocytes. PLoS ONE 2013, 8, e72776. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Glahder, J.; Wu, C.; Yu, H.; Nilsson, K.; Schwartz, S. hnRNP L controls HPV16 RNA polyadenylation and splicing in an Akt-kinase-dependent manner. Nucleic Acids Res. 2017, 45, 9654–9678. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 2014, 54, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.; Kelehan, P.; Lambkin, H.; Schwartz, S. Increased expression of cellular RNA-binding proteins in HPV-induced neoplasia and cervical cancer. J. Med. Virol. 2009, 81, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.; McFarlane, M.; Chuen-Im, T.; Milligan, S.G.; Millan, D.; Graham, S.V. RNA splicing factors regulated by HPV16 during cervical tumour progression. J. Pathol. 2009, 219, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, T.; Hernandez-Lopez, H.; MacDonald, A.I.; Bodily, J.M.; Graham, S.V. Human Papillomavirus E2 Regulates SRSF3 (SRp20) to Promote Capsid Protein Expression in Infected Differentiated Keratinocytes. J. Virol. 2016, 90, 5047–5058. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Windle, B.; Donaldson, M.M.; Caffarel, M.M.; Dornan, E.S.; Coleman, N.; Herzyk, P.; Henderson, S.C.; Wang, X.; Morgan, I.M. Regulation of human genome expression and RNA splicing by human papillomavirus 16 E2 protein. Virology 2014, 468–470, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High-Risk Alphapapillomavirus Oncogenes Impair the Homologous Recombination Pathway. J. Virol. 2017, 91, e01084-17. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Laimins, L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Laimins, L.A. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose. Methods Mol. Med. 2005, 119, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.F.; Ozbun, M.A.; Collins, A.; Holmgren, S.; Lee, D.; Nakahra, T. Using an imortalised cell line to study the HPV life cycle in organotypic “raft” cultures. Methods Mol. Med. 2005, 119, 141–155. [Google Scholar] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilsson, K.; Wu, C.; Schwartz, S. Role of the DNA Damage Response in Human Papillomavirus RNA Splicing and Polyadenylation. Int. J. Mol. Sci. 2018, 19, 1735. https://doi.org/10.3390/ijms19061735
Nilsson K, Wu C, Schwartz S. Role of the DNA Damage Response in Human Papillomavirus RNA Splicing and Polyadenylation. International Journal of Molecular Sciences. 2018; 19(6):1735. https://doi.org/10.3390/ijms19061735
Chicago/Turabian StyleNilsson, Kersti, Chengjun Wu, and Stefan Schwartz. 2018. "Role of the DNA Damage Response in Human Papillomavirus RNA Splicing and Polyadenylation" International Journal of Molecular Sciences 19, no. 6: 1735. https://doi.org/10.3390/ijms19061735
APA StyleNilsson, K., Wu, C., & Schwartz, S. (2018). Role of the DNA Damage Response in Human Papillomavirus RNA Splicing and Polyadenylation. International Journal of Molecular Sciences, 19(6), 1735. https://doi.org/10.3390/ijms19061735