The Impact of the Nitric Oxide (NO)/Soluble Guanylyl Cyclase (sGC) Signaling Cascade on Kidney Health and Disease: A Preclinical Perspective

 and
and
Abstract
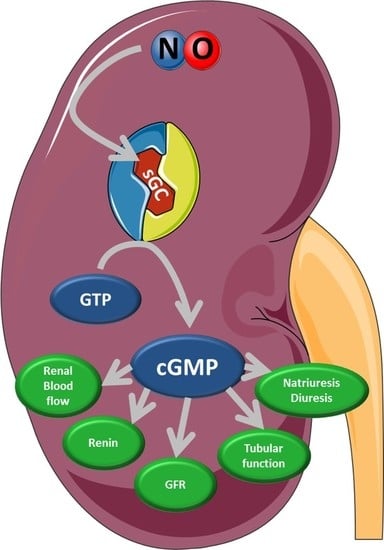
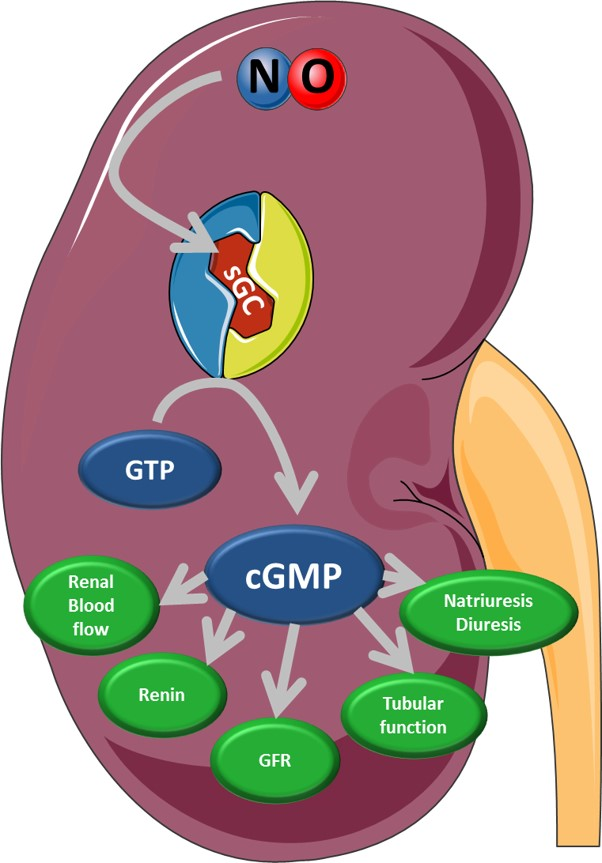
1. Summary
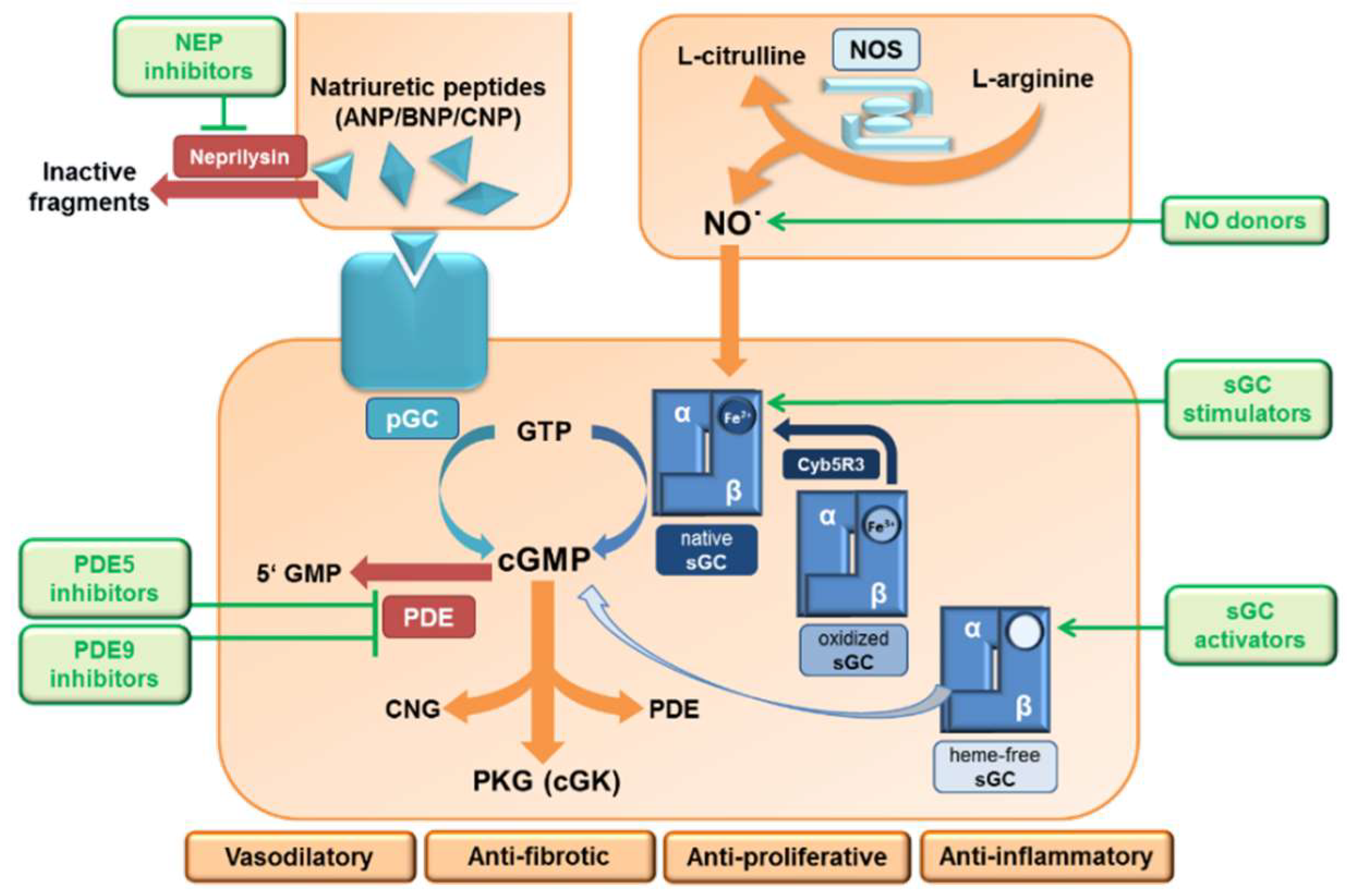
2. The NO/sGC/cGMP Signaling
3. The NPs/pGCs/cGMP Signaling
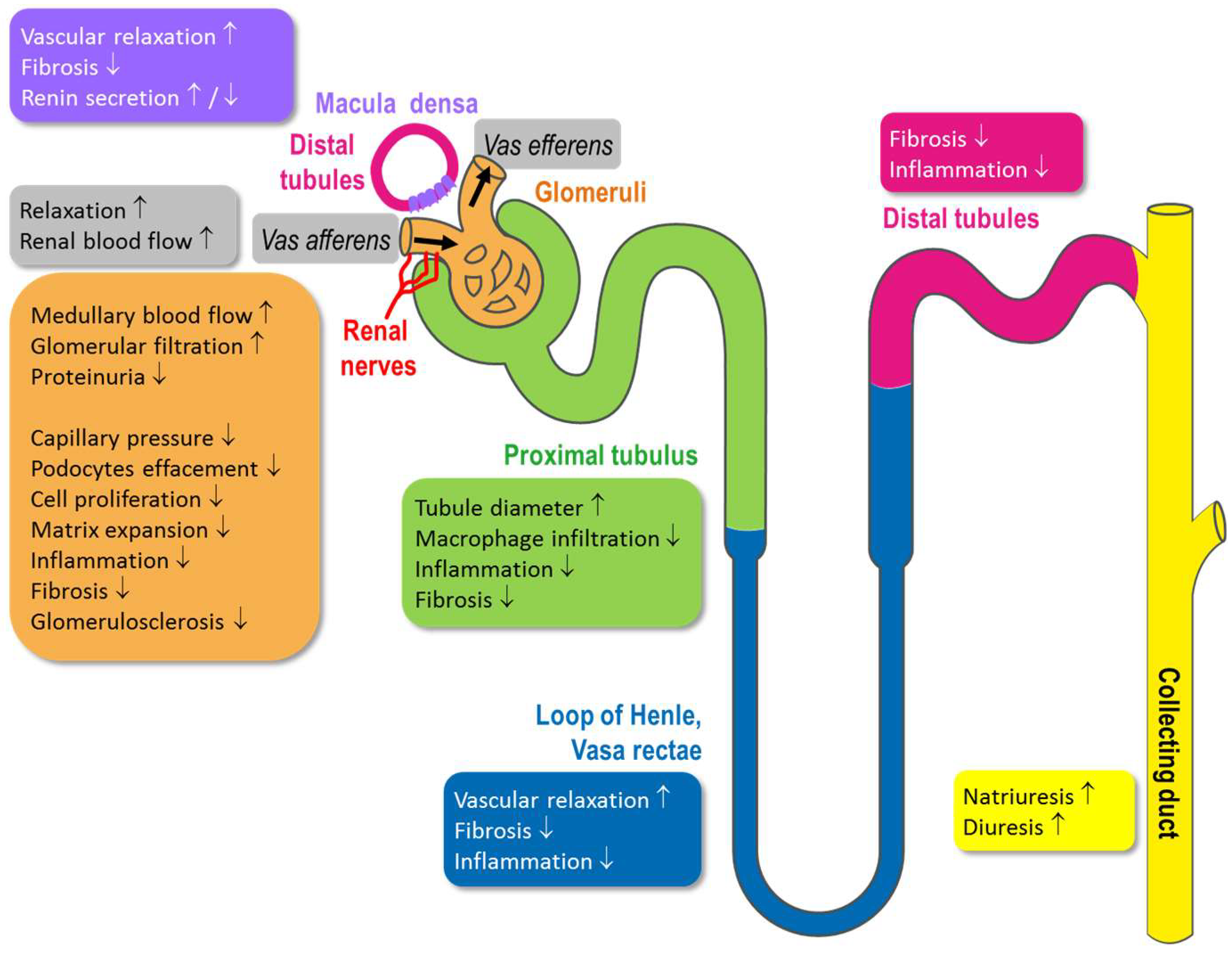
4. Physiological Function of Kidney Compartments and Regulation via NO/sGC/cGMP Signaling
5. Impact of the Impaired NO/sGC/cGMP Signaling on Kidney Function
6. Major Pharmacological Intervention Sites and Therapeutic Approaches on the NO/sGC Pathway
6.1. Nitrates
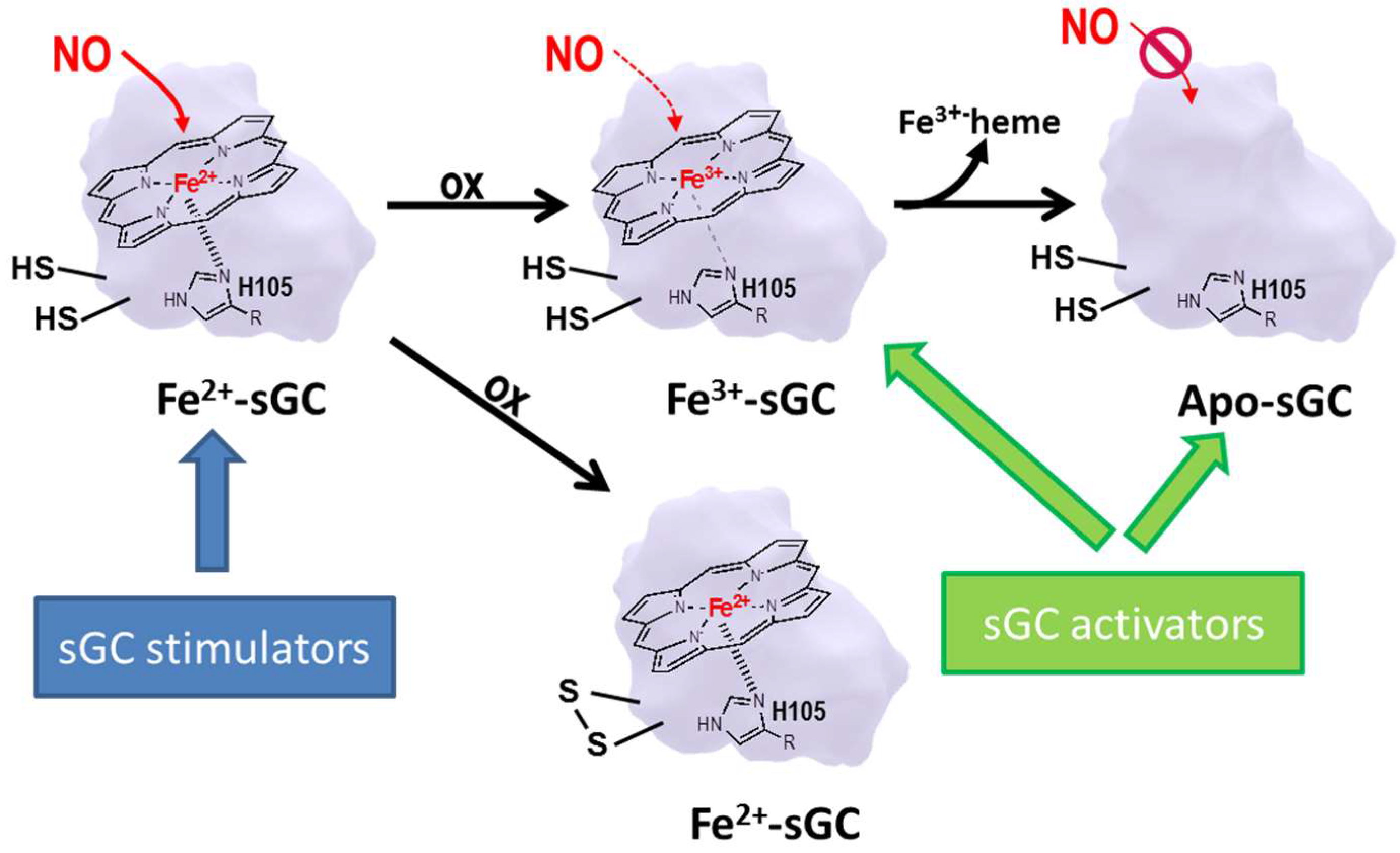
6.2. sGC Modulators
6.3. sGC Stimulators
6.4. sGC Activators
6.5. PDE5 and PDE9 Inhibitors
7. Reinforcing the NO/sGC/cGMP Axis as Treatment Option for Kidney Diseases
8. Outlook
Conflicts of Interest
References
- Beuve, A. Thiol-based redox modulation of soluble guanylyl cyclase, the nitric oxide receptor. Antioxid. Redox Signal. 2016, 26, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.M.; Nguyen, A.T.; Miller, M.P.; Hahn, S.A.; Sparacino-Watkins, C.; Jobbagy, S.; Carew, N.T.; Cantu-Medellin, N.; Wood, K.C.; Baty, C.J.; et al. Cytochrome b5 reductase 3 modulates soluble guanylate cyclase redox state and cgmp signaling. Circ. Res. 2017, 121, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.; Bosse, H.M.; Mundel, P. Topography of nitric oxide synthesis by localizing constitutive NO synthases in mammalian kidney. Am. J. Physiol. 1995, 268, F885–F898. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.L.; Sasser, J.M.; Hobbs, J.L.; Boriskie, A.; Pollock, D.M.; Carmines, P.K.; Pollock, J.S. Posttranslational regulation of NO synthase activity in the renal medulla of diabetic rats. Am. J. Physiol. Renal Physiol. 2005, 288, F82–F90. [Google Scholar] [CrossRef] [PubMed]
- Sasser, J.M.; Brinson, K.N.; Tipton, A.J.; Crislip, G.R.; Sullivan, J.C. Blood pressure, sex, and female sex hormones influence renal inner medullary nitric oxide synthase activity and expression in spontaneously hypertensive rats. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Mundel, P.; Gambaryan, S.; Bachmann, S.; Koesling, D.; Kriz, W. Immunolocalization of soluble guanylyl cyclase subunits in rat kidney. Histochem. Cell Biol. 1995, 103, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Theilig, F.; Bostanjoglo, M.; Pavenstadt, H.; Grupp, C.; Holland, G.; Slosarek, I.; Gressner, A.M.; Russwurm, M.; Koesling, D.; Bachmann, S. Cellular distribution and function of soluble guanylyl cyclase in rat kidney and liver. J. Am. Soc. Nephrol. 2001, 12, 2209–2220. [Google Scholar] [PubMed]
- Wolfertstetter, S.; Huettner, J.P.; Schlossmann, J. cGMP-dependent protein kinase inhibitors in health and disease. Pharmaceuticals 2013, 6, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Buglioni, A.; Burnett, J.C., Jr. Pathophysiology and the cardiorenal connection in heart failure. Circulating hormones: Biomarkers or mediators. Clin. Chim. Acta 2015, 443, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.A. The natriuretic peptide system: Kidney and cardiovascular effects. Curr. Opin. Nephrol. Hypertens. 2006, 15, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M. The Kidney; Saunders: Philadelphia, PA, USA, 2004. [Google Scholar]
- Ohishi, K.; Carmines, P.K.; Inscho, E.W.; Navar, L.G. EDRF-angiotensin II interactions in rat juxtamedullary afferent and efferent arterioles. Am. J. Physiol. Renal Physiol. 1992, 263, F900–F906. [Google Scholar] [CrossRef] [PubMed]
- Simons, M. The benefits of tubular proteinuria: An evolutionary perspective. J. Am. Soc. Nephrol. 2018, 29, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Abbate, M.; Remuzzi, G. Progression of renal injury toward interstitial inflammation and glomerular sclerosis is dependent on abnormal protein filtration. Nephrol. Dial. Transplant. 2015, 30, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-L.; Liu, L.; Barajas, L. Development of NOS-containing neuronal somata in the rat kidney. J. Auton. Nerv. Syst. 1996, 58, 81–88. [Google Scholar] [CrossRef]
- Friebe, A.; Sandner, P.; Schmidtko, A. Meeting report of the 8th International Conference on cGMP “cGMP: Generators, effectors, and therapeutic implications” at Bamberg, Germany, from June 23 to 25, 2017. Naunyn Schmiedebergs Arch. Pharmacol. 2017, 390, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P. From molecules to patients: Exploring the therapeutic role of soluble guanylate cyclase stimulators. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Baylis, C.; Qiu, C. Importance of nitric oxide in the control of renal hemodynamics. Kidney Int. 1996, 49, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Patzak, A.; Kleinmann, F.; Lai, E.Y.; Kupsch, E.; Skelweit, A.; Mrowka, R. Nitric oxide counteracts angiotensin II induced contraction in efferent arterioles in mice. Acta Physiol. Scand. 2004, 181, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Patzak, A.; Persson, A.E. Angiotensin II-nitric oxide interaction in the kidney. Curr. Opin. Nephrol. Hypertens. 2007, 16, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Kornfeld, M.; Ruan, X.; Arendshorst, W.J.; Kurtz, A. Nitric oxide/cAMP interactions in the control of rat renal vascular resistance. Circ. Res. 1999, 84, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Schricker, K.; Ritthaler, T.; Krämer, B.K.; Kurtz, A. Effect of endothelium-derived relaxing factor on renin secretion from isolated mouse renal juxtaglomerular cells. Acta Physiol. Scand. 1993, 149, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Polichnowski, A.J.; Licea-Vargas, H.; Picken, M.; Long, J.; Bisla, R.; Williamson, G.A.; Bidani, A.K.; Griffin, K.A. Glomerulosclerosis in the diet-induced obesity model correlates with sensitivity to nitric oxide inhibition but not glomerular hyperfiltration or hypertrophy. Am. J. Physiol. Renal Physiol. 2015, 309, F791–F799. [Google Scholar] [CrossRef] [PubMed]
- Bidani, A.K.; Polichnowski, A.J.; Loutzenhiser, R.; Griffin, K.A. Renal microvascular dysfunction, hypertension and CKD progression. Curr. Opin. Nephrol. Hypertens. 2013, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Thorup, C.; Erik, A.; Persson, G. Macula densa derived nitric oxide in regulation of glomerular capillary pressure. Kidney Int. 1996, 49, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.C.; Petrova, G.; Kurth, T.; Yang, C.; Bukowy, J.D.; Mattson, D.L.; Cowley, A.W., Jr. Increased perfusion pressure drives renal t-cell infiltration in the dahl salt-sensitive rat. Hypertension 2017, 70, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, Y.; DiPiero, A.; Hirt, E.; Brennaman, B.; Lockette, W. Vascular relaxation and cGMP in hypertension. Am. J. Physiol. Heart Circ. Physiol. 1988, 254, H163–H169. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, H.Y.; Roth, R.; Jain, S.; Heuser, J.E.; Shaw, A.S.; Miner, J.H. Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Boustany-Kari, C.M.; Harrison, P.C.; Chen, H.; Lincoln, K.A.; Qian, H.S.; Clifford, H.; Wang, H.; Zhang, X.; Gueneva-Boucheva, K.; Bosanac, T.; et al. A soluble guanylate cyclase activator inhibits the progression of diabetic nephropathy in the ZSF1 rat. J. Pharmacol. Exp. Ther. 2016, 356, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Brezis, M.; Heyman, S.N.; Dinour, D.; Epstein, F.H.; Rosen, S. Role of nitric oxide in renal medullary oxygenation. Studies in isolated and intact rat kidneys. J. Clin. Investig. 1991, 88, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Hohenstein, B.; Daniel, C.; Wagner, A.; Stasch, J.P.; Hugo, C. Stimulation of soluble guanylyl cyclase inhibits mesangial cell proliferation and matrix accumulation in experimental glomerulonephritis. Am. J. Physiol. Renal Physiol. 2005, 288, F685–F693. [Google Scholar] [CrossRef] [PubMed]
- Schinner, E.; Wetzl, V.; Schlossmann, J. Cyclic nucleotide signalling in kidney fibrosis. Int. J. Mol. Sci. 2015, 16, 2320–2351. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Berger, P.; Zenzmaier, C. The potential of sGC modulators for the treatment of age-related fibrosis: A mini-review. Gerontology 2017, 63, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Abboud, H.E. Mesangial cell biology. Exp. Cell Res. 2012, 318, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Maimaitiyiming, H.; Qi, X.; Norman, H.; Zhou, Q.; Wang, X.; Fu, J.; Wang, S. Increasing cGMP-dependent protein kinase activity attenuates unilateral ureteral obstruction-induced renal fibrosis. Am. J. Physiol. Renal Physiol. 2014, 306, F996–F1007. [Google Scholar] [CrossRef] [PubMed]
- Schinner, E.; Wetzl, V.; Schramm, A.; Kees, F.; Sandner, P.; Stasch, J.P.; Hofmann, F.; Schlossmann, J. Inhibition of the TGFβ signalling pathway by cGMP and cGMP-dependent kinase I in renal fibrosis. FEBS Open Bio 2017, 7, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Radovits, T.; Szabo, G.; Mozes, M.M.; Rosivall, L.; Kokeny, G. Selective phosphodiesterase-5 (PDE-5) inhibitor vardenafil ameliorates renal damage in type 1 diabetic rats by restoring cyclic 3′, 5′ guanosine monophosphate (cGMP) level in podocytes. Nephrol. Dial Transplant. 2013, 28, 1751–1761. [Google Scholar] [CrossRef] [PubMed]
- Helwig, J.J.; Yusufi, A.N.; Rebel, G.; Geiser, J.; Bollack, C.; Mandel, P. Distribution of enzymes of cGMP metabolism in glomeruli and tubules isolated from normal and nephrotic rat kidney cortex. Int. J. Biochem. 1980, 12, 209–214. [Google Scholar] [CrossRef]
- Dolinina, J.; Sverrisson, K.; Rippe, A.; Oberg, C.M.; Rippe, B. Nitric oxide synthase inhibition causes acute increases in glomerular permeability in vivo, dependent upon reactive oxygen species. Am. J. Physiol. Renal Physiol. 2016, 311, F984–F990. [Google Scholar] [CrossRef] [PubMed]
- Czirok, S.; Fang, L.; Radovits, T.; Szabó, G.; Szénási, G.; Rosivall, L.; Merkely, B.; Kökény, G. Cinaciguat ameliorates glomerular damage by reducing ERK1/2 activity and TGF-ß expression in type-1 diabetic rats. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.; Rowell, J.; Farinelli, F.; Gbadegesin, R.A.; Lavin, P.; Wu, G.; Homstad, A.; Malone, A.; Lindsey, T.; Jiang, R.; et al. Phosphodiesterase 5 inhibition ameliorates angiontensin II-induced podocyte dysmotility via the protein kinase G-mediated downregulation of TRPC6 activity. Am. J. Physiol. Renal Physiol. 2014, 306, F1442–F1450. [Google Scholar] [CrossRef] [PubMed]
- Tack, I.; Marin Castano, E.; Pecher, C.; Praddaude, F.; Bascands, J.L.; Bompart, G.; Ader, J.L.; Girolami, J.P. Endothelin increases NO-dependent cGMP production in isolated glomeruli but not in mesangial cells. Am. J. Physiol. 1997, 272, F31–F39. [Google Scholar] [CrossRef] [PubMed]
- Eitle, E.; Hiranyachattada, S.; Wang, H.; Harris, P.J. Inhibition of proximal tubular fluid absorption by nitric oxide and atrial natriuretic peptide in rat kidney. Am. J. Physiol. 1998, 274, C1075–C1080. [Google Scholar] [CrossRef] [PubMed]
- Shirai, A.; Yamazaki, O.; Horita, S.; Nakamura, M.; Satoh, N.; Yamada, H.; Suzuki, M.; Kudo, A.; Kawakami, H.; Hofmann, F.; et al. Angiotensin II dose-dependently stimulates human renal proximal tubule transport by the nitric oxide/guanosine 3′, 5′-cyclic monophosphate pathway. J. Am. Soc. Nephrol. 2014, 25, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Dousa, T.P. Cyclic-3′, 5′-nucleotide phosphodiesterase isozymes in cell biology and pathophysiology of the kidney. Kidney Int. 1999, 55, 29–62. [Google Scholar] [CrossRef] [PubMed]
- Van Aubel, R.A.; Smeets, P.H.; Peters, J.G.; Bindels, R.J.; Russel, F.G. The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: Putative efflux pump for urinary cAMP and cGMP. J. Am. Soc. Nephrol. 2002, 13, 595–603. [Google Scholar] [PubMed]
- Ciampolillo, F.; McCoy, D.E.; Green, R.B.; Karlson, K.H.; Dagenais, A.; Molday, R.S.; Stanton, B.A. Cell-specific expression of amiloride-sensitive, Na(+)-conducting ion channels in the kidney. Am. J. Physiol. Cell Physiol. 1996, 271, C1303–C1315. [Google Scholar] [CrossRef] [PubMed]
- Stoos, B.A.; Garcia, N.H.; Garvin, J.L. Nitric oxide inhibits sodium reabsorption in the isolated perfused cortical collecting duct. J. Am. Soc. Nephrol. 1995, 6, 89–94. [Google Scholar] [PubMed]
- Hyndman, K.A.; Mironova, E.V.; Giani, J.F.; Dugas, C.; Collins, J.; McDonough, A.A.; Stockand, J.D.; Pollock, J.S. Collecting duct nitric oxide synthase 1ß activation maintains sodium homeostasis during high sodium intake through suppression of aldosterone and renal angiotensin II pathways. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Stuart, D.; Pollock, J.S.; Takahishi, T.; Kohan, D.E. Collecting duct-specific knockout of nitric oxide synthase 3 impairs water excretion in a sex-dependent manner. Am. J. Physiol. Renal Physiol. 2016, 311, F1074–F1083. [Google Scholar] [CrossRef] [PubMed]
- Bouley, R.; Breton, S.; Sun, T.-X.; McLaughlin, M.; Nsumu, N.N.; Lin, H.Y.; Ausiello, D.A.; Brown, D. Nitric oxide and atrial natriuretic factor stimulate cGMP-dependent membrane insertion of aquaporin 2 in renal epithelial cells. J. Clin. Investig. 2000, 106, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Klokkers, J.; Langehanenberg, P.; Kemper, B.; Kosmeier, S.; Bally, G.V.; Riethmüller, C.; Wunder, F.; Sindic, A.; Pavenstädt, H.; Schlatter, E.; et al. Atrial natriuretic peptide and nitric oxide signaling antagonizes vasopressin-mediated water permeability in inner medullary collecting duct cells. Am. J. Physiol. Renal Physiol. 2009, 297, F693–F703. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, A. Renin release: Sites, mechanisms, and control. Annu. Rev. Physiol. 2011, 73, 377–399. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Pfeifer, A.; Ruth, P.; Hofmann, F.; Kurtz, A. Role of cGMP-kinase II in the control of renin secretion and renin expression. J. Clin. Investig. 1998, 102, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Eriguchi, M.; Tsuruya, K.; Haruyama, N.; Yamada, S.; Tanaka, S.; Suehiro, T.; Noguchi, H.; Masutani, K.; Torisu, K.; Kitazono, T. Renal denervation has blood pressure-independent protective effects on kidney and heart in a rat model of chronic kidney disease. Kidney Int. 2015, 87, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Armenia, A.; Munavvar, A.S.; Abdullah, N.A.; Helmi, A.; Johns, E.J. The contribution of adrenoceptor subtype(s) in the renal vasculature of diabetic spontaneously hypertensive rats. Br. J. Pharmacol. 2004, 142, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, H.; Yoshida, S.I.; Ishiguro, H.; Suzuki, S.; Yasuzaki, H.; Hashimoto, T.; Ishigami, T.; Hirawa, N.; Toya, Y.; Umemura, S.; et al. Arterial wall hypertrophy is ameliorated by alpha2-adrenergic receptor antagonist or aliskiren in kidneys of angiotensinogen-knockout mice. Clin. Exp. Nephrol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; D’Arrigo, G.; Leonardis, D.; Pizzini, P.; Postorino, M.; Tripepi, G.; Mallamaci, F.; van den Brand, J.; van Zuilen, A.; Wetzels, J.; et al. Neuropeptide Y and chronic kidney disease progression: A cohort study. Nephrol. Dial Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.J.; Trivedi, R.K.; Gao, J.; Li, Z.; Scarborough, A.L.; Goodchild, T.T.; Varner, K.J.; Xia, H.; Smart, F.W.; Kapusta, D.R.; et al. Renal sympathetic denervation protects the failing heart via inhibition of neprilysin activity in the kidney. J. Am. Coll. Cardiol. 2017, 70, 2139–2153. [Google Scholar] [CrossRef] [PubMed]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Progression after AKI: Understanding maladaptive repair processes to predict and identify therapeutic treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.M.; Stallons, L.J.; Kneff, J.E.; Alge, J.L.; Harmon, J.L.; Rahn, J.J.; Arthur, J.M.; Beeson, C.C.; Chan, S.L.; Schnellmann, R.G. Urinary mitochondrial DNA is a biomarker of mitochondrial disruption and renal dysfunction in acute kidney injury. Kidney Int. 2015, 88, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current understanding, key questions, and knowledge gaps. J. Am. Soc. Nephrol. 2016, 27, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Matejovic, M.; Ince, C.; Chawla, L.S.; Blantz, R.; Molitoris, B.A.; Rosner, M.H.; Okusa, M.D.; Kellum, J.A.; Ronco, C. Renal hemodynamics in AKI: In search of new treatment targets. J. Am. Soc. Nephrol. 2016, 27, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.M.; Wills, L.P.; Stallons, L.J.; Schnellmann, R.G. cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J. Pharmacol. Exp. Ther. 2013, 347, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, R.M.; Corum, D.; Beeson, C.C.; Schnellmann, R.G. Mitochondrial biogenesis as a pharmacological target: A new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 229–249. [Google Scholar] [CrossRef] [PubMed]
- Golshahi, J.; Nasri, H.; Gharipour, M. Contrast-induced nephropathy: A literature review. J. Nephropathol. 2014, 3, 51–56. [Google Scholar] [PubMed]
- Discigil, B.; Evora, P.R.B.; Pearson, P.J.; Viaro, F.; Rodrigues, A.J.; Schaff, H.V. Ionic radiocontrast inhibits endothelium-dependent vasodilation of the canine renal artery in vitro: Possible mechanism of renal failure following contrast medium infusion. Braz. J. Med. Biol. Res. 2004, 37, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Becker, E.M.; Alonso-Alija, C.; Apeler, H.; Dembowsky, K.; Feurer, A.; Gerzer, R.; Minuth, T.; Perzborn, E.; Pleiss, U.; et al. NO-independent regulatory site on soluble guanylate cyclase. Nature 2001, 410, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Denninger, J.W.; Schelvis, J.P.; Brandish, P.E.; Zhao, Y.; Babcock, G.T.; Marletta, M.A. Interaction of soluble guanylate cyclase with YC-1: Kinetic and resonance Raman studies. Biochemistry 2000, 39, 4191–4198. [Google Scholar] [CrossRef] [PubMed]
- Wales, J.A.; Chen, C.Y.; Breci, L.; Weichsel, A.; Bernier, S.G.; Sheppeck, J.E., 2nd; Solinga, R.; Nakai, T.; Renhowe, P.A.; Jung, J.; et al. Discovery of stimulator binding to a conserved pocket in the heme domain of soluble guanylyl cyclase. J. Biol. Chem. 2018, 293, 1850–1864. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Baskaran, P.; Ma, X.; Dunten, P.W.; Schaefer, M.; Stasch, J.P.; Beuve, A.; van den Akker, F. Structure of cinaciguat (BAY 58–2667) bound to Nostoc H-NOX domain reveals insights into heme-mimetic activation of the soluble guanylyl cyclase. J. Biol. Chem. 2010, 285, 22651–22657. [Google Scholar] [CrossRef] [PubMed]
- Alesutan, I.; Feger, M.; Tuffaha, R.; Castor, T.; Musculus, K.; Buehling, S.S.; Heine, C.L.; Kuro, O.M.; Pieske, B.; Schmidt, K.; et al. Augmentation of phosphate-induced osteo-/chondrogenic transformation of vascular smooth muscle cells by homoarginine. Cardiovasc. Res. 2016, 110, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Bongartz, L.G.; Braam, B.; Verhaar, M.C.; Cramer, M.J.; Goldschmeding, R.; Gaillard, C.A.; Steendijk, P.; Doevendans, P.A.; Joles, J.A. The nitric oxide donor molsidomine rescues cardiac function in rats with chronic kidney disease and cardiac dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H2037–H2045. [Google Scholar] [CrossRef] [PubMed]
- Attia, D.M.; Ni, Z.N.; Boer, P.; Attia, M.A.; Goldschmeding, R.; Koomans, H.A.; Vaziri, N.D.; Joles, J.A. Proteinuria is preceded by decreased nitric oxide synthesis and prevented by a NO donor in cholesterol-fed rats. Kidney Int. 2002, 61, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, S.; Ishima, Y.; Maeda, H.; Honda, N.; Bi, J.; Kinoshita, R.; Ikeda, M.; Iwao, Y.; Imafuku, T.; Nishida, K.; et al. Dual therapeutic effects of an albumin-based nitric oxide donor on 2 experimental models of chronic kidney disease. J. Pharm. Sci. 2018, 107, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.P.; Schlossmann, J.; Hocher, B. Renal effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Curr. Opin. Pharmacol. 2015, 21, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Tobin, J.V.; Zimmer, D.P.; Shea, C.; Germano, P.; Bernier, S.G.; Liu, G.; Long, K.; Miyashiro, J.; Ranganath, S.; Jacobson, S.; et al. Pharmacological characterization of iw-1973, a novel soluble guanylate cyclase stimulator with extensive tissue distribution, anti-hypertensive, anti-inflammatory, and anti-fibrotic effects in preclinical models of disease. J. Pharmacol. Exp. Ther. 2018, 365, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Follmann, M.; Ackerstaff, J.; Redlich, G.; Wunder, F.; Lang, D.; Kern, A.; Fey, P.; Griebenow, N.; Kroh, W.; Becker-Pelster, E.M.; et al. Discovery of the soluble guanylate cyclase stimulator vericiguat (BAY 1021189) for the treatment of chronic heart failure. J. Med. Chem. 2017, 60, 5146–5161. [Google Scholar] [CrossRef] [PubMed]
- Profy, A.T.; Shea, C.; Lonie, E.; Liu, G.; Milne, G.T.; Currie, M.G.; Masferrer, J.L. IW-1973, a Soluble Guanylate Cyclase Stimulator, Inhibits Progression of Diabetic Nephropathy in the ZSF1 Rat Model; The American Diabetes Association: Arlington, VA, USA, 2017; p. A134. [Google Scholar]
- Valdeci da Cunha, K.S.; Price, O.; Sinz, C.J.; Kim, R.M. Activation of soluble guanylate cyclase ameliorates the progression of glucose intolerance and nephropathy in obese ZSF1 rats. Circulation 2016, 134, A17305. [Google Scholar]
- Stancu, B.; Kramer, S.; Loof, T.; Mika, A.; Amann, K.; Neumayer, H.H.; Peters, H. Soluble guanylate cyclase stimulator BAY 41–8543 and female sex ameliorate uremic aortic remodeling in a rat model of mild uremia. J. Hypertens. 2015, 33, 1907–1920. [Google Scholar] [CrossRef] [PubMed]
- Nagasu, H.; Satoh, M.; Kidokoro, K.; Nishi, Y.; Channon, K.M.; Sasaki, T.; Kashihara, N. Endothelial dysfunction promotes the transition from compensatory renal hypertrophy to kidney injury after unilateral nephrectomy in mice. Am. J. Physiol. Renal Physiol. 2012, 302, F1402–F1408. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Zhou, Z.; Miura, H.; Papapetropoulos, A.; McCarthy, E.T.; Sharma, R.; Savin, V.J.; Lianos, E.A. ADMA injures the glomerular filtration barrier: Role of nitric oxide and superoxide. Am. J. Physiol. Renal Physiol. 2009, 296, F1386–F1395. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, G.; Costello-Boerrigter, L.C.; Cataliotti, A.; Tsuruda, T.; Harty, G.J.; Lapp, H.; Stasch, J.P.; Burnett, J.C., Jr. Cardiorenal and humoral properties of a novel direct soluble guanylate cyclase stimulator BAY 41–2272 in experimental congestive heart failure. Circulation 2003, 107, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Ramseyer, V.D.; Ortiz, P.A.; Carretero, O.A.; Garvin, J.L. Angiotensin II-mediated hypertension impairs nitric oxide-induced NKCC2 inhibition in thick ascending limbs. Am. J. Physiol. Renal Physiol. 2016, 310, F748–F754. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, C.O.; Alves, R.R.; de Oliveira, A.L.; Cruz, J.C.; de Franca-Silva, M.S.; Braga, V.A.; Balarini, C.M. Inhibition of PDE5 restores depressed baroreflex sensitivity in renovascular hypertensive rats. Front. Physiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Lauver, D.A.; Carey, E.G.; Bergin, I.L.; Lucchesi, B.R.; Gurm, H.S. Sildenafil citrate for prophylaxis of nephropathy in an animal model of contrast-induced acute kidney injury. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zheng, J.; Yao, X.; Weng, G.; Wu, L. Essential role of the cGMP/PKG signaling pathway in regulating the proliferation and survival of human renal carcinoma cells. Int. J. Mol. Med. 2014, 34, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Stegbauer, J.; Friedrich, S.; Potthoff, S.A.; Broekmans, K.; Cortese-Krott, M.M.; Quack, I.; Rump, L.C.; Koesling, D.; Mergia, E. Phosphodiesterase 5 attenuates the vasodilatory response in renovascular hypertension. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Citation | Compound | Disease | Treatment Groups | Results |
|---|---|---|---|---|
| NO donors | ||||
| Oshiro, S. et al., 2018 [75] | S-nitrosated human serum albumin (SNO-HSA) | Chronic kidney disease | Cisplatin-induced renal anemia model Saline SNO-HSA (240 nmol/kg of NO) Urinary ureter obstruction model Saline SNO-HSA (48 nmol/mouse of NO) | Treatment of animals with SNO-HSA |
| Alesutan, I. et al., 2016 [72] | Molsidomine | Vascular calcification in chronic kidney disease | Homoarginine + control Homoarginine + molsidomine (120 mg/L) | Molsidomine treatment reversed homoarginine-induced vascular calcification in mice compared to control treatment |
| Bongartz, L.G. et al., 2010 [73] | Molsidomine | Chronic kidney disease | Vehicle Molsidomine (120 mg/L) | Treatment with molsidomine resulted in a mild reduction in blood pressure and improved creatinine clearance. Furthermore, molsidomine treatment improved cardiac function. |
| Attia, D.M. et al., 2002 [74] | Molsidomine | Hypercholesterolemia | Vehicle Molsidomine (120 mg/L) | In hypercholesteremic rats, treatment with molsidomine reduces proteinuria and podocyte stress as well as glomerular and tubulo-interstitial injury compared to rats treated with the vehicle |
| sGC stimulators & sGC activators | ||||
| Stasch, J.P. et al., 2015 [76] | sGC stimulators and sGC activators | Review summarizing the effects of sGC stimulators and activators in preclinical models of kidney disease in papers published between 2004 and 2012. | ||
| Tobin et al., 2018 [77] | IW-1973 | Hypertension-induced kidney disease | Dahl rats + Vehicle Dahl rats + IW-1973 (1–10 mg/kg/d) | Treatment with IW-1973 reduced blood pressure prevented the progression of proteinuria and reduced renal fibrosis and markers of renal inflammation |
| Follmann, M. et al., 2017 [78] | Vericiguat | Chronic heart failure | Renin TG rats + L-NAME + Placebo Renin TG rats + L-NAME + Vericiguat (3 mg/kg) Renin TG rats + L-NAME + Vericiguat (10 mg/kg) | Treatment with vericiguat caused a reduction in KIM-1 and osteopontin expression, we all as a dose-dependent reduction in proteinuria |
| Profy, A.V. et al., 2018 [79] | IW-1973 | Diabetic nephropathy | Obese ZSF-1 rats + Vehicle Obese ZSF-1 rats + IW-1973 (1–10 mg/kg/d) | Treatment with IW-1973 reduced kidney weight, proteinuria, urine volume and fasting glucose levels. |
| Schinner, E. et al., 2017 [36] | BAY 41-8543 | Renal fibrosis post Unilateral Ureter Obstruction | Unilateral Ureter Obstruction (UUO) WT UUO WT + BAY 41-8543 UUO cGKI-KO UUO cGKI-KO + BAY 41-8543 | Post unilateral ureter obstruction, BAY41-8543 reduces mRNA expression of several biomarkers of fibrosis in the kidney of WT mice whereas the expression in the cGKI-KO remains unchanged |
| Cunha, V.D. et al., 2016 [80] | MRL-001 | Chronic kidney disease | ZSF-1 rats + Vehicle ZSF-1 rats + MRL-001 (1 mg/kg/d) | Compared to vehicle, treatment with MRL-001 attenuated markers of diabetic nephropathy, tubular damage, proteinuria and oxidative stress, and improved glucose tolerance |
| Boustany-Kari, C.M. et al., 2015 [29] | BI 703704 | Diabetic nephropathy | Obese ZSF-1 rats + Vehicle Obese ZSF-1 rats + BI 703704 (0.3–10 mg/kg/d) | Treatment with the sGC activator, BI 703704, reduced protein excretion, glomerulosclerosis and renal interstitial lesions in a dose-dependent manner, however it had an effect on BP and HR only with 10 mg/kg/d of BI 703704 |
| Stancu, B. et al., 2015 [81] | BAY 41-8543 | Arterial wall remodeling in a model of mild uremia | Sham Subtotally nephrectomized (SNX) SNX + BAY 41-8543 SNX + hydralazine | BAY 41-8543 ameliorates uremic aortic remodeling and stiffness in a blood-pressure independent manner |
| Nagasu, H. et al., 2012 [82] | BAY 41-2272 | Kidney injury post unilateral nephrectomy | Sham WT Uninephrectomy WT Sham eNOS KO Uninephrectomy eNOS KO Sham eNOS KO + BAY 41-2272 Uninephrectomy eNOS KO + BAY 41-2272 hPTECs treated with BAY 41-2272 hPTECs treated with GSNO | BAY 41-2272 induces compensatory renal hypertrophy and protects renal function in uninephrectomized eNOS KO mice Both BAY 41-2272 and GSNO activate the Akt-mToR pathway in hPTECs and are thus able to stimulate protein synthesis in proximal tubules |
| Sharma, M. et al., 2009 [83] | BAY 41-2272 | Chronic kidney disease | Vehicle ADMA ADMA + BAY 41-2272 | Bay 41-2272 attenuated ADMA-induced increases in albumin permeability in isolated glomeruli |
| Boerrigter, G. et al., 2003 [84] | BAY 41-2272 and nitroglycerin | Congestive heart failure | BAY 41-2272 (2 µg/kg/min) BAY 41-2272 (10 µg/kg/min) Nitroglycerin (1 µg/kg/min) Nitroglycerin (5 µg/kg/min) | Both, the administration of high dose BAY 41-2272 and nitroglycerin reduced mean arterial pressure, increased cardiac output and renal blood flow and maintain glomerular filtration rate. In addition, nitroglycerin treatment decreased right arterial pressure and pulmonary vascular resistance. |
| PDE inhibitors | ||||
| Ramseyer, V.D. et al., 2016 [85] | Vardenafil | Angiotensin II-induced hypertension | Saline-treated rats + vardenafil Ang II-treated rats + vardenafil | Treatment with vardenafil restores NO-mediated inhibition of NKCC2 activity and stimulation of cGMP production in isolated thick ascending limbs from Ang II-treated rats |
| Cavalcanti, C.O. et al., 2016 [86] | Sildenafil | Renovascular hypertension | 2-kidney-1-clip (2K1C) 2K1C + Sildenafil | Hypertensive rats treated with sildenafil had increased baroreflex sensitivity, decreased oxidative stress, were protected from autonomic imbalance and had an overall reduction in BP independent of changes in HR |
| Lauver, D.A. et al., 2014 [87] | Sildenafil | Contrast induced acute kidney injury (CIAKI) | CIAKI CIAKI + sildenafil (6 mg/kg) | Treatment with sildenafil in a rabbit model of CIAKI resulted in decreased levels of kidney histopathology, serum creatinine and electrolyte derangement. |
| Ren, Y. et al., 2014 [88] | PDE5 siRNA | Renal carcinoma | PDE5 siRNA | Suppression of PDE5 expression with PDE5 siRNA inhibits proliferation and survival of human renal carcinoma cells in vitro |
| Stegbauer, J. et al., 2013 [89] | Sildenafil | Renovascular hypertension | Wild type + 2KIC + sildenafil (100 mg/kg/d) NO GC1 KO + 2K1C + sildenafil | Sildenafil significantly reduced blood pressure in WT mice induced with 2K1C-hypertension, compared to NO GC1 KO mice. |
| Whitaker, R.M. et al., 2013 [64] | PDE3, PDE4 and PDE5 inhibitors | Acute kidney injury | In vitro Renal tubular epithelial cells treated with PDE3, PDE4 and PDE5 inhibitors In vivo Folic acid (FA)-induced AKI FA-induced AKI + sildenafil (0.3 mg/kg) | In vitro treatment with PDE3 and PDE5 inhibitors, but not PDE4 inhibitors induce mitochondrial biogenesis in renal proximal tubular epithelial cells, suggesting restoration of mitochondrial function post AKI. In addition, sildenafil treatment in mice with AKI also showed signs of mitochondrial biogenesis in the renal cortex |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnan, S.M.; Kraehling, J.R.; Eitner, F.; Bénardeau, A.; Sandner, P. The Impact of the Nitric Oxide (NO)/Soluble Guanylyl Cyclase (sGC) Signaling Cascade on Kidney Health and Disease: A Preclinical Perspective. Int. J. Mol. Sci. 2018, 19, 1712. https://doi.org/10.3390/ijms19061712
Krishnan SM, Kraehling JR, Eitner F, Bénardeau A, Sandner P. The Impact of the Nitric Oxide (NO)/Soluble Guanylyl Cyclase (sGC) Signaling Cascade on Kidney Health and Disease: A Preclinical Perspective. International Journal of Molecular Sciences. 2018; 19(6):1712. https://doi.org/10.3390/ijms19061712
Chicago/Turabian StyleKrishnan, Shalini M., Jan R. Kraehling, Frank Eitner, Agnès Bénardeau, and Peter Sandner. 2018. "The Impact of the Nitric Oxide (NO)/Soluble Guanylyl Cyclase (sGC) Signaling Cascade on Kidney Health and Disease: A Preclinical Perspective" International Journal of Molecular Sciences 19, no. 6: 1712. https://doi.org/10.3390/ijms19061712
APA StyleKrishnan, S. M., Kraehling, J. R., Eitner, F., Bénardeau, A., & Sandner, P. (2018). The Impact of the Nitric Oxide (NO)/Soluble Guanylyl Cyclase (sGC) Signaling Cascade on Kidney Health and Disease: A Preclinical Perspective. International Journal of Molecular Sciences, 19(6), 1712. https://doi.org/10.3390/ijms19061712