Validation of LDLr Activity as a Tool to Improve Genetic Diagnosis of Familial Hypercholesterolemia: A Retrospective on Functional Characterization of LDLr Variants

 ,
,
Abstract
1. Familial Hypercholesterolemia (FH)
2. FH Diagnosis
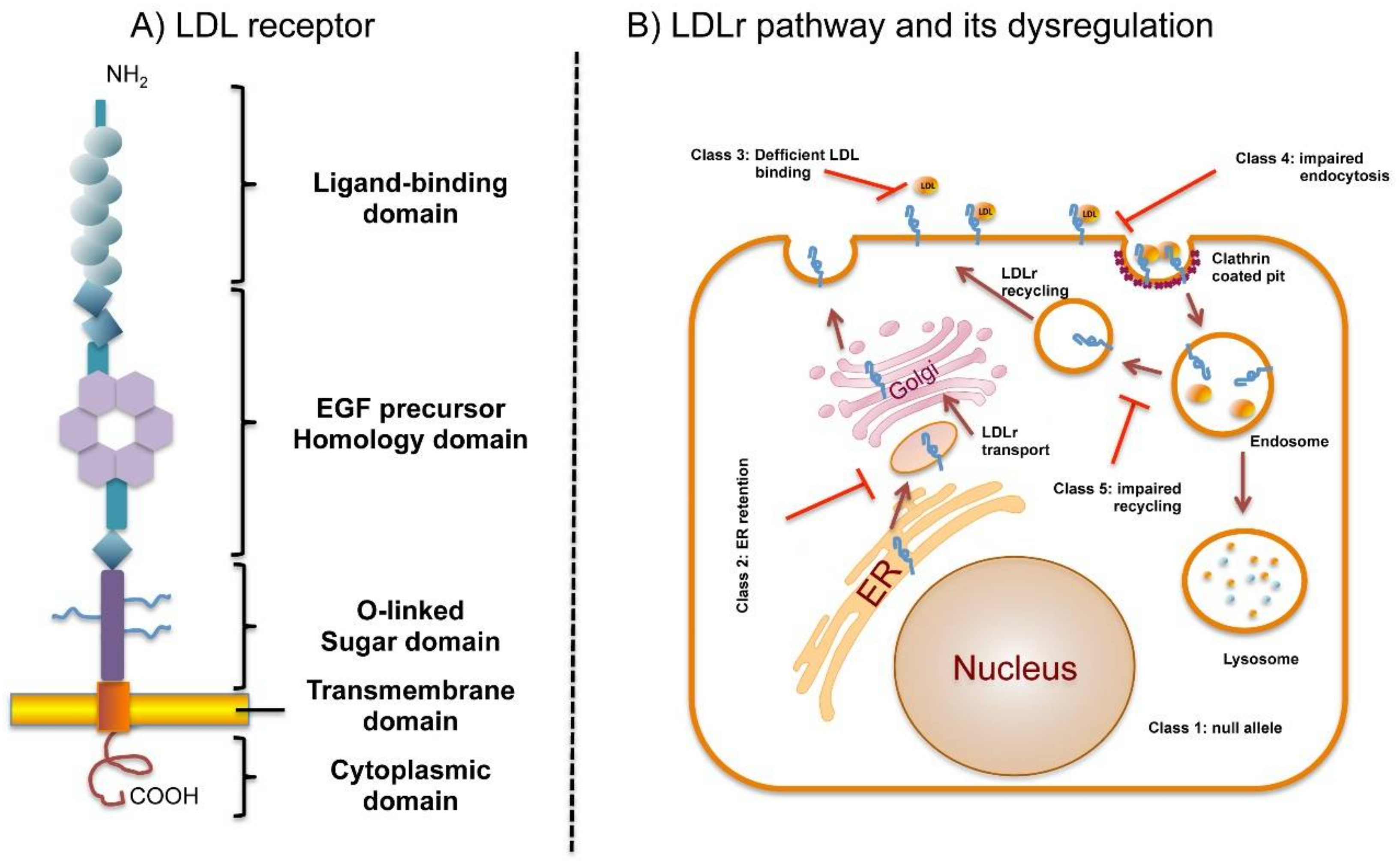
3. LDL Receptor
4. LDLr Pathway and Its Dysregulation by Defective Mutations
5. Determining the Pathogenicity of LDLr Variants
5.1. In Silico Analysis
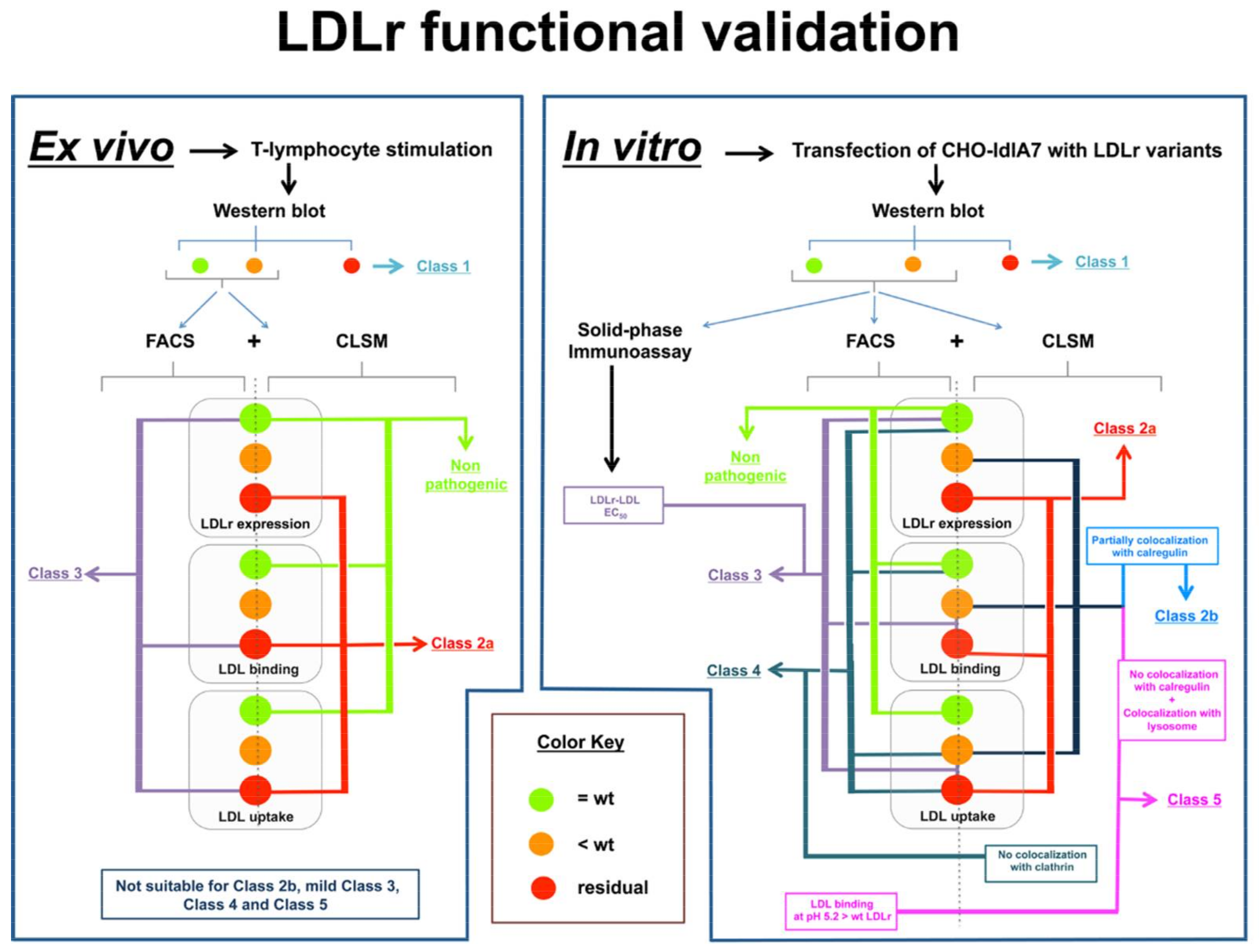
5.2. Functional Characterization of LDLr Variants
5.2.1. Ex vivo Functional Validation
5.2.2. In Vitro Functional Validation
5.2.2.1. Cell Transfection
5.2.2.2. Western Blot Analysis
5.2.2.3. Quantification of LDLr Expression by Flow Cytometry
5.2.2.4. Quantification of LDLR Activity by FACS
5.2.2.5. Confocal Laser Scanning Microscopy
5.2.2.6. LDL–LDLr Binding at Different pH
5.2.2.7. LDLr-LDL Affinity Assessment
6. ClinVar: Variant Pathogenicity Assignments based on LDLr Functional Characterization
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hopkins, P.N.; Toth, P.P.; Ballantyne, C.M.; Rader, D.J.; National Lipid Association Expert Panel on Familial, H. Familial hypercholesterolemias: Prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 2011, 5 (Suppl. 3), S9–S17. [Google Scholar] [CrossRef] [PubMed]
- Talmud, P.J.; Futema, M.; Humphries, S.E. The genetic architecture of the familial hyperlipidaemia syndromes: Rare mutations and common variants in multiple genes. Curr. Opin. Lipidol. 2014, 25, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Damgaard, D.; Jensen, J.M.; Larsen, M.L.; Soerensen, V.R.; Jensen, H.K.; Gregersen, N.; Jensen, L.G.; Faergeman, O. No genetic linkage or molecular evidence for involvement of the PCSK9, ARH or CYP7A1 genes in the Familial Hypercholesterolemia phenotype in a sample of Danish families without pathogenic mutations in the LDL receptor and apoB genes. Atherosclerosis 2004, 177, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.A.; McIlhatton, B.P.; Kirk, C.W.; Beattie, E.D.; Lyttle, K.; Hart, P.; Neely, R.D.; Young, I.S.; Nicholls, D.P. Genetic screening protocol for familial hypercholesterolemia which includes splicing defects gives an improved mutation detection rate. Atherosclerosis 2005, 182, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Cohen, J.; Hobbs, H.H. Monogenic hypercholesterolemia: New insights in pathogenesis and treatment. J. Clin. Investig. 2003, 111, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, S.W.; Kastelein, J.J.; Defesche, J.C. Update of the molecular basis of familial hypercholesterolemia in The Netherlands. Hum. Mutat. 2005, 26, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Hobbs, H.H.; Brown, M.S. Familial hypercholesterolemia. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Soutar, A.K. Intracellular transport of the low-density lipoprotein receptor. Biochem. Soc. Trans. 1996, 24, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.S.; Andersen, R.L.; Andersen, L.H.; O’Brien, E.C.; Kindt, I.; Shrader, P.; Vasandani, C.; Newman, C.B.; deGoma, E.M.; Baum, S.J.; et al. US physician practices for diagnosing familial hypercholesterolemia: Data from the CASCADE-FH registry. J. Clin. Lipidol. 2016, 10, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [PubMed]
- Neil, H.A.; Betteridge, D.J.; Broome, K.; Durrington, P.N.; Hawkins, M.M.; Humphries, S.E.; Mann, J.I.; Miller, J.P.; Thompson, G.R.; Thorogood, M.; et al. Mortality in treated heterozygous familial hypercholesterolaemia: Implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis 1999, 142, 105–112. [Google Scholar]
- Williams, R.R.; Hunt, S.C.; Schumacher, M.C.; Hegele, R.A.; Leppert, M.F.; Ludwig, E.H.; Hopkins, P.N. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am. J. Cardiol. 1993, 72, 171–176. [Google Scholar] [CrossRef]
- World Health Organization. Familial hypercholesterolemia (FH): Report of a Second WHO Consultation. Available online: http://apps.who.int/iris/bitstream/10665/66346/1/WHO_HGN_FH_CONS_99.2.pdf (accessed on 10 January 2018).
- Marks, D.; Wonderling, D.; Thorogood, M.; Lambert, H.; Humphries, S.E.; Neil, H.A. Cost effectiveness analysis of different approaches of screening for familial hypercholesterolaemia. BMJ 2002, 324, 1303. [Google Scholar] [CrossRef] [PubMed]
- Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ 1991, 303, 893–896. [Google Scholar]
- Abifadel, M.; Varret, M.; Rabes, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Civeira, F. International Panel on Management of Familial Hypercholesterolemia. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis 2004, 173, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Wallis, Y.; Payne, S.; McAnulty, C.; Bodmer, D.; Sister-mans, E.; Robertson, K.; Moore, D.; Abbs, S.; Deans, S.; Devereau, A. Practice Guidelines for the Evaluation of Pathogenicity and the Reporting of Sequence Variants in Clinical Molecular Genetics; Association for Clinical Genetic Science and the Dutch Society of Clinical Genetic Laboratory Specialists: London, UK, 2013. [Google Scholar]
- Hobbs, H.H.; Brown, M.S.; Goldstein, J.L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1992, 1, 445–466. [Google Scholar] [CrossRef] [PubMed]
- Strom, T.B.; Tveten, K.; Holla, O.L.; Cameron, J.; Berge, K.E.; Leren, T.P. The cytoplasmic domain is not involved in directing Class 5 mutant LDL receptors to lysosomal degradation. Biochem. Biophys. Res. Commun. 2011, 408, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Herz, J.; Goldstein, J.L. LDL-receptor structure. Calcium cages, acid baths and recycling receptors. Nature 1997, 388, 629–630. [Google Scholar] [CrossRef] [PubMed]
- Rudenko, G.; Henry, L.; Henderson, K.; Ichtchenko, K.; Brown, M.S.; Goldstein, J.L.; Deisenhofer, J. Structure of the LDL receptor extracellular domain at endosomal pH. Science 2002, 298, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Esser, V.; Limbird, L.E.; Brown, M.S.; Goldstein, J.L.; Russell, D.W. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J. Biol. Chem. 1988, 263, 13282–13290. [Google Scholar] [PubMed]
- Russell, D.W.; Brown, M.S.; Goldstein, J.L. Different combinations of cysteine-rich repeats mediate binding of low density lipoprotein receptor to two different proteins. J. Biol. Chem. 1989, 264, 21682–21688. [Google Scholar] [PubMed]
- Beglova, N.; Blacklow, S.C. The LDL receptor: How acid pulls the trigger. Trends Biochem. Sci. 2005, 30, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Lo Surdo, P.; Bottomley, M.J.; Calzetta, A.; Settembre, E.C.; Cirillo, A.; Pandit, S.; Ni, Y.G.; Hubbard, B.; Sitlani, A.; Carfi, A. Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 2011, 12, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Scanlon, M.J.; Djordjevic, J.T.; Kroon, P.A.; Smith, R. Three-dimensional structure of a cysteine-rich repeat from the low-density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 6334–6338. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mao, Y.; Narimatsu, Y.; Ye, Z.; Tian, W.; Goth, C.K.; Lira-Navarrete, E.; Pedersen, N.B.; Benito-Vicente, A.; Martin, C.; et al. Site-specific O-glycosylation of members of the low-density lipoprotein receptor superfamily enhances ligand interactions. J. Biol. Chem. 2018, 293, 7408–7422. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.G.; Goldstein, J.L.; Sudhof, T.C.; Anderson, R.G.; Russell, D.W.; Brown, M.S. Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature 1987, 326, 760–765. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Meng, W.; Takagi, J.; Eck, M.J.; Springer, T.A.; Blacklow, S.C. Implications for familial hypercholesterolemia from the structure of the LDL receptor YWTD-EGF domain pair. Nat. Struct. Biol. 2001, 8, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.G.; Elhammer, A.; Russell, D.W.; Schneider, W.J.; Kornfeld, S.; Brown, M.S.; Goldstein, J.L. Deletion of clustered O-linked carbohydrates does not impair function of low density lipoprotein receptor in transfected fibroblasts. J. Biol. Chem. 1986, 261, 2828–2838. [Google Scholar] [PubMed]
- Yokode, M.; Pathak, R.K.; Hammer, R.E.; Brown, M.S.; Goldstein, J.L.; Anderson, R.G. Cytoplasmic sequence required for basolateral targeting of LDL receptor in livers of transgenic mice. J. Cell Biol. 1992, 117, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.G.; Lehrman, M.A.; Russell, D.W.; Anderson, R.G.; Brown, M.S.; Goldstein, J.L. The J.D. mutation in familial hypercholesterolemia: Amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell 1986, 45, 15–24. [Google Scholar] [CrossRef]
- Lehrman, M.A.; Goldstein, J.L.; Brown, M.S.; Russell, D.W.; Schneider, W.J. Internalization-defective LDL receptors produced by genes with nonsense and frameshift mutations that truncate the cytoplasmic domain. Cell 1985, 41, 735–743. [Google Scholar] [CrossRef]
- Dawson, P.A.; Hofmann, S.L.; van der Westhuyzen, D.R.; Sudhof, T.C.; Brown, M.S.; Goldstein, J.L. Sterol-dependent repression of low density lipoprotein receptor promoter mediated by 16-base pair sequence adjacent to binding site for transcription factor Sp1. J. Biol. Chem. 1988, 263, 3372–3379. [Google Scholar] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Goedeke, L.; Rotllan, N.; Canfran-Duque, A.; Aranda, J.F.; Ramirez, C.M.; Araldi, E.; Lin, C.S.; Anderson, N.N.; Wagschal, A.; de Cabo, R.; et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat. Med. 2015, 21, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Goldstein, J.L.; Brown, M.S. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J. Biol. Chem. 1990, 265, 3116–3123. [Google Scholar] [PubMed]
- Goldstein, J.L.; Brown, M.S. The LDL receptor and the regulation of cellular cholesterol metabolism. J. Cell Sci. Suppl. 1985, 3, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, H.H.; Russell, D.W.; Brown, M.S.; Goldstein, J.L. The LDL receptor locus in familial hypercholesterolemia: Mutational analysis of a membrane protein. Annu. Rev. Genet. 1990, 24, 133–170. [Google Scholar] [CrossRef] [PubMed]
- Tolleshaug, H.; Goldstein, J.L.; Schneider, W.J.; Brown, M.S. Posttranslational processing of the LDL receptor and its genetic disruption in familial hypercholesterolemia. Cell 1982, 30, 715–724. [Google Scholar] [CrossRef]
- Strom, T.B.; Laerdahl, J.K.; Leren, T.P. Mutation p.L799R in the LDLR, which affects the transmembrane domain of the LDLR, prevents membrane insertion and causes secretion of the mutant LDLR. Hum. Mol. Genet. 2015, 24, 5836–5844. [Google Scholar] [CrossRef] [PubMed]
- Futema, M.; Plagnol, V.; Li, K.; Whittall, R.A.; Neil, H.A.; Seed, M.; Simon Broome, C.; Bertolini, S.; Calandra, S.; Descamps, O.S.; et al. Whole exome sequencing of familial hypercholesterolaemia patients negative for LDLR/APOB/PCSK9 mutations. J. Med. Genet. 2014, 51, 537–544. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Khafizov, K.; Madrid-Aliste, C.; Almo, S.C.; Fiser, A. Trends in structural coverage of the protein universe and the impact of the Protein Structure Initiative. Proc. Natl. Acad. Sci. USA 2014, 111, 3733–3738. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. Nature of the protein universe. Proc. Natl. Acad. Sci. USA 2009, 106, 11079–11084. [Google Scholar] [CrossRef] [PubMed]
- Vandrovcova, J.; Thomas, E.R.; Atanur, S.S.; Norsworthy, P.J.; Neuwirth, C.; Tan, Y.; Kasperaviciute, D.; Biggs, J.; Game, L.; Mueller, M.; et al. The use of next-generation sequencing in clinical diagnosis of familial hypercholesterolemia. Genet. Med. 2013, 15, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Fredman, D.; Siegfried, M.; Yuan, Y.P.; Bork, P.; Lehvaslaiho, H.; Brookes, A.J. HGVbase: A human sequence variation database emphasizing data quality and a broad spectrum of data sources. Nucleic Acids Res. 2002, 30, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J. Biol. Chem. 1974, 249, 5153–5162. [Google Scholar] [PubMed]
- Chan, P.; Jones, C.; Lafreniere, R.; Parsons, H.G. Surface expression of low density lipoprotein receptor in EBV-transformed lymphocytes: Characterization and use for studying familial hypercholesterolemia. Atherosclerosis 1997, 131, 149–160. [Google Scholar] [CrossRef]
- Holla, O.L.; Nakken, S.; Mattingsdal, M.; Ranheim, T.; Berge, K.E.; Defesche, J.C.; Leren, T.P. Effects of intronic mutations in the LDLR gene on pre-mRNA splicing: Comparison of wet-lab and bioinformatics analyses. Mol. Genet. Metab. 2009, 96, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Di Taranto, M.D.; Mirabelli, P.; D’Agostino, M.N.; Iannuzzi, A.; Marotta, G.; Gentile, M.; Raia, M.; Di Noto, R.; Del Vecchio, L.; et al. An improved method on stimulated T-lymphocytes to functionally characterize novel and known LDLR mutations. J. Lipid Res. 2011, 52, 2095–2100. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.C.; Lafreniere, R.; Parsons, H.G. Lovastatin increases surface low density lipoprotein receptor expression by retarding the receptor internalization rate in proliferating lymphocytes. Biochem. Biophys. Res. Commun. 1997, 235, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, N.; Iwata, S.; Ichikawa, T.; Fujinami, T. Assessment of functional low-density-lipoprotein receptors on lymphocytes by a simplified method using culture medium with lipoprotein-free fetal calf serum and pravastatin. Clin. Biochem. 1992, 25, 368–370. [Google Scholar] [CrossRef]
- Tada, H.; Kawashiri, M.A.; Noguchi, T.; Mori, M.; Tsuchida, M.; Takata, M.; Nohara, A.; Inazu, A.; Kobayashi, J.; Yachie, A.; et al. A novel method for determining functional LDL receptor activity in familial hypercholesterolemia: Application of the CD3/CD28 assay in lymphocytes. Clin. Chim. Acta 2009, 400, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Van der Westhuyzen, D.R.; Coetzee, G.A.; Demasius, I.P.; Harley, E.H.; Gevers, W.; Baker, S.G.; Seftel, H.C. Low density lipoprotein receptor mutations in South African homozygous familial hypercholesterolemic patients. Arteriosclerosis 1984, 4, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, A.; Palacios, L.; Stef, M.; Tejedor, D.; Uribe, K.B.; Oleaga, A.; Irigoyen, L.; Torres, B.; Ostolaza, H.; Martin, C. Functional characterization of splicing and ligand-binding domain variants in the LDL receptor. Hum. Mutat. 2012, 33, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Raungaard, B.; Heath, F.; Brorholt-Petersen, J.U.; Jensen, H.K.; Faergeman, O. Flow cytometric assessment of LDL receptor activity in peripheral blood mononuclear cells compared to gene mutation detection in diagnosis of heterozygous familial hypercholesterolemia. Cytometry 1999, 36, 52–59. [Google Scholar] [CrossRef]
- Etxebarria, A.; Benito-Vicente, A.; Palacios, L.; Stef, M.; Cenarro, A.; Civeira, F.; Ostolaza, H.; Martin, C. Functional characterization and classification of frequent low-density lipoprotein receptor variants. Hum. Mutat. 2015, 36, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, A.; Benito-Vicente, A.; Alves, A.C.; Ostolaza, H.; Bourbon, M.; Martin, C. Advantages and versatility of fluorescence-based methodology to characterize the functionality of LDLR and class mutation assignment. PLoS ONE 2014, 9, e112677. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Alves, A.C.; Patel, D.; Malho, R.; Soutar, A.K.; Bourbon, M. In vitro functional characterization of missense mutations in the LDLR gene. Atherosclerosis 2012, 225, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Benito-Vicente, A.; Alves, A.C.; Etxebarria, A.; Medeiros, A.M.; Martin, C.; Bourbon, M. The importance of an integrated analysis of clinical, molecular, and functional data for the genetic diagnosis of familial hypercholesterolemia. Genet. Med. 2015, 17, 980–988. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Benito-Vicente, A.; Tang, L.; Etxebarria, A.; Cui, W.; Uribe, K.B.; Pan, X.D.; Ostolaza, H.; Yang, S.W.; Zhou, Y.J.; et al. Analysis of LDLR variants from homozygous FH patients carrying multiple mutations in the LDLR gene. Atherosclerosis 2017, 263, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, A.; Benito-Vicente, A.; Stef, M.; Ostolaza, H.; Palacios, L.; Martin, C. Activity-associated effect of LDL receptor missense variants located in the cysteine-rich repeats. Atherosclerosis 2015, 238, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Di Taranto, M.D.; Benito-Vicente, A.; Giacobbe, C.; Uribe, K.B.; Rubba, P.; Etxebarria, A.; Guardamagna, O.; Gentile, M.; Martin, C.; Fortunato, G. Identification and in vitro characterization of two new PCSK9 Gain of Function variants found in patients with Familial Hypercholesterolemia. Sci. Rep. 2017, 7, 15282. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wu, W.F.; Sun, L.Y.; Chen, P.P.; Wang, W.; Benito-Vicente, A.; Zhang, F.; Pan, X.D.; Cui, W.; Yang, S.W.; et al. The use of targeted exome sequencing in genetic diagnosis of young patients with severe hypercholesterolemia. Sci. Rep. 2016, 6, 36823. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Knight, B.L.; Soutar, A.K. Changes in the metabolism of modified and unmodified low-density lipoproteins during the maturation of cultured blood monocyte-macrophages from normal and homozygous familial hypercholesterolaemic subjects. Eur. J. Biochem. 1982, 125, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Langenhoven, E.; Warnich, L.; Thiart, R.; Rubinsztein, D.C.; van der Westhuyzen, D.R.; Marais, A.D.; Kotze, M.J. Two novel point mutations causing receptor-negative familial hypercholesterolemia in a South African Indian homozygote. Atherosclerosis 1996, 125, 111–119. [Google Scholar] [CrossRef]
- Cassanelli, S.; Bertolini, S.; Rolleri, M.; De Stefano, F.; Casarino, L.; Elicio, N.; Naselli, A.; Calandra, S. A ‘de novo’ point mutation of the low-density lipoprotein receptor gene in an Italian subject with primary hypercholesterolemia. Clin. Genet. 1998, 53, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, S.; Cassanelli, S.; Garuti, R.; Ghisellini, M.; Simone, M.L.; Rolleri, M.; Masturzo, P.; Calandra, S. Analysis of LDL receptor gene mutations in Italian patients with homozygous familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Jialal, I.; Leitersdorf, E.; Coetzee, G.A.; van der Westhuyzen, D.R. Identification of two new LDL-receptor mutations causing homozygous familial hypercholesterolemia in a South African of Indian origin. Biochim. Biophys. Acta 1993, 1182, 75–82. [Google Scholar] [CrossRef]
- Romano, M.; Di Taranto, M.D.; D’Agostino, M.N.; Marotta, G.; Gentile, M.; Abate, G.; Mirabelli, P.; Di Noto, R.; Del Vecchio, L.; Rubba, P.; et al. Identification and functional characterization of LDLR mutations in familial hypercholesterolemia patients from Southern Italy. Atherosclerosis 2010, 210, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Slimane, M.N.; Lestavel, S.; Clavey, V.; Maatouk, F.; Ben Fahrat, M.H.; Fruchart, J.C.; Hammami, M.; Benlian, P. CYS127S (FH-Kairouan) and D245N (FH-Tozeur) mutations in the LDL receptor gene in Tunisian families with familial hypercholesterolaemia. J. Med. Genet. 2002, 39, e74. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, H.H.; Leitersdorf, E.; Leffert, C.C.; Cryer, D.R.; Brown, M.S.; Goldstein, J.L. Evidence for a dominant gene that suppresses hypercholesterolemia in a family with defective low density lipoprotein receptors. J. Clin. Investig. 1989, 84, 656–364. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.S.; Scharnagl, H.; Nissen, H.; Schurmann, C.; Matern, D.; Nauck, M.A.; Wieland, H.; Marz, W. FH-Freiburg: A novel missense mutation (C317Y) in growth factor repeat A of the low density lipoprotein receptor gene in a German patient with homozygous familial hypercholesterolemia. Atherosclerosis 2000, 151, 525–534. [Google Scholar] [CrossRef]
- Assouline, L.; Leitersdorf, E.; Lambert, M.; Reshef, A.; Feoli-Fonseca, J.C.; Levy, E. Identification of two novel LDL receptor gene defects in French-Canadian pediatric population: Mutational analysis and biochemical studies. Hum. Mutat. 1997, 9, 555–562. [Google Scholar] [CrossRef]
- Webb, J.C.; Sun, X.M.; McCarthy, S.N.; Neuwirth, C.; Thompson, G.R.; Knight, B.L.; Soutar, A.K. Characterization of mutations in the low density lipoprotein (LDL)-receptor gene in patients with homozygous familial hypercholesterolemia, and frequency of these mutations in FH patients in the United Kingdom. J. Lipid Res. 1996, 37, 368–381. [Google Scholar] [PubMed]
- Miyake, Y.; Yamamura, T.; Sakai, N.; Miyata, T.; Kokubo, Y.; Yamamoto, A. Update of Japanese common LDLR gene mutations and their phenotypes: Mild type mutation L547V might predominate in the Japanese population. Atherosclerosis 2009, 203, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Leitersdorf, E.; Van der Westhuyzen, D.R.; Coetzee, G.A.; Hobbs, H.H. Two common low density lipoprotein receptor gene mutations cause familial hypercholesterolemia in Afrikaners. J. Clin. Investig. 1989, 84, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.M.; Patel, D.D.; Knight, B.L.; Soutar, A.K. Comparison of the genetic defect with LDL-receptor activity in cultured cells from patients with a clinical diagnosis of heterozygous familial hypercholesterolemia. The Familial Hypercholesterolaemia Regression Study Group. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3092–3101. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Tietge, U.J.; Buettner, J.; Barg-Hock, H.; Offner, G.; Schweitzer, S.; Dedoussis, G.V.; Rodeck, B.; Kallfelz, H.C.; Schlitt, H.J.; et al. Liver transplantation in a subject with familial hypercholesterolemia carrying the homozygous p.W577R LDL-receptor gene mutation. Clin. Transplant. 2008, 22, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Hattori, H.; Hirayama, T.; Nobe, Y.; Nagano, M.; Kujiraoka, T.; Egashira, T.; Ishii, J.; Tsuji, M.; Emi, M. Eight novel mutations and functional impairments of the LDL receptor in familial hypercholesterolemia in the north of Japan. J. Hum. Genet. 2002, 47, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Leitersdorf, E.; Tobin, E.J.; Davignon, J.; Hobbs, H.H. Common low-density lipoprotein receptor mutations in the French Canadian population. J. Clin. Investig. 1990, 85, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Soutar, A.K.; Knight, B.L.; Patel, D.D. Identification of a point mutation in growth factor repeat C of the low density lipoprotein-receptor gene in a patient with homozygous familial hypercholesterolemia that affects ligand binding and intracellular movement of receptors. Proc. Natl. Acad. Sci. USA 1989, 86, 4166–4170. [Google Scholar] [CrossRef] [PubMed]
- Bourbon, M.; Duarte, M.A.; Alves, A.C.; Medeiros, A.M.; Marques, L.; Soutar, A.K. Genetic diagnosis of familial hypercholesterolaemia: The importance of functional analysis of potential splice-site mutations. J. Med. Genet. 2009, 46, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Mak, Y.T.; Pang, C.P.; Tomlinson, B.; Zhang, J.; Chan, Y.S.; Mak, T.W.; Masarei, J.R. Mutations in the low-density lipoprotein receptor gene in Chinese familial hypercholesterolemia patients. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1600–1605. [Google Scholar] [CrossRef] [PubMed]
- Pavlouskova, J.; Reblova, K.; Tichy, L.; Freiberger, T.; Fajkusova, L. Functional analysis of the p.(Leu15Pro) and p.(Gly20Arg) sequence changes in the signal sequence of LDL receptor. Atherosclerosis 2016, 250, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Pan, J.P.; Tai, D.Y.; Huang, A.C.; Li, P.H.; Ho, H.L.; Hsieh, H.L.; Chou, S.C.; Lin, W.L.; Lo, E.; et al. Identification and characterization of LDL receptor gene mutations in hyperlipidemic Chinese. J. Lipid Res. 2003, 44, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.C.; Sun, X.M.; Patel, D.D.; McCarthy, S.N.; Knight, B.L.; Soutar, A.K. Characterization of two new point mutations in the low density lipoprotein receptor genes of an English patient with homozygous familial hypercholesterolemia. J. Lipid Res. 1992, 33, 689–698. [Google Scholar] [PubMed]
- Khoo, K.L.; van Acker, P.; Defesche, J.C.; Tan, H.; van de Kerkhof, L.; Heijnen-van Eijk, S.J.; Kastelein, J.J.; Deslypere, J.P. Low-density lipoprotein receptor gene mutations in a Southeast Asian population with familial hypercholesterolemia. Clin. Genet. 2000, 58, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Leitersdorf, E.; Reshef, A.; Meiner, V.; Dann, E.J.; Beigel, Y.; van Roggen, F.G.; van der Westhuyzen, D.R.; Coetzee, G.A. A missense mutation in the low density lipoprotein receptor gene causes familial hypercholesterolemia in Sephardic Jews. Hum. Genet. 1993, 91, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Thormaehlen, A.S.; Schuberth, C.; Won, H.H.; Blattmann, P.; Joggerst-Thomalla, B.; Theiss, S.; Asselta, R.; Duga, S.; Merlini, P.A.; Ardissino, D.; et al. Systematic cell-based phenotyping of missense alleles empowers rare variant association studies: A case for LDLR and myocardial infarction. PLoS Genet. 2015, 11, e1004855. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, S.; Sun, L.; Pan, X.; Yang, S.; Wang, L. Functional characterization of two low-density lipoprotein receptor gene mutations in two Chinese patients with familial hypercholesterolemia. PLoS ONE 2014, 9, e92703. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, U.; Abrahamson, M.; Floren, C.H.; Tollig, H.; Wettrell, G.; Nilsson, G.; Sun, X.M.; Soutar, A.K.; Nilsson-Ehle, P. An individual with a healthy phenotype in spite of a pathogenic LDL receptor mutation (C240F). Clin. Genet. 1999, 55, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, U.; Abrahamson, M.; Sveger, T.; Sun, X.M.; Soutar, A.K.; Nilsson-Ehle, P. Expression of an LDL receptor allele with two different mutations (E256K and I402T). Mol. Pathol. 2000, 53, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Martin de Llano, J.J.; Fuertes, G.; Andreu, E.J.; Puig, O.; Chaves, F.J.; Soutar, A.K.; Armengod, M.E.; Knecht, E. A single point mutation in the low-density lipoprotein receptor switches the degradation of its mature protein from the proteasome to the lysosome. Int. J. Biochem. Cell Biol. 2006, 38, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Michaely, P. Role of an intramolecular contact on lipoprotein uptake by the LDL receptor. Biochim. Biophys. Acta 2011, 1811, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.M.; Patel, D.D.; Webb, J.C.; Knight, B.L.; Fan, L.M.; Cai, H.J.; Soutar, A.K. Familial hypercholesterolemia in China. Identification of mutations in the LDL-receptor gene that result in a receptor-negative phenotype. Arterioscler. Thromb. Vasc. Biol. 1994, 14, 85–94. [Google Scholar] [CrossRef]
- Jensen, H.K.; Jensen, T.G.; Faergeman, O.; Jensen, L.G.; Andresen, B.S.; Corydon, M.J.; Andreasen, P.H.; Hansen, P.S.; Heath, F.; Bolund, L.; et al. Two mutations in the same low-density lipoprotein receptor allele act in synergy to reduce receptor function in heterozygous familial hypercholesterolemia. Hum. Mutat. 1997, 9, 437–444. [Google Scholar] [CrossRef]
- Alves, A.C.; Etxebarria, A.; Medeiros, A.M.; Benito-Vicente, A.; Thedrez, A.; Passard, M.; Croyal, M.; Martin, C.; Lambert, G.; Bourbon, M. Characterization of the first PCSK9 gain of function homozygote. J. Am. Coll. Cardiol. 2015, 66, 2152–2154. [Google Scholar] [CrossRef] [PubMed]
- Cenarro, A.; Etxebarria, A.; de Castro-Oros, I.; Stef, M.; Bea, A.M.; Palacios, L.; Mateo-Gallego, R.; Benito-Vicente, A.; Ostolaza, H.; Tejedor, T.; et al. The p.Leu167del Mutation in APOE Gene Causes Autosomal Dominant Hypercholesterolemia by Down-regulation of LDL Receptor Expression in Hepatocytes. J. Clin. Endocrinol. Metab. 2016, 101, 2113–2121. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Higuero, J.A.; Benito-Vicente, A.; Etxebarria, A.; Milicua, J.C.; Ostolaza, H.; Arrondo, J.L.; Martin, C. Structural changes induced by acidic pH in human apolipoprotein B-100. Sci. Rep. 2016, 6, 36324. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Higuero, J.A.; Etxebarria, A.; Benito-Vicente, A.; Alves, A.C.; Arrondo, J.L.; Ostolaza, H.; Bourbon, M.; Martin, C. Structural analysis of APOB variants, p.(Arg3527Gln), p.(Arg1164Thr) and p.(Gln4494del), causing Familial Hypercholesterolaemia provides novel insights into variant pathogenicity. Sci. Rep. 2015, 5, 18184. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Differences between Functional Validation Methodologies | |
|---|---|
| Radioactivity | Fluorescence |
| Highly reproducible | Highly reproducible |
| Highly sensitive activity measurements | Highly sensitive activity measurements |
| Stable labeling | Stable labeling |
| Risk of exposure to radioisotopes | Nonradioisotopes used |
| Ethical considerations regarding waste elimination | In combination with CLSM allow LDLr classification |
| Noncompatible with CLSM | |
| Functional Validated and Classified LDLr Variants | Classification | LDLr Activity | Reference |
|---|---|---|---|
| c.226G>T p.(Gly76Trp) | Nonpathogenic | 100% | [72] |
| c. 292G>A (p.Gly98Ser) | Nonpathogenic | 100% | [73] |
| c.346T>C (p.Cys116Arg) | Class 3 | 25% | [74] |
| c.464G>A (p.Cys155Tyr) | Class 3 | <20% | [69] |
| c.502G>A (p.Asp168Asn) | Class 3 | 40% | [74] |
| c.514G>A (p.Asp172Asn) | Class 3 | <2% | [74] |
| c.769C>T (p.Arg257Trp) | Nonpathogenic | 100% | [74] |
| c.806G>A (p.Gly269Asp) | Nonpathogenic | 100% | [67] |
| c.829G>A (p.Glu277Lys) | Nonpathogenic | 100% | [72] |
| c.862G>A (p.Glu288Lys) | Class 3 | 60% | [67] |
| c. 890A>C (p.Asn297Thr) | Nonpathogenic | 100% | [73] |
| c.895G>A (p.Ala299Thr) | Class 3 | 60% | [67] |
| c.898A>G (p.Arg300Gly) | Class 3 | 60% | [74] |
| c.902A>G (p.Asp301Gly) | Class 3 | 40% | [74] |
| c.1216C>T (p.Arg406Trp) | Class 2b or 5 | 60% | [72] |
| c.1246C>T (p.Arg416Trp) | Class 5 | 60% | [69] |
| c.1285G>C (p.Val429Leu) | Class 2a | <10% | [70] |
| c.1322T>C (p.Ile441Thr) | Class 2a | <10% | [72] |
| c.1336 C>G (p.Leu446Val) | Nonpathogenic | 100% | [75] |
| c.1361C>A (p.Thr454Asn) | Class 5 | 60% | [69] |
| c.1468T>C (p.Trp490Arg) | Class 2a | <10% | [70] |
| c.1633G>T (p.Gly545Trp) | Class 2a | <10% | [72] |
| c.1723G>T (p.Leu575Phe) | Class 2 | 60% | [76] |
| c.1729T>G (p.Trp577Gly) | Class 2a | <10% | [69] |
| c.1744C>T (p.Leu582Phe) | Class 2 | 60% | [76] |
| c.1942T >C (p.Ser648Pro) | Class 2b | <25% | [70] |
| c.2053C>T (p.Pro685Ser) | Class 2b | <75% | [70] |
| c.2475C>A (p.Asn825Lys) | Class 4 | 60% | [69] |
| c.2575G>A (p.Val859Met) | Nonpathogenic | 100% | [72] |
| Ex Vivo | |||
|---|---|---|---|
| Functional validated LDLr variants | LDLr activity | Method | Reference |
| c.1A>T (p.Met1Leu) | residual | Radioactivity | [79] |
| c.28T>A (p.Trp10Arg) | 40% | Radioactivity | [80] |
| c.81C>G (p.Cys27Trp) | 15–30% | Radioactivity | [20] |
| c.265T>C (p.Cys89Arg) | <5% Comp Htz | Radioactivity | [81] |
| c.268G>T (p.Asp90Tyr) | not determined | Radioactivity | [82] |
| c.407A>T (p.Asp136Val) | 76% Htz | Fluorescence | [83] |
| c.418G>A (p.Glu140Lys) | 30% Comp Htz | Radioactivity | [82] |
| c.443G>C (p.Cys148Ser) | 2% | Radioactivity | [84] |
| c.530C>T (p.Ser177Leu) | <2% | Radioactivity | [85] |
| c.590G>T (p.Cys197Phe) | <2% Comp Htz | Radioactivity | [20] |
| c.590G>A (p.Cys197Tyr) | <2% Comp Htz | Radioactivity | [20] |
| c.662A>G (p.Asp221Gly) | <2% Comp Htz | Radioactivity | [20] |
| c.670G>A (p.Asp224Asn) | <2% | Radioactivity | [20] |
| c.676T>C (p.Ser226Pro) | <2% | Radioactivity | [20] |
| c.681C>G (p.Asp227Glu) | <2% | Radioactivity | [20] |
| c.682G>C (p.Glu228Gln) | 2–5% Comp Htz | Radioactivity | [20] |
| c.796G>A (p.Asp266Asn) | <2% | Radioactivity | [84] |
| c.798T>A (p.Asp266Glu) | 15–30% | Radioactivity | [20] |
| c.910G>A (p.Asp304Asn) | 5–15% | Radioactivity | [20] |
| c.917C>T (p.Ser306Leu) | 2–5% Comp Htz | Radioactivity | [20] |
| c.953G>A (p.Cys318Arg) | 2–5% | Radioactivity | [20] |
| c.974G>A (p.Cys325Tyr) | <64% | Fluorescence | [62] |
| c.1003G>A (p.Gly335Ser) | 30–40% Htz | Radioactivity | [20] |
| c.1013G>A (p.Cys338Tyr) | <10% | Radioactivity | [86] |
| c.1027G>A (p.Gly343Ser) | 15–30% Comp Htz | Radioactivity | [20] |
| c.1055G>A (p.Cys352Tyr) | 15–30% Comp htz | Radioactivity | [20] |
| c.1056C>G (p.Cys352Trp) | 9% | Radioactivity | [81] |
| c.1090T>C (p.Cys364Arg) | 15–30% | Radioactivity | [20] |
| c.1124A>G (p.Tyr375Cys) | <40% | Radioactivity | [87] |
| c.1135T>C (p.Cys379Arg) | 15–30% | Radioactivity | [20] |
| c.1222G>A (p.Glu408Lys) | 5–10% | Radioactivity | [88] |
| c.1252G>A (p.Glu418Lys) | <70 Comp Htz | Radioactivity | [89] |
| c.1285G>A (p.Val429Met) | <2% | Radioactivity | [90] |
| c.1291G>A (p.Ala431Thr) | 5–15% | Radioactivity | [42] |
| c.1297G>C (p.Asp433His) | <10% | Radioactivity | [89] |
| c.1301C>A (p.Thr434Lys) | 5–15% Comp Htz | Radioactivity | [20] |
| c.1432G>A (p.Gly478Arg) | 2–5% Comp Htz | Radioactivity | [20] |
| c.1444G>A (p.Asp482Asn) | 15% Comp Htz | Radioactivity | [88] |
| c.1567G>A (p.Val523Met) | 15–30% | Radioac+Fluores. | [42,81] |
| c.1618G>A (p.Ala540Thr) | <50% | Radioactivity | [91] |
| c.1637G>A (p.Gly546Asp) | <2% | Radioactivity | [20] |
| c.1646G>A (p.Gly549Asp) | <2% | Radioactivity | [42] |
| c.1694G>T (p.Gly565Val) | <2% | Radioactivity | [20] |
| c.1702C>G (p.Leu568Val) | 25% | Radioactivity | [89] |
| c.1729T>C (p.Trp577Arg) | <5% | Fluorescence | [92] |
| c.1731G>A (p.Trp577Cys) | 64% | Fluorescence | [93] |
| c.1735G>A (p.Asp579Asn) | <2% Comp Htz | Radioactivity | [20] |
| c.1775G>A (p.Gly592Glu) | <5% Comp Htz | Radioactivity | [20] |
| c.1796T>C (p.Leu599Ser) | 5–15% | Radioactivity | [20] |
| c.2000G>A (p.Cys667Tyr) | <2% | Radioactivity | [94] |
| c.2054C>T (p.Pro685Leu) | 15–30% | Radioactivity | [95] |
| c.2177C>T (p.Thr726Ile) | 15–30% Comp Htz | Fluorescence | [20] |
| c.2389G>T (p.Val797Leu) | not determined | Other techniques | [96] |
| c.2389G>A (p.Val797Met) | not determined | Other techniques | [97] |
| c.2479G>A (p.Val827Ile) | 15–30% Comp Htz | Radioactivity | [20] |
| In vitro | |||
| Functional validated LDLr variants | LDLr activity | Method | Reference |
| c.58G>A (p.Gly20Arg) | 100% | Fluorescence | [98] |
| c.226G>T (p.Gly76Trp) | 100% | Fluorescence | [72] |
| c.259T>G (p.Trp87Gly) | 25–100% | Radioactivity | [94] |
| c.268G>A (p.Asp90Asn) | 55% | Fluorescence | [99] |
| c.301G>A (p.Glu101Lys) | 15–30% | Radioactivity | [100] |
| c.344G>A (p.Arg115His) | 64% | Fluorescence | [101] |
| c.346T>C (p.Cys116Arg) | 25% | Fluorescence | [74] |
| c.464G>A (p.Cys155Tyr) | <20% | Fluorescence | [74] |
| c.502G>A (p.Asp168Asn) | 40% | Fluorescence | [74] |
| c.502G>C (p.Asp168His) | <2% | Radioactivity | [102] |
| c.514G > A (p.Asp172Asn) | 40% | Fluorescence | [74] |
| c.589T>C (p.Cys197Arg) | <10% | fluorescence | [103] |
| c.665G>T (p.Cys222Phe) | 33% | Fluorescence | [104] |
| c.769C>T (p.Arg257Trp) | 100% | Fluorescence | [74] |
| c.782G>T (p.Cys261Phe) | <20% | Radioactivity | [105] |
| c.806G>A (p.Gly269Asp) | 100% | Fluorescence | [67] |
| c.829G>A (p.Glu277Lys) | 100% | Radioactivity | [106] |
| c.862G>A (p.Glu288Lys) | 60% | Fluorescence | [67] |
| c.895G>A (p.Ala299Thr) | 60% | Fluorescence | [67] |
| c.898A>G (p.Arg300Gly) | 60% | Fluorescence | [74] |
| c.902A>G (p.Asp301Gly) | 40% | Fluorescence | [74] |
| c.986G>A (p.Cys329Tyr) | 31% | Fluorescence | [99] |
| c.1072T>C (p.Cys358Arg) | 67–72% | Fluorescence | [93] |
| c.1136G>A (p.Cys379Tyr) | <40% | Radioactivity | [107] |
| c.1186G>A (p.Gly396Ser) | 100% | Radioac+Fluores. | [108] |
| c.1216C>T (p.Arg406Trp) | 60% | Fluorescence | [72] |
| c.1246C>T (p.Arg416Trp) | 60% | Fluorescence | [69] |
| c.1268T>C (p.Ile423Thr) | 54% | Radioactivity | [99] |
| c.1285G>C (p.Val429Leu) | <10% | Radioactivity | [71] |
| c.1322T>C (p.Ile441Thr) | <10% | Fluorescence | [72] |
| c.1361C>A (p.Thr454Asn) | 60% | Fluorescence | [69] |
| c.1468T>C (p.Trp490Arg) | <10% | Radioactivity | [71] |
| c.1633G>T (p.Gly545Trp) | <10% | Fluorescence | [72] |
| c.1664T>C (p.Leu555Pro) | <2% | Radioactivity | [109] |
| c.1690A>C (p.Asn564His) | 100% | Fluorescence | [110] |
| c.1729T>G (p.Trp577Gly) | <10% | Fluorescence | [69] |
| c.1744C>T (p.Leu582Phe) | 60% | Fluorescence | [76] |
| c.1747C>T (p.His583Tyr) | <60% | Radioactivity | [108] |
| c.1942T>C (p.Ser648Pro) | <25% | Radioactivity | [71] |
| c.2053C>T (p.Pro685Ser) | <75% | Radioactivity | [71] |
| c.2093G>T (p.Cys698Phe) | <10% | Fluorescence | [72] |
| c.2396T>G (p.Leu799Arg) | residual | Other techniques | [44] |
| c.2475C>A (p.Asn825Lys) | 60% | Fluorescence | [69] |
| c.2483A>G (p.Tyr828Cys) | <2% Comp Htz | Radioactivity | [33] |
| c.2575G>A (p.Val859Met) | 100% | Radioactivity | [71] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Validation of LDLr Activity as a Tool to Improve Genetic Diagnosis of Familial Hypercholesterolemia: A Retrospective on Functional Characterization of LDLr Variants. Int. J. Mol. Sci. 2018, 19, 1676. https://doi.org/10.3390/ijms19061676
Benito-Vicente A, Uribe KB, Jebari S, Galicia-Garcia U, Ostolaza H, Martin C. Validation of LDLr Activity as a Tool to Improve Genetic Diagnosis of Familial Hypercholesterolemia: A Retrospective on Functional Characterization of LDLr Variants. International Journal of Molecular Sciences. 2018; 19(6):1676. https://doi.org/10.3390/ijms19061676
Chicago/Turabian StyleBenito-Vicente, Asier, Kepa B. Uribe, Shifa Jebari, Unai Galicia-Garcia, Helena Ostolaza, and Cesar Martin. 2018. "Validation of LDLr Activity as a Tool to Improve Genetic Diagnosis of Familial Hypercholesterolemia: A Retrospective on Functional Characterization of LDLr Variants" International Journal of Molecular Sciences 19, no. 6: 1676. https://doi.org/10.3390/ijms19061676
APA StyleBenito-Vicente, A., Uribe, K. B., Jebari, S., Galicia-Garcia, U., Ostolaza, H., & Martin, C. (2018). Validation of LDLr Activity as a Tool to Improve Genetic Diagnosis of Familial Hypercholesterolemia: A Retrospective on Functional Characterization of LDLr Variants. International Journal of Molecular Sciences, 19(6), 1676. https://doi.org/10.3390/ijms19061676