A New Perspective: Mitochondrial Stat3 as a Regulator for Lymphocyte Function

{kind=link}
{kind=link}
Abstract
1. Introduction: Revisiting the Role of Stat3 25 Years Later
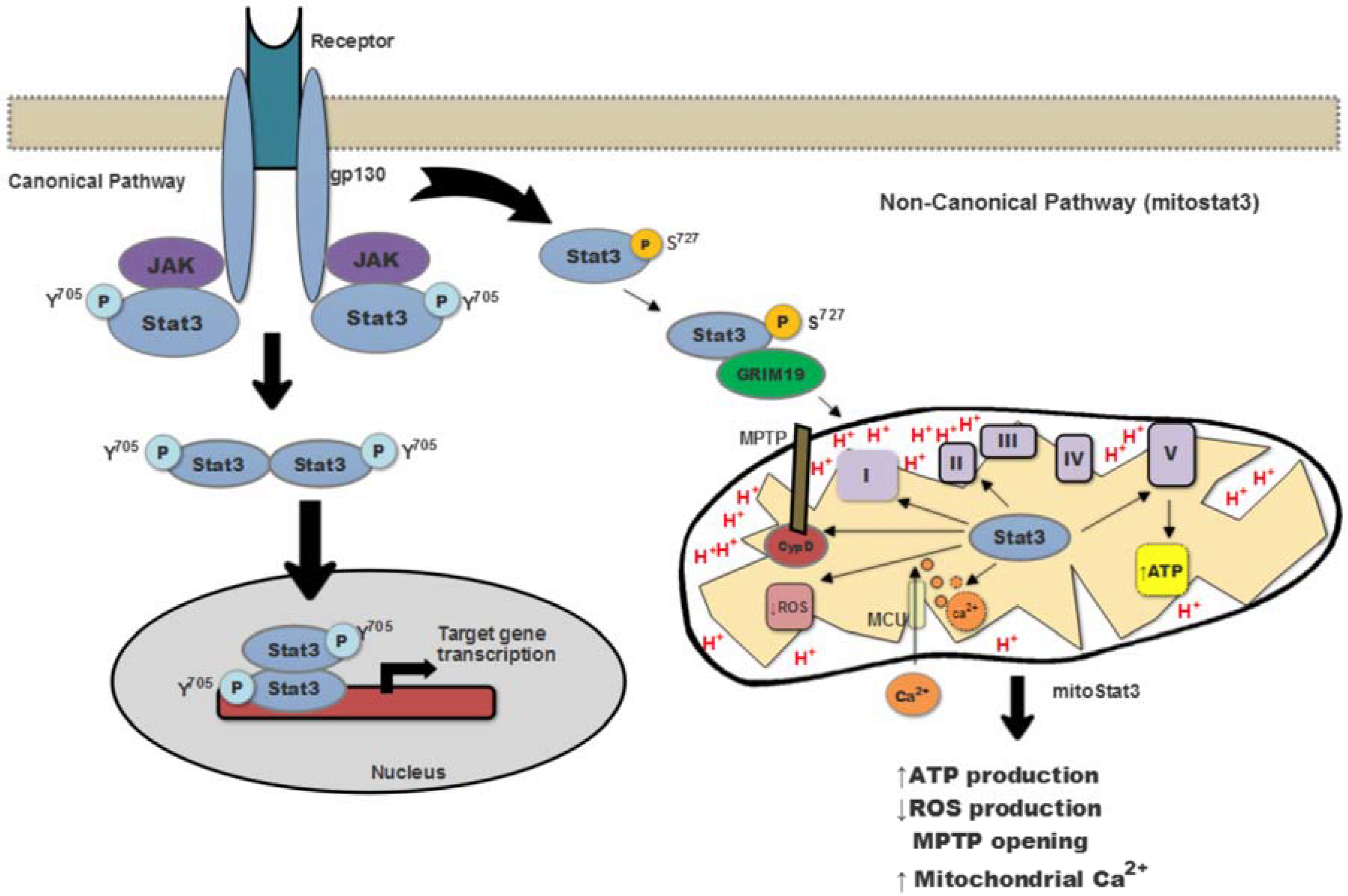
2. Mitochondrial Stat3 as a Modulator of Mitochondria Function
3. Stat3 as a Modulator of Gene Transcription in T and B Lymphocytes
3.1. Stat3 in CD4 Cell Function
3.2. Stat3 in CD8 Cell Function
3.3. Stat3 in B Cell Function
4. Mitochondrial Stat3 in T and B Cells: What We Know and What We Need to Know
5. Need for an Unconventional Design Thinking Involving mitoStat3 for Novel Therapies in Inflammatory Diseases
Funding
Acknowledgments
Conflicts of Interest
References
- Lutticken, C.; Wegenka, U.M.; Yuan, J.; Buschmann, J.; Schindler, C.; Ziemiecki, A.; Harpur, A.G.; Wilks, A.F.; Yasukawa, K.; Taga, T.; et al. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science 1994, 263, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Vignali, D.A. STAT heterodimers in immunity: A mixed message or a unique signal? Jakstat 2013, 2, e23060. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef] [PubMed]
- Niemand, C.; Nimmesgern, A.; Haan, S.; Fischer, P.; Schaper, F.; Rossaint, R.; Heinrich, P.C.; Muller-Newen, G. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J. Immunol. 2003, 170, 3263–3272. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr. Opin. Pharmacol. 2006, 6, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Raeber, M.E.; Zurbuchen, Y.; Impellizzieri, D.; Boyman, O. The role of cytokines in T-cell memory in health and disease. Immunol. Rev. 2018, 283, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Howlett, G.J.; Discolo, G.; Yasukawa, K.; Hammacher, A.; Moritz, R.L.; Simpson, R.J. High affinity interleukin-6 receptor is a hexameric complex consisting of two molecules each of interleukin-6, interleukin-6 receptor, and gp-130. J. Biol. Chem. 1994, 269, 23286–23289. [Google Scholar] [PubMed]
- Silver, J.S.; Hunter, C.A. gp130 at the nexus of inflammation, autoimmunity, and cancer. J. Leukoc. Biol. 2010, 88, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Lufei, C.; Ma, J.; Huang, G.; Zhang, T.; Novotny-Diermayr, V.; Ong, C.T.; Cao, X. GRIM-19, a death-regulatory gene product, suppresses Stat3 activity via functional interaction. EMBO J. 2003, 22, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; D’Angeli, L.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Watson, C.J.; et al. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [PubMed]
- Sarafian, T.A.; Montes, C.; Imura, T.; Qi, J.; Coppola, G.; Geschwind, D.H.; Sofroniew, M.V. Disruption of astrocyte STAT3 signaling decreases mitochondrial function and increases oxidative stress in vitro. PLoS ONE 2010, 5, e9532. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Lirussi, D.; Thornton, T.M.; Jelley-Gibbs, D.M.; Diehl, S.A.; Case, L.K.; Madesh, M.; Taatjes, D.J.; Teuscher, C.; Haynes, L.; et al. Mitochondrial Ca2+ and membrane potential, an alternative pathway for interleukin 6 to regulate CD4 cell effector function. Elife 2015, 4, e06376. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Rincon, M. Mitochondrial Stat3, the need for design thinking. Int. J. Biol. Sci. 2016, 12, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 2010, 105, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.A.; Hyun, M.; Cantwell, M.; Raza, A.; Mertens, C.; Raje, V.; Sisler, J.; Tracy, E.; Torres-Odio, S.; Gispert, S.; et al. Stress-induced dynamic regulation of mitochondrial STAT3 and its association with cyclophilin D reduce mitochondrial ROS production. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef] [PubMed]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, G.G.; Huang, L.; Alston, N.; Ouyang, N.; Vrankova, K.; Mattheolabakis, G.; Constantinides, P.P.; Rigas, B. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS ONE 2013, 8, e61532. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.S.; Liang, J.J.; Wang, Y.; Zhao, X.J.; Xu, L.; Xu, Y.Y.; Zou, Q.C.; Zhang, J.M.; Tu, C.E.; Cui, Y.G.; et al. STAT3 undergoes acetylation-dependent mitochondrial translocation to regulate pyruvate metabolism. Sci. Rep. 2016, 6, 39517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Sheng, M.; Wang, J.; Teng, T.; Sun, Y.; Yang, Q.; Xu, Z. Zinc improves mitochondrial respiratory function and prevents mitochondrial ROS generation at reperfusion by phosphorylating STAT3 at Ser(727). J. Mol. Cell. Cardiol. 2018, 118, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Capron, C.; Jondeau, K.; Casetti, L.; Jalbert, V.; Costa, C.; Verhoeyen, E.; Masse, J.M.; Coppo, P.; Bene, M.C.; Bourdoncle, P.; et al. Viability and stress protection of chronic lymphoid leukemia cells involves overactivation of mitochondrial phosphoSTAT3Ser727. Cell Death Dis. 2014, 5, e1451. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, e4924–e4933. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Too, H.P. Mitochondrial localized STAT3 is involved in ngf induced neurite outgrowth. PLoS ONE 2011, 6, e21680. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Too, H.P. Gdnf family ligand dependent STAT3 activation is mediated by specific alternatively spliced isoforms of GFRalpha2 and RET. Biochim. Biophys. Acta 2013, 1833, 2789–2802. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ribeiro, M.; Bray, E.R.; Lee, D.H.; Yungher, B.J.; Mehta, S.T.; Thakor, K.A.; Diaz, F.; Lee, J.K.; Moraes, C.T.; et al. Enhanced transcriptional activity and mitochondrial localization of STAT3 co-induce axon regrowth in the adult central nervous system. Cell Rep. 2016, 15, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Lin, C.Y.; Benveniste, E.N.; Lee, Y.S. Mitochondrial STAT3 is negatively regulated by SOCS3 and upregulated after spinal cord injury. Exp. Neurol. 2016, 284, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M.; DeLeo, F.R.; Elloumi, H.Z.; Hsu, A.P.; Uzel, G.; Brodsky, N.; Freeman, A.F.; Demidowich, A.; Davis, J.; Turner, M.L.; et al. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 2007, 357, 1608–1619. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, S.E.; Haapaniemi, E.; Russell, M.A.; Caswell, R.; Allen, H.L.; De Franco, E.; McDonald, T.J.; Rajala, H.; Ramelius, A.; Barton, J.; et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat. Genet. 2014, 46, 812–814. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, Y.; Saito, M.; Tsuchiya, S.; Tsuge, I.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; Pasic, S.; Stojkovic, O.; et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007, 448, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.F.; Holland, S.M. The hyper-IgE syndromes. Immunol. Allergy Clin. N. Am. 2008, 28, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Haapaniemi, E.M.; Kaustio, M.; Rajala, H.L.; van Adrichem, A.J.; Kainulainen, L.; Glumoff, V.; Doffinger, R.; Kuusanmaki, H.; Heiskanen-Kosma, T.; Trotta, L.; et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood 2015, 125, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Basso, A.S.; Cheroutre, H.; Mucida, D. More stories on Th17 cells. Cell Res. 2009, 19, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Chew, G.Y.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Cypowyj, S.; Picard, C.; Marodi, L.; Casanova, J.L.; Puel, A. Immunity to infection in IL-17-deficient mice and humans. Eur. J. Immunol. 2012, 42, 2246–2254. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Paul, W.E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.K.; Andraski, A.B.; Spolski, R.; Li, P.; Kazemian, M.; Oh, J.; Samsel, L.; Swanson, P.A., 2nd; McGavern, D.B.; Sampaio, E.P.; et al. Opposing roles of STAT1 and STAT3 in IL-21 function in CD4+ T cells. Proc. Natl. Acad. Sci. USA 2015, 112, 9394–9399. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.; Anguita, J.; Hoffmeyer, A.; Zapton, T.; Ihle, J.N.; Fikrig, E.; Rincon, M. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity 2000, 13, 805–815. [Google Scholar] [CrossRef]
- Nishihara, M.; Ogura, H.; Ueda, N.; Tsuruoka, M.; Kitabayashi, C.; Tsuji, F.; Aono, H.; Ishihara, K.; Huseby, E.; Betz, U.A.; et al. IL-6-gp130-STAT3 in T cells directs the development of IL-17+ Th with a minimum effect on that of Treg in the steady state. Int. Immunol. 2007, 19, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M.; Anguita, J.; Nakamura, T.; Fikrig, E.; Flavell, R.A. Interleukin (IL)-6 directs the differentiation of IL-4-producing CD4+ T cells. J. Exp. Med. 1997, 185, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.; Chow, C.W.; Weiss, L.; Palmetshofer, A.; Twardzik, T.; Rounds, L.; Serfling, E.; Davis, R.J.; Anguita, J.; Rincon, M. Induction of NFATc2 expression by interleukin 6 promotes t helper type 2 differentiation. J. Exp. Med. 2002, 196, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bryant, V.L.; Ma, C.S.; Avery, D.T.; Li, Y.; Good, K.L.; Corcoran, L.M.; de Waal Malefyt, R.; Tangye, S.G. Cytokine-mediated regulation of human b cell differentiation into Ig-secreting cells: Predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J. Immunol. 2007, 179, 8180–8190. [Google Scholar] [CrossRef] [PubMed]
- Linterman, M.A.; Beaton, L.; Yu, D.; Ramiscal, R.R.; Srivastava, M.; Hogan, J.J.; Verma, N.K.; Smyth, M.J.; Rigby, R.J.; Vinuesa, C.G. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J. Exp. Med. 2010, 207, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Batten, M.; Ramamoorthi, N.; Kljavin, N.M.; Ma, C.S.; Cox, J.H.; Dengler, H.S.; Danilenko, D.M.; Caplazi, P.; Wong, M.; Fulcher, D.A.; et al. IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J. Exp. Med. 2010, 207, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Dienz, O.; Eaton, S.M.; Bond, J.P.; Neveu, W.; Moquin, D.; Noubade, R.; Briso, E.M.; Charland, C.; Leonard, W.J.; Ciliberto, G.; et al. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J. Exp. Med. 2009, 206, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.A.; Schmidlin, H.; Nagasawa, M.; Blom, B.; Spits, H. IL-6 triggers IL-21 production by human CD4+ T cells to drive STAT3-dependent plasma cell differentiation in B cells. Immunol. Cell Biol. 2012, 90, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.I.; Chung, Y.; Hwang, D.; Yang, X.O.; Kang, H.S.; Ma, L.; Wang, Y.H.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 2008, 29, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Leonard, W.J.; Wan, C.K. IL-21 signaling in immunity. F1000Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Siegel, A.M.; Heimall, J.; Freeman, A.F.; Hsu, A.P.; Brittain, E.; Brenchley, J.M.; Douek, D.C.; Fahle, G.H.; Cohen, J.I.; Holland, S.M.; et al. A critical role for STAT3 transcription factor signaling in the development and maintenance of human t cell memory. Immunity 2011, 35, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Masters, A.R.; Fortner, K.A.; Champagne, D.P.; Yanguas-Casas, N.; Silberger, D.J.; Weaver, C.T.; Haynes, L.; Rincon, M. IL-6 promotes the differentiation of a subset of naive CD8+ T cells into IL-21-producing B helper CD8+ T cells. J. Exp. Med. 2016, 213, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; van Besouw, N.M.; Shi, Y.; Hoogduijn, M.J.; Wang, L.; Baan, C.C. The biological effects of IL-21 signaling on B-cell-mediated responses in organ transplantation. Front. Immunol. 2016, 7, 319. [Google Scholar] [CrossRef] [PubMed]
- Fornek, J.L.; Tygrett, L.T.; Waldschmidt, T.J.; Poli, V.; Rickert, R.C.; Kansas, G.S. Critical role for Stat3 in T-dependent terminal differentiation of IgG B cells. Blood 2006, 107, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.A.; Schmidlin, H.; Nagasawa, M.; van Haren, S.D.; Kwakkenbos, M.J.; Yasuda, E.; Beaumont, T.; Scheeren, F.A.; Spits, H. STAT3-mediated up-regulation of BLIMP1 is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J. Immunol. 2008, 180, 4805–4815. [Google Scholar] [CrossRef] [PubMed]
- Avery, D.T.; Deenick, E.K.; Ma, C.S.; Suryani, S.; Simpson, N.; Chew, G.Y.; Chan, T.D.; Palendira, U.; Bustamante, J.; Boisson-Dupuis, S.; et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J. Exp. Med. 2010, 207, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Deenick, E.K.; Avery, D.T.; Chan, A.; Berglund, L.J.; Ives, M.L.; Moens, L.; Stoddard, J.L.; Bustamante, J.; Boisson-Dupuis, S.; Tsumura, M.; et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J. Exp. Med. 2013, 210, 2739–2753. [Google Scholar] [CrossRef] [PubMed]
- Carbone, G.; Wilson, A.; Diehl, S.A.; Bunn, J.; Cooper, S.M.; Rincon, M. Interleukin-6 receptor blockade selectively reduces IL-21 production by CD4 T cells and IgG4 autoantibodies in rheumatoid arthritis. Int. J. Biol. Sci. 2013, 9, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Frezza, C.; Soriano, M.E.; Varanita, T.; Quintana-Cabrera, R.; Corrado, M.; Cipolat, S.; Costa, V.; Casarin, A.; Gomes, L.C.; et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 2013, 155, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Hackenbrock, C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J. Cell Biol. 1966, 30, 269–297. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell 2016, 167, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Wu, M.; Guo, R.; Yan, K.; Lei, J.; Gao, N.; Yang, M. The architecture of the mammalian respirasome. Nature 2016, 537, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Althoff, T.; Mills, D.J.; Popot, J.L.; Kuhlbrandt, W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 2011, 30, 4652–4664. [Google Scholar] [CrossRef] [PubMed]
- Vukotic, M.; Oeljeklaus, S.; Wiese, S.; Vogtle, F.N.; Meisinger, C.; Meyer, H.E.; Zieseniss, A.; Katschinski, D.M.; Jans, D.C.; Jakobs, S.; et al. Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab. 2012, 15, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Lastres, D.; Fontanesi, F.; Garcia-Consuegra, I.; Martin, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 2012, 15, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Taylor, E.B.; Dephoure, N.; Heo, J.M.; Tonhato, A.; Papandreou, I.; Nath, N.; Denko, N.C.; Gygi, S.P.; Rutter, J. Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 2012, 15, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Shulman, Z.; Gitlin, A.D.; Weinstein, J.S.; Lainez, B.; Esplugues, E.; Flavell, R.A.; Craft, J.E.; Nussenzweig, M.C. Dynamic signaling by t follicular helper cells during germinal center B cell selection. Science 2014, 345, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.H.; Verkhratsky, A. Calcium and ATP control multiple vital functions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef] [PubMed]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the complex interplay between T cell metabolism and function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Allard, E.L.; Hardy, M.P.; Leignadier, J.; Marquis, M.; Rooney, J.; Lehoux, D.; Labrecque, N. Overexpression of IL-21 promotes massive CD8+ memory T cell accumulation. Eur. J. Immunol. 2007, 37, 3069–3077. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Spolski, R.; Finkelstein, S.E.; Oh, S.; Kovanen, P.E.; Hinrichs, C.S.; Pise-Masison, C.A.; Radonovich, M.F.; Brady, J.N.; Restifo, N.P.; et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J. Exp. Med. 2005, 201, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and approaches of targeting STAT3 for cancer treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.; Etemadi, N.; Hollande, F.; Ernst, M.; Buchert, M. The JAK/STAT3 axis: A comprehensive drug target for solid malignancies. Semin. Cancer Biol. 2017, 45, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Leong, P.L.; Andrews, G.A.; Johnson, D.E.; Dyer, K.F.; Xi, S.; Mai, J.C.; Robbins, P.D.; Gadiparthi, S.; Burke, N.A.; Watkins, S.F.; et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. USA 2003, 100, 4138–4143. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Expert Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yue, P.; Page, B.D.; Li, T.; Zhao, W.; Namanja, A.T.; Paladino, D.; Zhao, J.; Chen, Y.; Gunning, P.T.; et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc. Natl. Acad. Sci. USA 2012, 109, 9623–9628. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Kim, J.S.; Zhang, S.; Yuan, J.; Huang, M.; Glenn, M.; Haura, E.; Sebti, S.; Hamilton, A.D.; Jove, R. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol. Cancer Ther. 2004, 3, 261–269. [Google Scholar] [PubMed]
- Bharadwaj, U.; Eckols, T.K.; Xu, X.; Kasembeli, M.M.; Chen, Y.; Adachi, M.; Song, Y.; Mo, Q.; Lai, S.Y.; Tweardy, D.J. Small-molecule inhibition of STAT3 in radioresistant head and neck squamous cell carcinoma. Oncotarget 2016, 7, 26307–26330. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Uchida, T.; Terui, Y.; Hayakawa, F.; Kobayashi, Y.; Taniwaki, M.; Takamatsu, Y.; Naoe, T.; Tobinai, K.; Munakata, W.; et al. Phase i study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015, 106, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.; Soo, R.A.; Tan, D.S.; Lee, S.C.; Lim, J.S.; Marban, P.C.; Kong, L.R.; Lee, Y.J.; Wang, L.Z.; Thuya, W.L.; et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015, 26, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, K.; Takaishi, M.; Nakajima, K.; Ikeda, M.; Kanda, T.; Tarutani, M.; Iiyama, T.; Asao, N.; DiGiovanni, J.; Sano, S. Stat3 as a therapeutic target for the treatment of psoriasis: A clinical feasibility study with STA-21, a Stat3 inhibitor. J. Investig. Dermatol. 2011, 131, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Rubbert-Roth, A.; Furst, D.E.; Nebesky, J.M.; Jin, A.; Berber, E. A review of recent advances using tocilizumab in the treatment of rheumatic diseases. Rheumatol. Ther. 2018, 5, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Tofacitinib: A review in rheumatoid arthritis. Drugs 2017, 77, 1987–2001. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rincon, M.; Pereira, F.V. A New Perspective: Mitochondrial Stat3 as a Regulator for Lymphocyte Function. Int. J. Mol. Sci. 2018, 19, 1656. https://doi.org/10.3390/ijms19061656
Rincon M, Pereira FV. A New Perspective: Mitochondrial Stat3 as a Regulator for Lymphocyte Function. International Journal of Molecular Sciences. 2018; 19(6):1656. https://doi.org/10.3390/ijms19061656
Chicago/Turabian StyleRincon, Mercedes, and Felipe Valença Pereira. 2018. "A New Perspective: Mitochondrial Stat3 as a Regulator for Lymphocyte Function" International Journal of Molecular Sciences 19, no. 6: 1656. https://doi.org/10.3390/ijms19061656
APA StyleRincon, M., & Pereira, F. V. (2018). A New Perspective: Mitochondrial Stat3 as a Regulator for Lymphocyte Function. International Journal of Molecular Sciences, 19(6), 1656. https://doi.org/10.3390/ijms19061656