GBS Mapping and Analysis of Genes Conserved between Gossypium tomentosum and Gossypium hirsutum Cotton Cultivars that Respond to Drought Stress at the Seedling Stage of the BC2F2 Generation

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
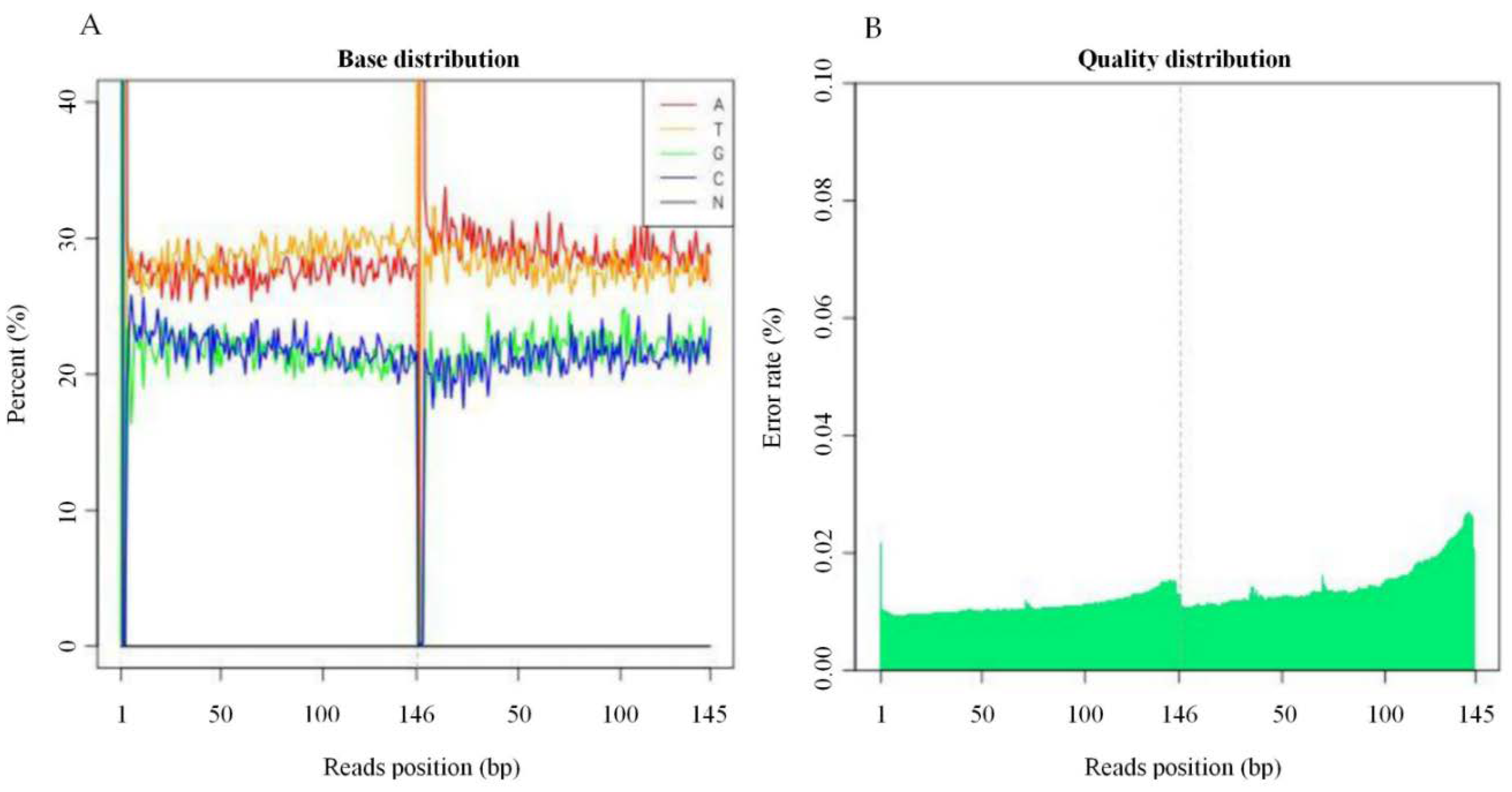
2.1. Sequencing Base Content Distribution, and Error Rate Distribution Statistics
2.2. Raw Sequencing Data Filtering Mechanism, SNP Detection, and Annotation
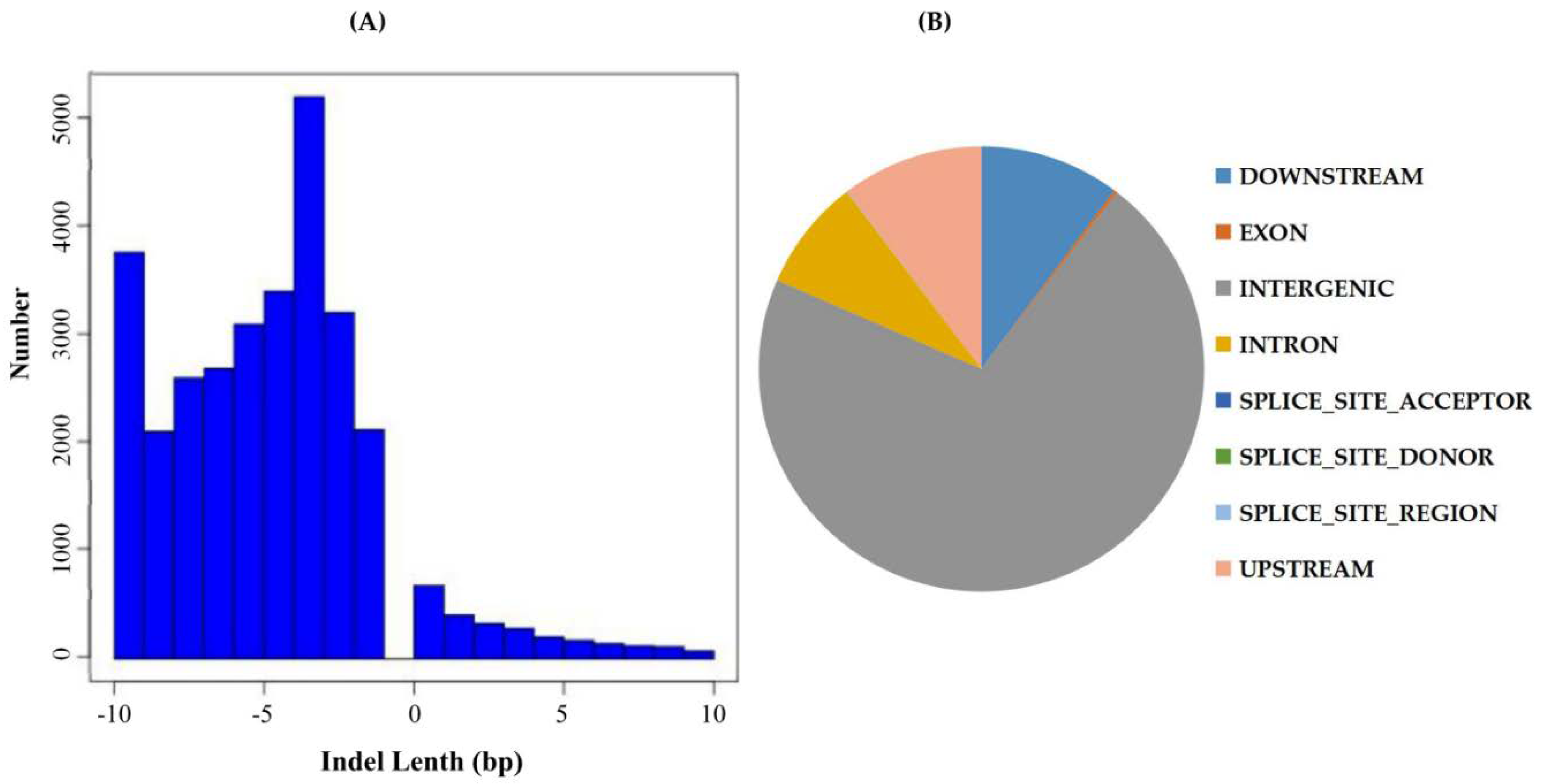
2.3. Indel Detection, Annotation, and Location Distribution
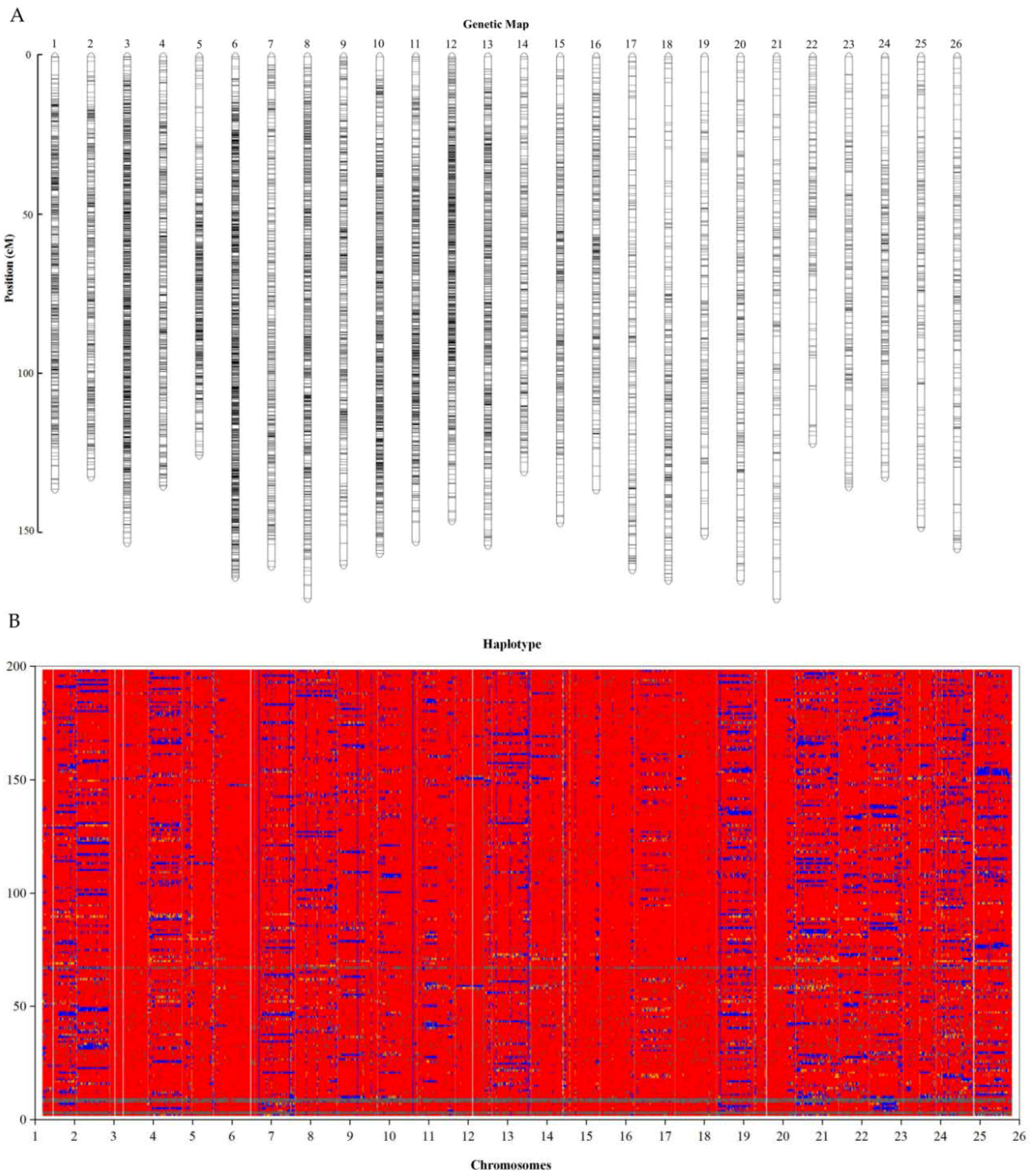
2.4. Genetic Map Construction
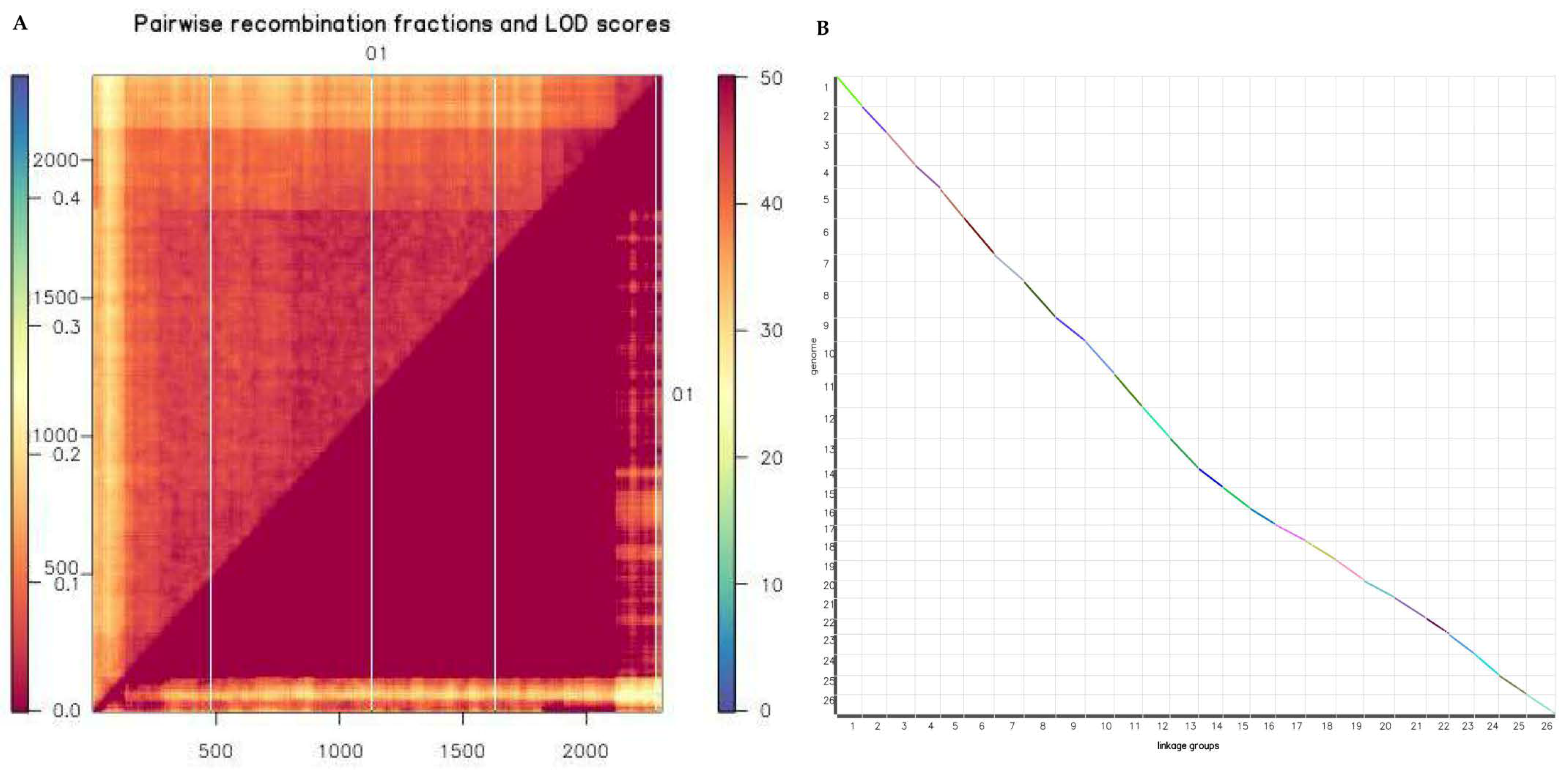
2.5. Fine Genetic Linkage Map Construction using the GBS-SNP Markers
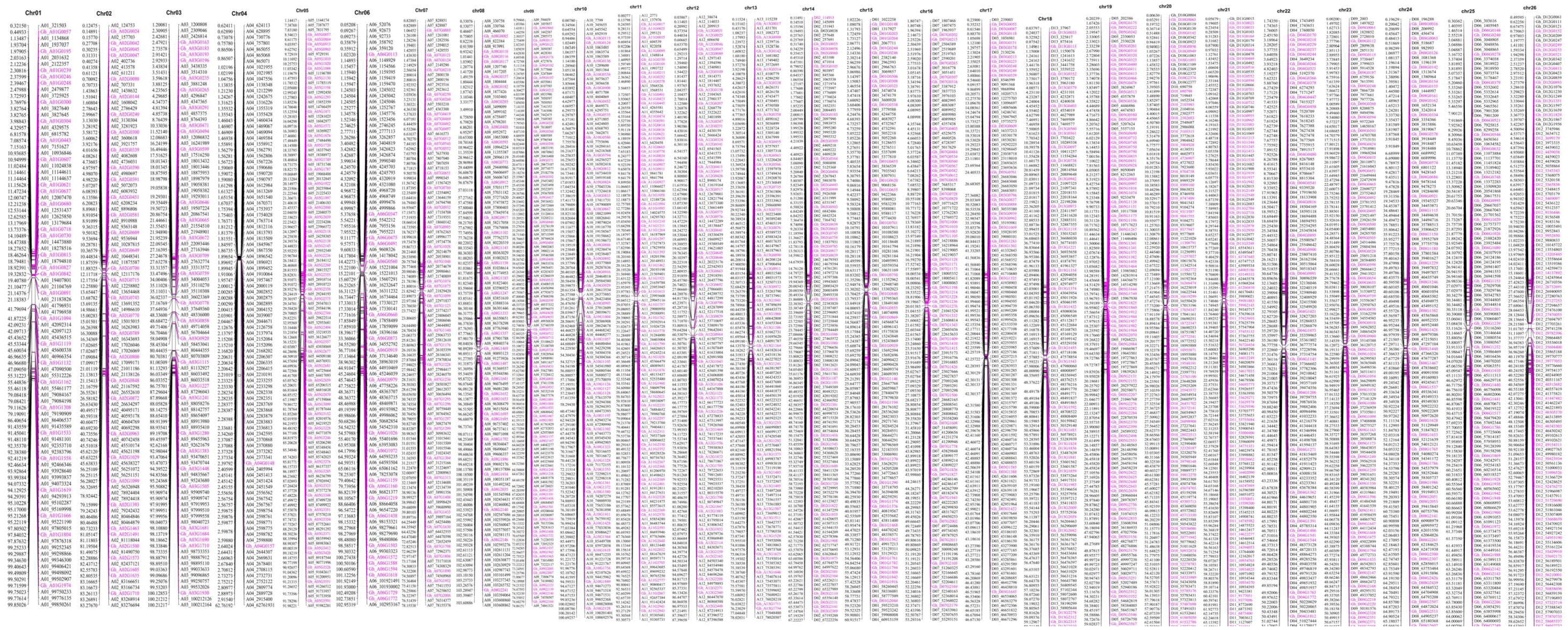
2.6. Evaluation of Reorganization Relationship and Collinearity of Genetic Maps and Genomes
2.7. Gene Mining within the GBS Marker Regions of the Mapping Population
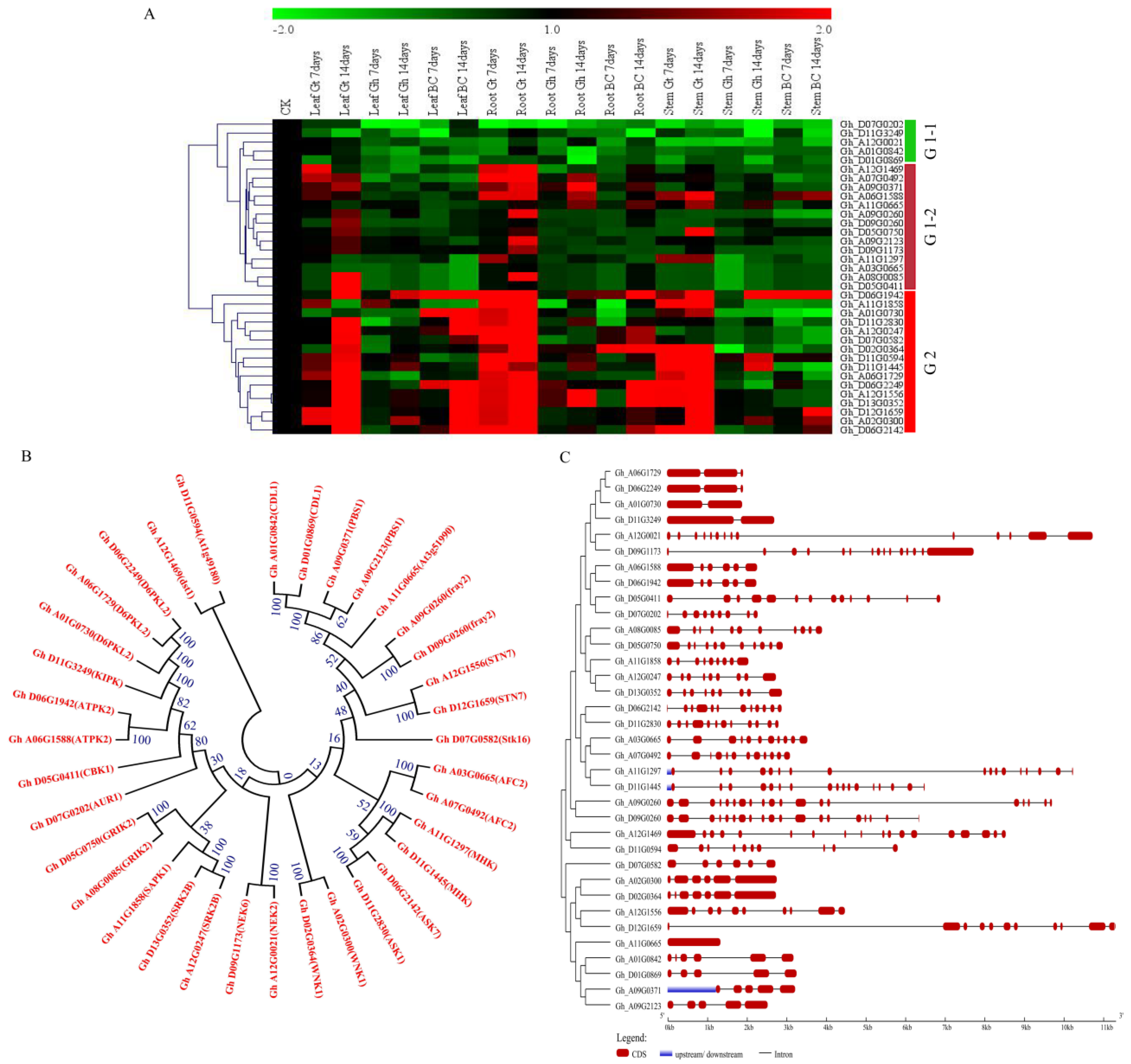
2.8. Chromosomes Mapping of the Genes Mined for the Dominant Domain, Pkinase
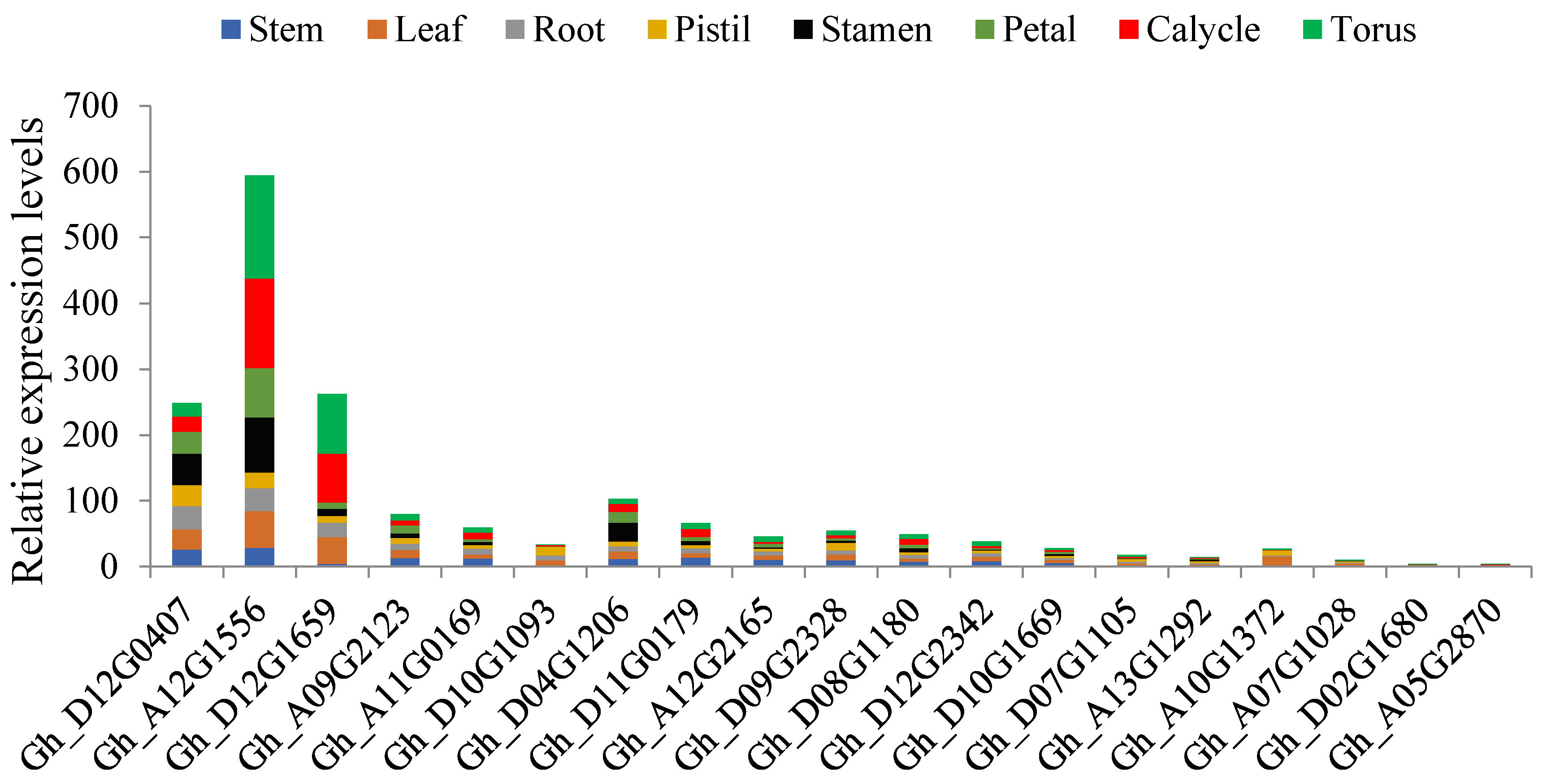
2.9. RNA Sequence Data of the Genes of the Pkinase Domain
2.10. miRNA Target Analysis of the 271 Dominant Genes
2.11. Cis Element Analysis
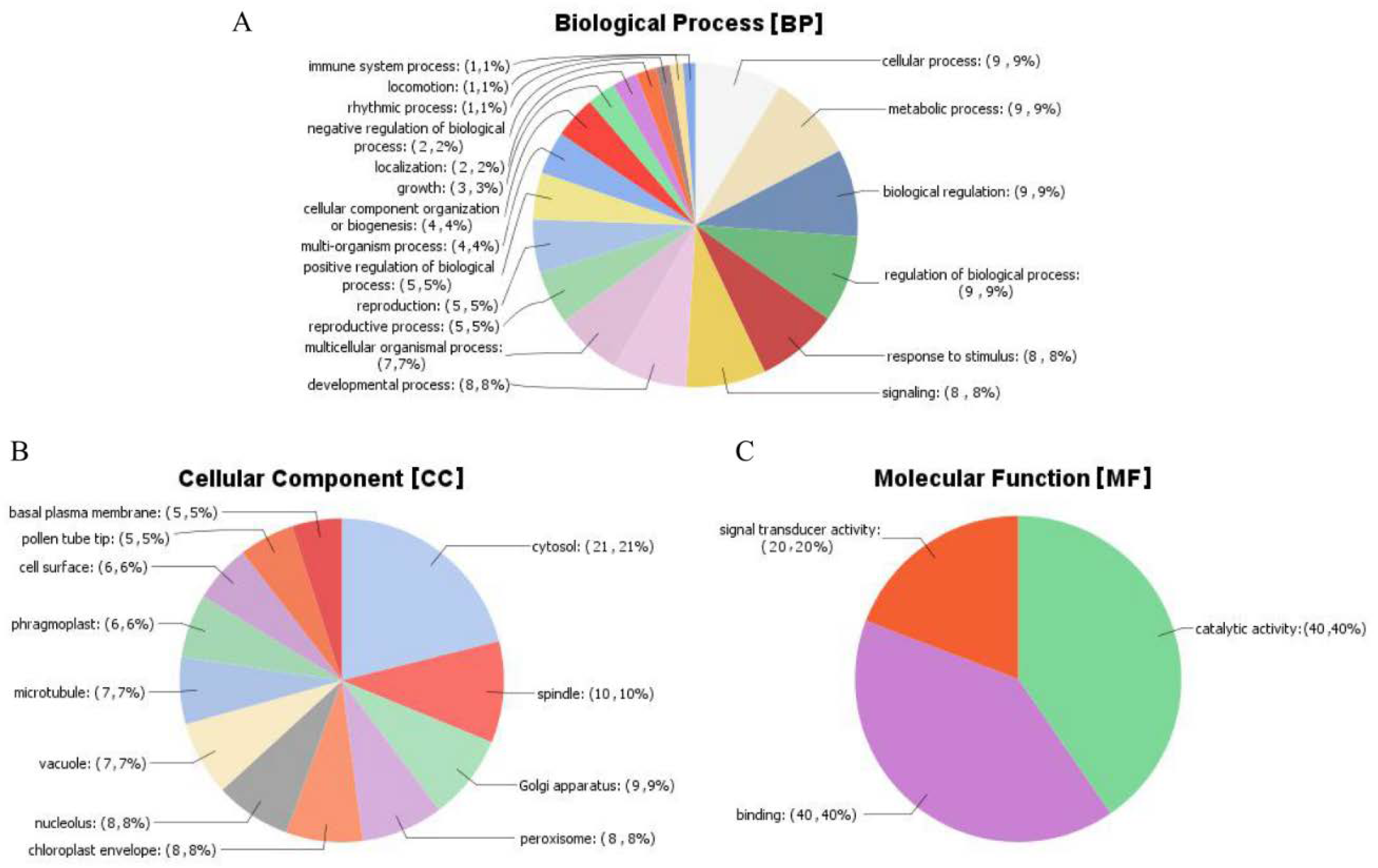
2.12. Gene Ontology (GO) Analysis of the Mined Genes
2.13. qRT-PCR Validation of the Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Development of Plant Materials
4.2. Sample Collection, Extraction, Quantification and Quality Determination of DNA
4.3. The GBS Library Preparation, Sequencing, and SNP Genotyping
4.4. Linkage Map Construction
4.5. Gene Mining and Functional Characterization
4.6. miRNA Target and Promoter Analysis
4.7. RNA Sequence Analysis of the Mined Genes and qRT-PCR Validation of Key Genes under Drought Stress Condition
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| GBS | Genotyping by sequencing |
| WGS | Whole genome shotgun |
| GO | Gene ontology |
| PCD | Programmed cell death |
| ABA | Abscisic acid |
| MAS | Marker assisted selection |
| MPS | Massively parallel sequencing |
| NGS | Next generation sequencing |
References
- Renny-Byfield, S.; Page, J.T.; Udall, J.A.; Sanders, W.S.; Peterson, D.G.; Arick, M.A.; Grover, C.E.; Wendel, J.F. Independent Domestication of Two Old World Cotton Species. Genome Biol. Evol. 2016, 8, 1940–1947. [Google Scholar] [CrossRef] [PubMed]
- Bowman, D.T. Public Cotton Breeders—Do We Need Them? J. Cotton Sci. 1999, 3, 139–152. [Google Scholar]
- Abdurakhmonov, I.Y.; Kohel, R.J.; Yu, J.Z.; Pepper, A.E.; Abdullaev, A.A.; Kushanov, F.N.; Salakhutdinov, I.B.; Buriev, Z.T.; Saha, S.; Scheffler, B.E.; et al. Molecular diversity and association mapping of fiber quality traits in exotic G. hirsutum L. germplasm. Genomics 2008, 92, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Dabbert, T.A.; Gore, M.A. Challenges and Perspectives on Improving Heat and Drought Stress Resilience in Cotton. Breed. Genet. 2014, 409, 393–409. [Google Scholar]
- Zhang, J.; Percy, R.G.; McCarty, J.C. Introgression genetics and breeding between Upland and Pima cotton: A review. Euphytica 2014, 198, 1–12. [Google Scholar] [CrossRef]
- Peng, Z.; He, S.; Gong, W.; Sun, J.; Pan, Z.; Xu, F.; Lu, Y.; Du, X. Comprehensive analysis of differentially expressed genes and transcriptional regulation induced by salt stress in two contrasting cotton genotypes. BMC Genom. 2014, 15, 760. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, H.D.; Yadav, D.; Dronavalli, N.; Gowda, C.L.L.; Singh, S. Mini core germplasm collections for infusing genetic diversity in plant breeding programs. Electron. J. Plant Breed. 2010, 1, 1294–1309. [Google Scholar]
- Petrov, P.; Petrova, A.; Dimitrov, I.; Tashev, T.; Olsovska, K.; Brestic, M.; Misheva, S. Relationships between leaf morpho-anatomy, water status and cell membrane stability in leaves of wheat seedlings subjected to severe soil drought. J. Agron. Crop Sci. 2017, 219–227. [Google Scholar] [CrossRef]
- Ullah, A.; Sun, H.; Yang, X.; Zhang, X. Drought coping strategies in cotton: Increased crop per drop. Plant Biotechnol. J. 2017, 15, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Maxted, N.; Kell, S.; Toledo, Á.; Dulloo, E.; Heywood, V.; Hodgkin, T.; Hunter, D.; Guarino, L.; Jarvis, A.; Ford-Lloyd, B. A global approach to crop wild relative conservation: Securing the gene pool for food and agriculture. Kew Bull. 2010, 65, 561–576. [Google Scholar] [CrossRef]
- Atwell, B.J.; Wang, H.; Scafaro, A.P. Could abiotic stress tolerance in wild relatives of rice be used to improve Oryza sativa? Plant Sci. 2014, 215–216, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Valkoun, J.J. Wheat pre-breeding using wild progenitors. Euphytica 2001, 119, 17–23. [Google Scholar] [CrossRef]
- Fang, D.D.; Jenkins, J.N.; Deng, D.D.; McCarty, J.C.; Li, P.; Wu, J. Quantitative trait loci analysis of fiber quality traits using a random-mated recombinant inbred population in Upland cotton (Gossypium hirsutum L.). BMC Genom. 2014, 15, 397. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, K.; Li, S.; Yu, S.; Zhai, H.; Wu, M.; Li, X.; Fan, S.; Song, M.; Yang, D.; et al. Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Theor. Appl. Genet. 2013, 126. [Google Scholar] [CrossRef] [PubMed]
- Lacape, J.M.; Gawrysiak, G.; Cao, T.V.; Viot, C.; Llewellyn, D.; Liu, S.; Jacobs, J.; Becker, D.; Barroso, P.A.V.; de Assunção, J.H.; et al. Mapping QTLs for traits related to phenology, morphology and yield components in an inter-specific Gossypium hirsutum × G. barbadense cotton RIL population. Field. Crops Res. 2013, 144, 256–267. [Google Scholar] [CrossRef]
- DeJoode, D.R.; Wendel, J.F. Genetic Diversity and Origin of the Hawaiian Islands Cotton, Gossypium tomentosum. Am. J. Bot. 1992, 79, 1311–1319. [Google Scholar] [CrossRef]
- Lehman, A.; Pender, R.; Morden, C.; Wieczorek, A.M. Assessment of Persistence of Hybrids between Alien Pima Cotton, Gossypium barbadense (Malvaceae), and Endemic Hawaiian Cotton, G. tomentosum, in Hawai’i1. Pac. Sci. 2014, 68, 85–96. [Google Scholar] [CrossRef]
- Oluoch, G.; Zheng, J.; Wang, X.; Khan, M.K.R.; Zhou, Z.; Cai, X.; Wang, C.; Wang, Y.; Li, X.; Wang, H.; et al. QTL mapping for salt tolerance at seedling stage in the interspecific cross of Gossypium tomentosum with Gossypium hirsutum. Euphytica 2016, 209, 223–235. [Google Scholar] [CrossRef]
- Hou, M.; Cai, C.; Zhang, S.; Guo, W.; Zhang, T.; Zhou, B. Construction of microsatellite-based linkage map and mapping of nectarilessness and hairiness genes in Gossypium tomentosum. J. Genet. 2013, 92, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.K.R.; Chen, H.; Zhou, Z.; Ilyas, M.K.; Wang, X.; Cai, X.; Wang, C.; Liu, F.; Wang, K. Genome Wide SSR High Density Genetic Map Construction from an Interspecific Cross of Gossypium hirsutum × Gossypium tomentosum. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.J.; Caligari, P.D.S. Efficiencies of F2 and backcross generations for bulked segregant analysis using dominant markers. Crop Sci. 2000, 40, 626–630. [Google Scholar] [CrossRef]
- Hospital, F. Selection in backcross programmes. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Miah, G.; Rafii, M.Y.; Ismail, M.R.; Puteh, A.B.; Rahim, H.A.; Latif, M.A. Recurrent parent genome recovery analysis in a marker-assisted backcrossing program of rice (Oryza sativa L.). C. R. Biol. 2015, 338, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Diers, B.W.; Hyten, D.L.; Rouf Mian, M.A.; Shannon, J.G.; Nelson, R.L. Identification of positive yield QTL alleles from exotic soybean germplasm in two backcross populations. Theor. Appl. Genet. 2012, 125, 1353–1369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ni, D.; Yi, C.; Wang, X.; Wang, Z.Y.; Yang, J. Lowering grain amylose content in backcross offsprings of indica rice variety 057 by molecular marker-assisted selection. Rice Sci. 2005, 12, 157–162. [Google Scholar]
- Li, J.Z.; Huang, X.Q.; Heinrichs, F.; Ganal, M.W.; Röder, M.S. Analysis of QTLs for yield components, agronomic traits, and disease resistance in an advanced backcross population of spring barley. Genome 2006, 49, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, N.; Miklas, P.; Reiser, J.; Coyne, D. Backcross breeding for improved resistance to common bacterial blight in pinto bean (Phaseolus vulgaris L.). Plant Breed. 2005, 124, 282–287. [Google Scholar] [CrossRef]
- Behera, T.K.; Staub, J.E.; Behera, S.; Delannay, I.Y.; Chen, J.F. Marker-assisted backcross selection in an interspecific Cucumis population broadens the genetic base of cucumber (Cucumis sativus L.). Euphytica 2011, 178, 261–272. [Google Scholar] [CrossRef]
- Tucker, T.; Marra, M.; Friedman, J.M. Massively Parallel Sequencing: The Next Big Thing in Genetic Medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef] [PubMed]
- De Summa, S.; Malerba, G.; Pinto, R.; Mori, A.; Mijatovic, V.; Tommasi, S. GATK hard filtering: Tunable parameters to improve variant calling for next generation sequencing targeted gene panel data. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Kosman, E.; Leonard, K.J. Similarity coefficients for molecular markers in studies of genetic relationships between individuals for haploid, diploid, and polyploid species. Mol. Ecol. 2005, 14, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Ajawatanawong, P.; Baldauf, S.L. Evolution of protein indels in plants, animals and fungi. BMC Evol. Biol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Bansal, V.; Libiger, O. A probabilistic method for the detection and genotyping of small indels from population-scale sequence data. Bioinformatics 2011, 27, 2047–2053. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, S.B.; Goode, D.L.; Kvikstad, E.; Albers, C.A.; Zhang, Z.D.; Mu, X.J.; Ananda, G.; Howie, B.; Karczewski, K.J.; Smith, K.S.; et al. The origin, evolution, and functional impact of short insertion-deletion variants identified in 179 human genomes. Genome Res. 2013, 23, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, R.; Li, H.; Lu, J.; Li, Y.; Bolund, L.; Schierup, M.H.; Wang, J. SOAPindel: Efficient identification of indels from short paired reads. Genome Res. 2013, 23, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wang, Y.; Brudno, M. PRISM: Pair-read informed split-read mapping for base-pair level detection of insertion, deletion and structural variants. Bioinformatics 2012, 28, 2576–2583. [Google Scholar] [CrossRef] [PubMed]
- Abyzov, A.; Urban, A.E.; Snyder, M.; Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011, 21, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Zhang, X.; Borevitz, J.O.; Li, W.H.; Shiu, S.H. A large number of novel coding small open reading frames in the intergenic regions of the Arabidopsis thaliana genome are transcribed and/or under purifying selection. Genome Res. 2007, 17, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Lu, C.; Xiao, G.; Zou, C.; Kohel, R.J.; Ma, Z.; Shang, H.; Ma, X.; Wu, J.; et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 2015, 33, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Jeong, H.J.; Yang, H.B.; Kang, S.M.; Kwon, J.K.; Kim, S.; Choi, D.; Kang, B.C. An ultra-high-density bin map facilitates high-throughput QTL mapping of horticultural traits in pepper (Capsicum annuum). DNA Res. 2016, 23, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Kohel, R.J.; Smith, C.W. The construction of a tetraploid cotton genome wide comprehensive reference map. Genomics 2010, 95, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Pascual, L.; Desplat, N.; Huang, B.E.; Desgroux, A.; Bruguier, L.; Bouchet, J.P.; Le, Q.H.; Chauchard, B.; Verschave, P.; Causse, M. Potential of a tomato MAGIC population to decipher the genetic control of quantitative traits and detect causal variants in the resequencing era. Plant Biotechnol. J. 2015, 13, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Tang, Z.; Wang, M.; Gao, W.; Tu, L.; Jin, X.; Chen, L.; He, Y.; Zhang, L.; Zhu, L.; et al. The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 2016, 5, 17662. [Google Scholar] [CrossRef] [PubMed]
- Dahab, A.A.; Saeed, M.; Hamza, N.B.; Babiker, B. Linkage disequilibrium and association mapping of drought tolerance in cotton (Gossypium hirsutum L.) germplasm population from diverse regions of Pakistan. Afr. J. Biotechnol. 2016, 15, 2603–2612. [Google Scholar] [CrossRef]
- Sciences, P.; Saleem, M.A.; Malik, T.A.; Shakeel, A.; Ashraf, M. QTL Mapping for Some Important Drought Tolerant Traits in Upland. J. Anim. Plant Sci. 2015, 25, 502–509. [Google Scholar]
- Rong, J.; Feltus, F.A.; Waghmare, V.N.; Pierce, G.J.; Chee, P.W.; Draye, X.; Saranga, Y.; Wright, R.J.; Wilkins, T.A.; May, O.L.; et al. Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 2007, 176, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Scheeff, E.D.; Bourne, P.E. Structural Evolution of the Protein Kinase–Like Superfamily. PLoS Comput. Biol. 2005, 1, e49. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Guo, W.; Zhu, X.; Yuan, Y.; Yu, J.Z.; Kohel, R.J.; Zhang, T. Molecular mapping of QTLs for fiber qualities in three diverse lines in Upland cotton using SSR markers. Mol. Breed. 2005, 15, 169–181. [Google Scholar] [CrossRef]
- Boss, I.W.; Renne, R. Viral miRNAs: Tools for immune evasion. Curr. Opin. Microbiol. 2010, 13, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and Their Regulatory Roles in Plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.A.; Declerck, M.; Sorin, C.; Hartmann, C.; Crespi, M.; Lelandais-Brière, C. MicroRNAs as regulators of root development and architecture. Plant Mol. Biol. 2011, 77, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R. Role of microRNAs in biotic and abiotic stress responses in crop plants. Appl. Biochem. Biotechnol. 2014, 174, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Liang, R.; Ge, L.; Li, W.; Xiao, H.; Lin, H.; Ruan, K.; Jin, Y. Identification of drought-induced microRNAs in rice. Biochem. Biophys. Res. Commun. 2007, 354, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Marone, D.; Russo, M.A.; Laidò, G.; De Leonardis, A.M.; Mastrangelo, A.M. Plant nucleotide binding site-leucine-rich repeat (NBS-LRR) genes: Active guardians in host defense responses. Int. J. Mol. Sci. 2013, 14, 7302–7326. [Google Scholar] [CrossRef] [PubMed]
- Yoon, E.K.; Yang, J.H.; Lim, J.; Kim, S.H.; Kim, S.K.; Lee, W.S. Auxin regulation of the microRNA390-dependent transacting small interfering RNA pathway in Arabidopsis lateral root development. Nucleic Acids Res. 2009, 38, 1382–1391. [Google Scholar] [CrossRef] [PubMed]
- Crombach, A.; Hogeweg, P. Evolution of evolvability in gene regulatory networks. PLoS Comput. Biol. 2008, 4. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and validation of promoters and cis-acting regulatory elements. Plant Sci. 2014, 217–218, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci. 2005, 10, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.E.; Chaitanya, K.V. Photosynthesis and antioxidative defense mechanisms in deciphering drought stress tolerance of crop plants. Biol. Plant. 2016, 60, 201–218. [Google Scholar] [CrossRef]
- Williams, B.; Verchot, J.; Dickman, M.B. When supply does not meet demand-ER stress and plant programmed cell death. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Gechev, T.S.; Van Breusegem, F.; Stone, J.M.; Denev, I.; Laloi, C. Reactive oxygen species as signals that modulate plant stress responses and programmed cell death. BioEssays 2006, 28, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Preuss, M.L.; Delmer, D.P.; Liu, B. The cotton kinesin-like calmodulin-binding protein associates with cortical microtubules in cotton fibers. Plant Physiol. 2003, 132, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Ghelis, T.; Bolbach, G.; Clodic, G.; Habricot, Y.; Miginiac, E.; Sotta, B.; Jeannette, E. Protein Tyrosine Kinases and Protein Tyrosine Phosphatases Are Involved in Abscisic Acid-Dependent Processes in Arabidopsis Seeds and Suspension Cells. Plant Physiol. 2008, 148, 1668–1680. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, S.; Kobayashi, M.; Taji, T.; Naramoto, M.; Seki, M.; Kato, T.; Tabata, S.; Kakubari, Y.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis. Plant J. 2001, 27, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Testerink, C.; Munnik, T. Phosphatidic acid: A multifunctional stress signaling lipid in plants. Trends Plant Sci. 2005, 10, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Debnath, M.; Pandey, M.; Bisen, P.S. An Omics Approach to Understand the Plant Abiotic Stress. OMICS 2011, 15, 739–762. [Google Scholar] [CrossRef] [PubMed]
- Duncker, B.P.; Davies, P.L.; Walker, V.K. Introns boost transgene expression in Drosophila melanogaster. Mol. Gen. Genet. 1997, 254, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Guo, P.; Baum, M.; Grando, S.; Ceccarelli, S.; Bai, G.; Li, R.; Von Korff, M.; Varshney, R.K.; Graner, A.; Valkoun, J. Differentially expressed genes between drought-tolerant and drought-sensitive barley genotypes in response to drought stress during the reproductive stage. J. Exp. Bot. 2009, 60, 3531–3544. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.D.; Nelson, J.C. Advanced backcross QTL analysis: A method for the simultaneous discovery and transfer of valuable QTLs from unadapted germplasm into elite breeding lines. Theor. Appl. Genet. 1996, 92, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.J.; Gyenis, L.; Bossolini, E.; Hayes, P.M.; Matus, I.; Smith, K.P.; Steffenson, B.J.; Tuberosa, R.; Muehlbauer, G.J. Validation of quantitative trait loci for multiple disease resistance in barley using advanced backcross lines developed with a wild barley. Crop Sci. 2006, 46, 1179–1186. [Google Scholar] [CrossRef]
- Wang, B.; Liu, L.; Zhang, D.; Zhuang, Z.; Guo, H.; Qiao, X.; Wei, L.; Rong, J.; May, O.L.; Paterson, A.H.; et al. A Genetic Map between Gossypium hirsutum and the Brazilian Endemic G. mustelinum and Its Application to QTL Mapping. G3 2016, 6, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Waghmare, V.N.; Rong, J.; Rogers, C.J.; Pierce, G.J.; Wendel, J.F.; Paterson, A.H. Genetic mapping of a cross between Gossypium hirsutum (cotton) and the Hawaiian endemic, Gossypium tomentosum. Theor. Appl. Genet. 2005, 111, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, V.S.K.; Reddy, T.P.; Reddy, V.D.; Rao, K.V. Current status of genetic engineering in cotton (Gossypium hirsutum L.): An assessment. Crit. Rev. Biotechnol. 2012, 8551, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kulik, A.; Wawer, I.; Krzywińska, E.; Bucholc, M.; Dobrowolska, G. SnRK2 Protein Kinases—Key Regulators of Plant Response to Abiotic Stresses. OMICS 2011, 15, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Diédhiou, C.J.; Popova, O.V.; Dietz, K.J.; Golldack, D. The SNF1-type serine-threonine protein kinase SAPK4 regulates stress-responsive gene expression in rice. BMC Plant Biol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Swiderski, M.R.; Innes, R.W. The Arabidopsis PBS1 resistance gene encodes a member of a novel protein kinase subfamily. Plant J. 2001, 26, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Kirungu, J.N.; Deng, Y.; Cai, X.; Magwanga, R.O.; Zhou, Z.; Wang, X.; Wang, Y.; Zhang, Z.; Wang, K.; Liu, F. Simple sequence repeat (SSR) genetic linkage map of D genome diploid cotton derived from an interspecific cross between Gossypium davidsonii and Gossypium klotzschianum. Int. J. Mol. Sci. 2018, 19, 204. [Google Scholar] [CrossRef] [PubMed]
- Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Lu, H.; Wang, X.; Cai, X.; Zhou, Z.; Zhang, Z.; Salih, H.; Wang, K.; et al. Characterization of the late embryogenesis abundant (LEA) proteins family and their role in drought stress tolerance in Upland cotton. BMC Genet. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Nuruzzaman, M.; Sharoni, A.M.; Kikuchi, S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- De Souza, E.E.; Meirelles, G.V.; Godoy, B.B.; Perez, A.M.; Smetana, J.H.C.; Doxsey, S.J.; McComb, M.E.; Costello, C.E.; Whelan, S.A.; Kobarg, J. Characterization of the human NEK7 interactome suggests catalytic and regulatory properties distinct from those of NEK6. J. Proteome Res. 2014, 13, 4074–4090. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Fujita, M.; Satoh, R.; Maruyama, K.; Parvez, M.M.; Seki, M.; Hiratsu, K.; Ohme-Takagi, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AREB1 is a transcription activator of novel ABRE-dependent ABA signaling that enhances drought stress tolerance in Arabidopsis. Plant Cell 2005, 17, 3470–3488. [Google Scholar] [CrossRef] [PubMed]
- Tikkanen, M.; Grieco, M.; Kangasjarvi, S.; Aro, E.-M. Thylakoid Protein Phosphorylation in Higher Plant Chloroplasts Optimizes Electron Transfer under Fluctuating Light. Plant Physiol. 2010, 152, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Gururani, M.A.; Venkatesh, J.; Tran, L.S.P. Regulation of photosynthesis during abiotic stress-induced photoinhibition. Mol. Plant 2015, 8, 1304–1320. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H. Redox Homeostasis and Antioxidant Signaling: A Metabolic Interface between Stress Perception and Physiological Responses. Plant Cell 2005, 17, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. J. Bot. 2012, 2012, 1–26. [Google Scholar] [CrossRef]
- Rahman, M.; Ullah, I.; Ahsraf, M.; Stewart, J.M.; Zafar, Y. Genotypic variation for drought tolerance in cotton. Agron. Sustain. Dev. 2002, 28, 439–447. [Google Scholar] [CrossRef]
- Edelman, M.; Mattoo, A.K. D1-protein dynamics in photosystem II: The lingering enigma. Photosynth. Res. 2008, 98, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hoehenwarter, W. Changes in the Phosphoproteome and Metabolome link Early Signaling Events to Rearrangement of Photosynthesis and Central Metabolism in Salinity and Oxidative Stress Response in Arabidopsis. Plant Physiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Giardi, M.T.; Cona, A.; Geiken, B.; Kucera, T.; Masojidek, J.; Mattoo, A.K. Long-term drought stress induces structural and functional reorganization of photosystem II. Planta 1996, 199, 118–125. [Google Scholar] [CrossRef]
- Betterle, N.; Ballottari, M.; Baginsky, S.; Bassi, R. High Light-Dependent Phosphorylation of Photosystem II Inner Antenna CP29 in Monocots Is STN7 Independent and Enhances Nonphotochemical Quenching. Plant Physiol. 2015, 167, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Wilfinger, W.W.; Mackey, K.; Chomczynski, P. 260/280 and 260/230 Ratios NanoDrop ® ND-1000 and ND-8000 8-Sample Spectrophotometers. BioTechniques 1997, 22, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Invitrogen QubitTM dsDNA HS Assay Kits. Thermo Fish. 2015. Available online: https://www.thermofisher.com/order/catalog/product/Q32851 (accessed on 5 March 2018).
- Bartram, A.K.; Lynch, M.D.J.; Stearns, J.C.; Moreno-Hagelsieb, G.; Neufeld, J.D. Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl. Environ. Microbiol. 2011, 77, 3846–3852. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map (SAM) Format and SAMtools 1000 Genome Project Data Processing Subgroup. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Paten, B.; Novak, A.; Haussler, D. Mapping to a Reference Genome Structure. arXiv, 2014; arXiv:1404.5010. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Abreu, P.C.; Greenberg, D.A.; Hodge, S.E. Direct Power Comparisons between Simple LOD Scores and NPL Scores for Linkage Analysis in Complex Diseases. Am. J. Hum. Genet. 1999, 65, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Van Ooijen, J.W.; Voorrips, R.E. JoinMap 3.0—A Software for the Calculation of Genetic Linkage Maps Completely; Plant Research International: Wageningen, The Netherlands, 2001; pp. 1–51. [Google Scholar]
- Kosambi, D.D. the Estimation of Map Distances From Recombination Values. Ann. Eugen. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- Donovan, L.; Linton, M.; Richards, J. Predawn plant water potential does not necessarily equilibrate with soil water potential under well-watered conditions. Oecologia 2001, 129, 328–335. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkages | Markers (P. map) | Markers (G. map) | G. map Size (cM) | Largest Gap (cM) | Smallest Gap (cM) | <1 cM | 1–5 cM | 5–10 cM | 10–20 cM | >20 cM | Ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LG1_chrA01 | 2306 | 772 | 185.4601 | 19.828 | 0.1706 | 767 | 2 | 0 | 1 | 0 | 0.99 |
| LG2_chrA02 | 2368 | 707 | 133.6326 | 2.1472 | 0.1527 | 705 | 1 | 0 | 0 | 0 | 1 |
| LG3_chrA03 | 221 | 70 | 183.6242 | 60.9522 | 0.7464 | 44 | 19 | 4 | 0 | 2 | 0.63 |
| LG4_chrA04 | 2057 | 803 | 184.7308 | 1.3336 | 0.1704 | 800 | 2 | 0 | 0 | 0 | 1 |
| LG5_chrA05 | 778 | 285 | 180.1844 | 10.5478 | 0.3584 | 247 | 35 | 0 | 2 | 0 | 0.87 |
| LG6_chrA06 | 193 | 88 | 135.7902 | 15.3278 | 0.4373 | 48 | 36 | 2 | 1 | 0 | 0.55 |
| LG7_chrA07 | 1572 | 474 | 146.5589 | 4.5868 | 0.2307 | 467 | 6 | 0 | 0 | 0 | 0.99 |
| LG8_chrA08 | 1664 | 542 | 164.4535 | 13.5821 | 0.1979 | 536 | 4 | 0 | 1 | 0 | 0.99 |
| LG9_chrA09 | 1054 | 230 | 176.1339 | 3.8988 | 0.6234 | 190 | 39 | 0 | 0 | 0 | 0.83 |
| LG10_chrA10 | 1139 | 380 | 184.0143 | 2.227 | 0.3827 | 361 | 18 | 0 | 0 | 0 | 0.95 |
| LG11_chrA11 | 1650 | 480 | 182.8387 | 20.4651 | 0.2612 | 474 | 4 | 0 | 0 | 1 | 0.99 |
| LG12_chrA12 | 353 | 113 | 131.5913 | 8.1154 | 0.6518 | 71 | 40 | 1 | 0 | 0 | 0.63 |
| LG13_chrA13 | 1285 | 354 | 159.996 | 9.8116 | 0.3271 | 350 | 1 | 2 | 0 | 0 | 0.99 |
| At_sub Total | 16,640 | 6318 | 2149.009 | 60.9522 | 0.1527 | 5060 | 207 | 9 | 5 | 3 | 0.8 |
| LG14_chrD01 | 198 | 45 | 151.7771 | 145.8727 | 0.104742 | 43 | 0 | 0 | 0 | 1 | 0.96 |
| LG15_chrD02 | 237 | 85 | 129.8661 | 32.0047 | 0.75454 | 60 | 21 | 2 | 0 | 1 | 0.71 |
| LG16_chrD03 | 161 | 79 | 131.4133 | 17.51484 | 0.861499 | 44 | 31 | 2 | 1 | 0 | 0.56 |
| LG17_chrD04 | 365 | 129 | 151.0376 | 8.6138 | 0.841359 | 98 | 27 | 3 | 0 | 0 | 0.76 |
| LG18_chrD05 | 109 | 70 | 173.9945 | 78.08868 | 0.570595 | 33 | 34 | 0 | 1 | 0 | 0.47 |
| LG19_chrD06 | 2419 | 947 | 158.7206 | 0.81108 | 0.12249 | 499 | 0 | 0 | 0 | 0 | 0.53 |
| LG20_chrD07 | 393 | 164 | 159.8483 | 5.9477 | 0.617165 | 96 | 66 | 1 | 0 | 0 | 0.59 |
| LG21_chrD08 | 1918 | 824 | 178.4512 | 0.85601 | 0.164057 | 823 | 0 | 0 | 0 | 1 | 1 |
| LG22_chrD09 | 852 | 293 | 149.2598 | 101.1146 | 0.131356 | 291 | 0 | 0 | 0 | 1 | 0.99 |
| LG23_chrD10 | 1854 | 705 | 137.6682 | 2.3434 | 0.136199 | 700 | 4 | 0 | 0 | 0 | 0.99 |
| LG24_chrD11 | 691 | 218 | 186.8999 | 2.0093 | 0.714943 | 173 | 44 | 0 | 0 | 0 | 0.79 |
| LG25_chrD12 | 1593 | 558 | 133.5851 | 1.5444 | 0.178899 | 555 | 2 | 0 | 0 | 0 | 0.99 |
| LG26_chrD13 | 1230 | 455 | 199.7287 | 17.6884 | 0.300543 | 446 | 7 | 0 | 1 | 0 | 0.98 |
| Dt_sub total | 12,020 | 4572 | 2042.25 | 145.8727 | 0.104742 | 3861 | 236 | 8 | 3 | 4 | 0.84 |
| At/Dt Totals | 28,660 | 10,888 | 4191.259 | 145.8727 | 0.104742 | 8921 | 443 | 17 | 8 | 7 | 0.82 |
| Chr. | Number of Markers | Cover Length (Mb) | Chr. Length (Mb) | Coverage (%) | Density (marker/Mb) | Genes in Reference Genome/chr | Mined Genes/Chro. | % of Mined Genes/Chro. | Number of Domains |
|---|---|---|---|---|---|---|---|---|---|
| LG1_chrA01 | 2306 | 99.85026 | 99.8847 | 100 | 23.1 | 2162 | 1946 | 90 | 800 |
| LG2_chrA02 | 2368 | 83.27669 | 83.447906 | 99.8 | 28.4 | 1824 | 1692 | 92.8 | 673 |
| LG3_chrA03 | 221 | 100.2122 | 100.26305 | 99.9 | 2.21 | 2187 | 1860 | 85 | 771 |
| LG4_chrA04 | 2057 | 62.76193 | 62.913772 | 99.8 | 32.8 | 1491 | 1210 | 81.2 | 102 |
| LG5_chrA05 | 778 | 91.9822 | 92.047023 | 99.9 | 8.46 | 4026 | 2900 | 72 | 997 |
| LG6_chrA06 | 193 | 102.9532 | 103.17044 | 99.8 | 1.87 | 2119 | 1788 | 84.4 | 757 |
| LG7_chrA07 | 1572 | 78.15538 | 78.251018 | 99.9 | 20.1 | 2369 | 2078 | 87.7 | 831 |
| LG8_chrA08 | 1664 | 103.6089 | 103.62634 | 100 | 16.1 | 2571 | 2233 | 86.9 | 869 |
| LG9_chrA09 | 1054 | 74.86152 | 74.999931 | 99.8 | 14.1 | 2532 | 2168 | 85.6 | 873 |
| LG10_chrA10 | 1139 | 100.6926 | 100.8666 | 99.8 | 11.3 | 2357 | 2176 | 92.3 | 897 |
| LG11_chrA11 | 1650 | 93.30573 | 93.316192 | 100 | 17.7 | 3305 | 2947 | 89.2 | 993 |
| LG12_chrA12 | 353 | 87.39659 | 87.484866 | 99.9 | 4.04 | 2733 | 2498 | 91.4 | 938 |
| LG13_chrA13 | 1285 | 78.02031 | 79.961121 | 97.6 | 16.5 | 2356 | 1829 | 77.6 | 777 |
| At-sub Total | 16,640 | 1157.077 | 1160.23 | 99.7 | 14.4 | 32,032 | 17,106 | 85.9 | 3007 |
| LG14_chrD01 | 198 | 60.91516 | 61.456009 | 99.1 | 3.25 | 2383 | 2063 | 86.6 | 800 |
| LG15_chrD02 | 237 | 67.22226 | 67.284553 | 99.9 | 3.53 | 2448 | 2356 | 96.2 | 845 |
| LG16_chrD03 | 161 | 46.6713 | 46.690656 | 100 | 3.45 | 1860 | 1632 | 87.7 | 681 |
| LG17_chrD04 | 365 | 51.27027 | 51.45413 | 99.6 | 7.12 | 2040 | 1765 | 86.5 | 121 |
| LG18_chrD05 | 109 | 60.2436 | 61.933047 | 97.3 | 1.81 | 3942 | 3610 | 91.6 | 1128 |
| LG19_chrD06 | 2419 | 64.09113 | 64.294643 | 99.7 | 37.7 | 2394 | 2231 | 93.2 | 844 |
| LG20_chrD07 | 393 | 55.29315 | 55.312611 | 100 | 7.11 | 2503 | 2191 | 87.5 | 854 |
| LG21_chrD08 | 1918 | 65.83381 | 65.894135 | 99.9 | 29.1 | 2765 | 2621 | 94.8 | 951 |
| LG22_chrD09 | 852 | 50.90761 | 50.995436 | 99.8 | 16.7 | 2493 | 2401 | 96.3 | 921 |
| LG23_chrD10 | 1854 | 62.78548 | 63.374666 | 99.1 | 29.5 | 2646 | 2378 | 89.9 | 888 |
| LG24_chrD11 | 691 | 65.54361 | 66.087774 | 99.2 | 10.5 | 3539 | 3250 | 91.8 | 1038 |
| LG25_chrD12 | 1593 | 58.94278 | 59.109837 | 99.7 | 27 | 2838 | 2581 | 90.9 | 931 |
| LG26_chrD13 | 1230 | 59.8794 | 60.534298 | 98.9 | 20.5 | 2551 | 2367 | 92.8 | 922 |
| Dt-sub total | 12,020 | 769.6 | 774.4 | 99.4 | 15.6 | 34,402 | 15,786 | 91.2 | 3134 |
| At/Dt Totals | 28,660 | 1926.7 | 1934.7 | 99.6 | 14.9 | 66,434 | 32,892 | 88.5 | 6141 |
| Factor or Site Name | Signal Sequence | Function | Number of Genes |
|---|---|---|---|
| MYCATERD1 | CATGTG | water-stress responsiveness | 123 |
| AGCBOXNPGLB | AGCCGCC | stress signal-response factors | 32 |
| ASF1MOTIFCAMV | TGACG | Response to abiotic and biotic stress | 123 |
| AUXREPSIAA4 | KGTCCCAT | Response to abiotic and biotic stress | 16 |
| ATHB1ATCONSENSUS | CAATWATTG | Response to abiotic and biotic stress | 10 |
| ATHB5ATCORE | CAATNATTG | Response to abiotic and biotic stress | 10 |
| AUXRETGA1GMGH3 | TGACGTAA | Response to abiotic and biotic stress | 3 |
| ACGTATERD1 | ACGT | Early responsive to dehydration | 214 |
| CCAATBOX1 | CCAAT | Promoter of heat shock protein | 251 |
| MYBST1 | GGATA | Plant MYB binding site | 233 |
| MYBPZM | CCWACC | Plant MYB binding site | 176 |
| MYBPLANT | MACCWAMC | Plant MYB binding site | 79 |
| GCCCORE | GCCGCC | pathogen-responsive genes | 65 |
| CPBCSPOR | TATTAG | NADPH-protochlorophyllide reductase | 127 |
| CCA1ATLHCB1 | AAMAATCT | myb-related transcription factor | 24 |
| MYB2CONSENSUSAT | YAACKG | MYB recognition site/dehydration-responsive | 236 |
| MYBCOREATCYCB1 | AACGG | Myb core/M-phase-specific expression | 207 |
| MYBGAHV | TAACAAA | Myb binding site | 79 |
| LTRECOREATCOR15 | CCGAC | Low-temperature-responsive element | 171 |
| LTRE1HVBLT49 | CCGAAA | Low-temperature-responsive element | 159 |
| LTREATLTI78 | ACCGACA | Low-temperature-responsive element | 30 |
| INRNTPSADB | YTCANTYY | Light-responsive transcription | 229 |
| EBOXBNNAPA | CANNTG | Light-responsive and tissue-specific activation | 260 |
| GT1CORE | GGTTAA | Light-dependent transcriptional activation | 91 |
| IBOXCORE | GATAA | Light regulation | 248 |
| IBOX | GATAAG | Light regulation | 158 |
| IBOXCORENT | GATAAGR | Light regulation | 135 |
| GLMHVCHORD | RTGASTCAT | Involved in the nitrogen response | 10 |
| GMHDLGMVSPB | CATTAATTAG | Involved in the nitrogen response | 2 |
| TAAAGSTKST1 | TAAAG | Guard cell-specific gene expression | 254 |
| GT1CONSENSUS | GRWAAW | GT-1 binding site in many light-regulated genes | 260 |
| AGATCONSENSUS | TTWCCWWWWNNGGWW | Function in flower development | 3 |
| BOXIIPCCHS | ACGTGGC | Essential for light regulation | 9 |
| CRTDREHVCBF2 | GTCGAC | Regulation of low-temperature responsive genes | 33 |
| CTRMCAMV35S | TCTCTCTCT | Regulation of low-temperature responsive genes | 13 |
| ABRELATERD1 | ACGTG | Early responsive to dehydration | 157 |
| ABREOSRAB21 | ACGTSSSC | Early responsive to dehydration | 3 |
| DRECRTCOREAT | RCCGAC | Drought/high-light/cold responsive | 109 |
| DRE1COREZMRAB17 | ACCGAGA | Drought response | 102 |
| BIHD1OS | TGTCA | Disease resistance responses | 242 |
| MYB1AT | WAACCA | Dehydration-responsive gene | 236 |
| MYB2AT | TAACTG | Dehydration-responsive gene | 120 |
| MYB1LEPR | GTTAGTT | Dehydration-responsive gene | 29 |
| MYB26PS | GTTAGGTT | Dehydration-responsive gene | 10 |
| CBFHV | RYCGAC | Dehydration-responsive element (DRE) | 179 |
| MYCCONSENSUSAT | CANNTG | Dehydration-responsive | 260 |
| MYBCORE | CNGTTR | Dehydration/Water stress | 258 |
| MYCATRD22 | CACATG | Dehydration/Water stress | 123 |
| MYBATRD22 | CTAACCA | Dehydration/Water stress | 19 |
| AGMOTIFNTMYB2 | AGATCCAA | Defense-related gene | 49 |
| CARGATCONSENSUS | CCWWWWWWGG | Component of the vernalization (low-temperature) | 32 |
| TBOXATGAPB | ACTTTG | Chloroplast glyceraldehyde-3-phosphate dehydrogenase(GADPH) | 187 |
| GATABOX | GATA | Chlorophyll a/b binding protein/light regulations | 260 |
| CMSRE1IBSPOA | TGGACGG | Carbohydrate Metabolite Signal Responsive Element 1 | 18 |
| TCA1MOTIF | TCATCTTCTT | Salicylic acid/stress induced | 26 |
| ACGTABREMOTIFA2OSEM | ACGTGKC | ABA-responsive expression | 27 |
| ABREATCONSENSUS | YACGTGGC | ABA-responsive elements (ABREs) | 5 |
| ABRECE1HVA22 | TGCCACCGG | ABA-responsive elements (ABREs) | 3 |
| ABREATRD22 | RYACGTGGYR | ABA-responsive elements (ABREs) | 2 |
| ABREBZMRAB28 | TCCACGTCTC | ABA-responsive elements (ABREs) | 2 |
| ABREDISTBBNNAPA | GCCACTTGTC | ABA-responsive elements (ABREs) | 1 |
| WRKY71OS | TGAC | A transcriptional repressor of the gibberellin signaling pathway | 260 |
| E2F1OSPCNA | GCGGGAAA | Involved in transcriptional activation in actively dividing cells and tissue | 5 |
| DPBFCOREDCDC3 | ACACNNG | Abscisic acid response gene | 202 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magwanga, R.O.; Lu, P.; Kirungu, J.N.; Diouf, L.; Dong, Q.; Hu, Y.; Cai, X.; Xu, Y.; Hou, Y.; Zhou, Z.; et al. GBS Mapping and Analysis of Genes Conserved between Gossypium tomentosum and Gossypium hirsutum Cotton Cultivars that Respond to Drought Stress at the Seedling Stage of the BC2F2 Generation. Int. J. Mol. Sci. 2018, 19, 1614. https://doi.org/10.3390/ijms19061614
Magwanga RO, Lu P, Kirungu JN, Diouf L, Dong Q, Hu Y, Cai X, Xu Y, Hou Y, Zhou Z, et al. GBS Mapping and Analysis of Genes Conserved between Gossypium tomentosum and Gossypium hirsutum Cotton Cultivars that Respond to Drought Stress at the Seedling Stage of the BC2F2 Generation. International Journal of Molecular Sciences. 2018; 19(6):1614. https://doi.org/10.3390/ijms19061614
Chicago/Turabian StyleMagwanga, Richard Odongo, Pu Lu, Joy Nyangasi Kirungu, Latyr Diouf, Qi Dong, Yangguang Hu, Xiaoyan Cai, Yanchao Xu, Yuqing Hou, Zhongli Zhou, and et al. 2018. "GBS Mapping and Analysis of Genes Conserved between Gossypium tomentosum and Gossypium hirsutum Cotton Cultivars that Respond to Drought Stress at the Seedling Stage of the BC2F2 Generation" International Journal of Molecular Sciences 19, no. 6: 1614. https://doi.org/10.3390/ijms19061614
APA StyleMagwanga, R. O., Lu, P., Kirungu, J. N., Diouf, L., Dong, Q., Hu, Y., Cai, X., Xu, Y., Hou, Y., Zhou, Z., Wang, X., Wang, K., & Liu, F. (2018). GBS Mapping and Analysis of Genes Conserved between Gossypium tomentosum and Gossypium hirsutum Cotton Cultivars that Respond to Drought Stress at the Seedling Stage of the BC2F2 Generation. International Journal of Molecular Sciences, 19(6), 1614. https://doi.org/10.3390/ijms19061614