Hyaluronic Acid Influence on Normal and Osteoarthritic Tissue-Engineered Cartilage

Abstract
:1. Introduction
2. Results
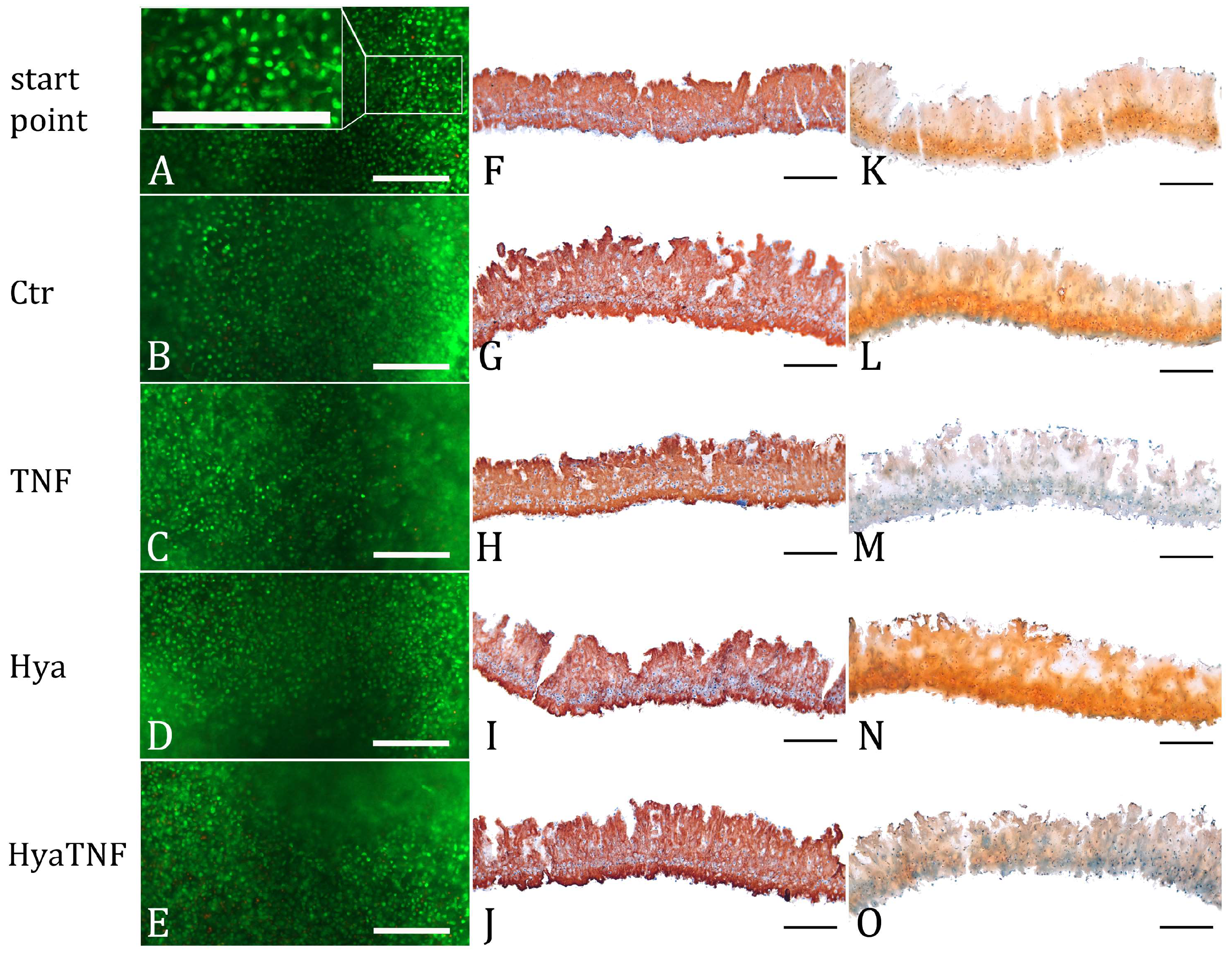
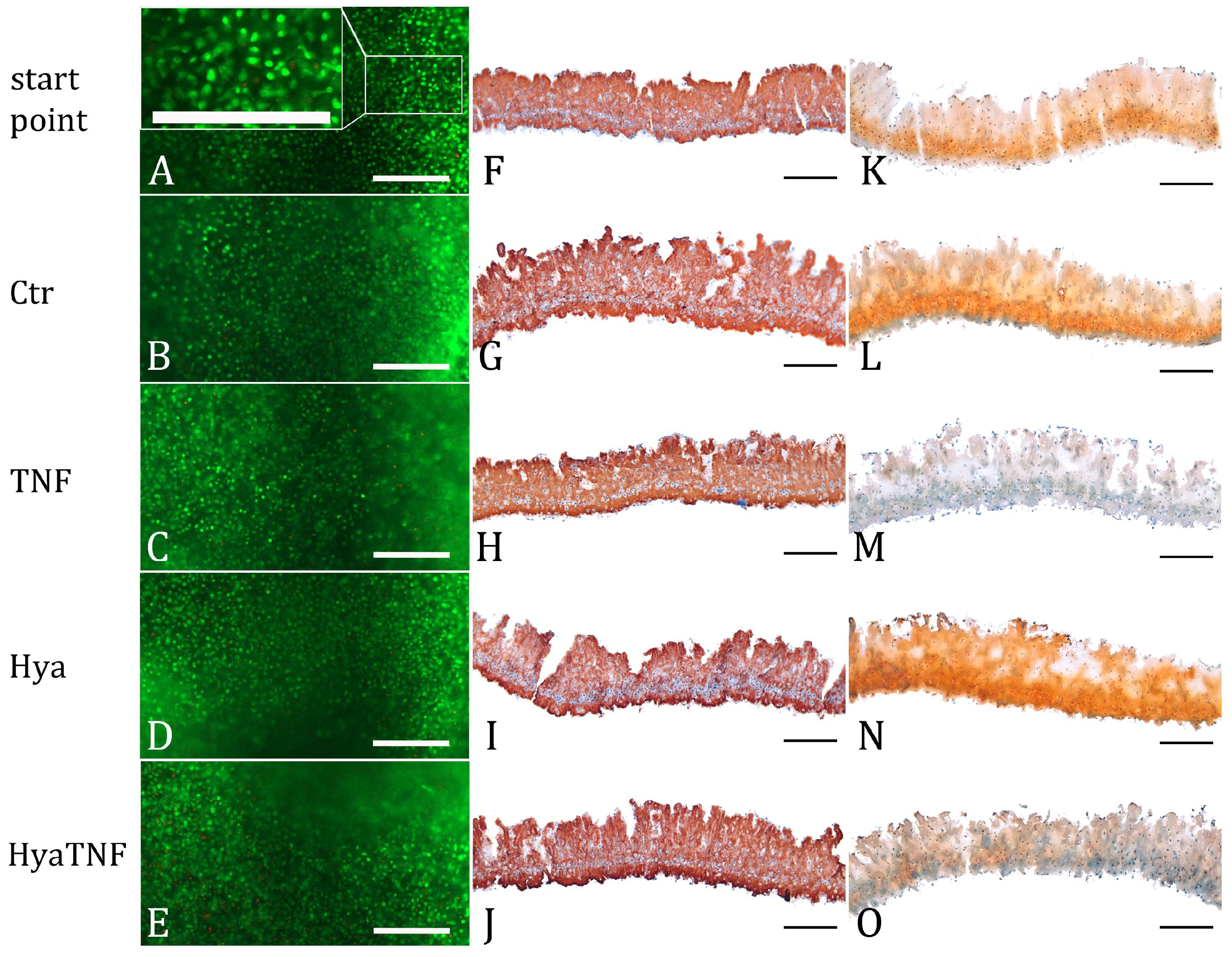
2.1. Effect of Hyaluronic Acid on Chondrocyte Viability and Extracellular Matrix Formation
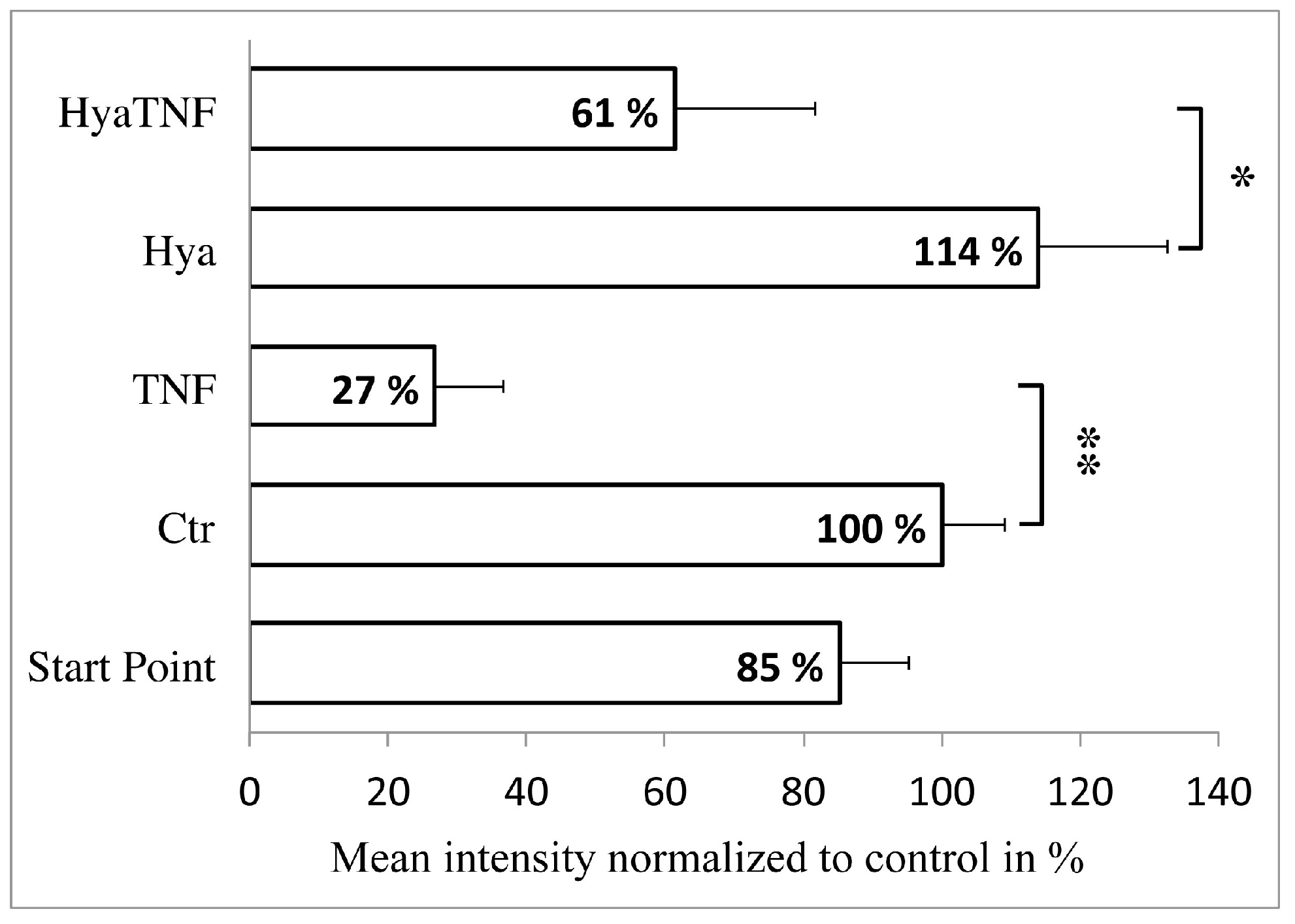
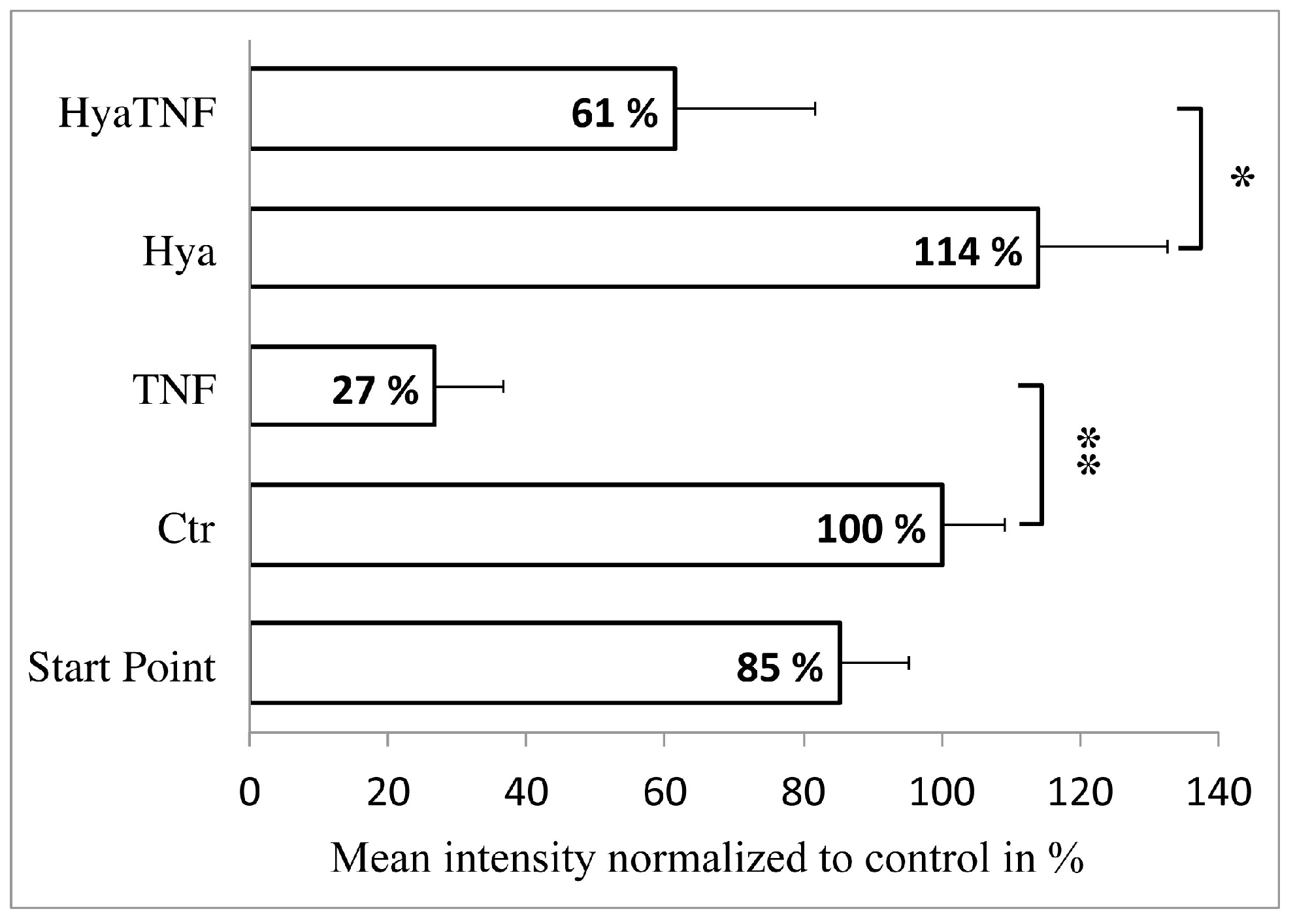
2.2. In Vitro Model Verification by Cartilage-Related Markers
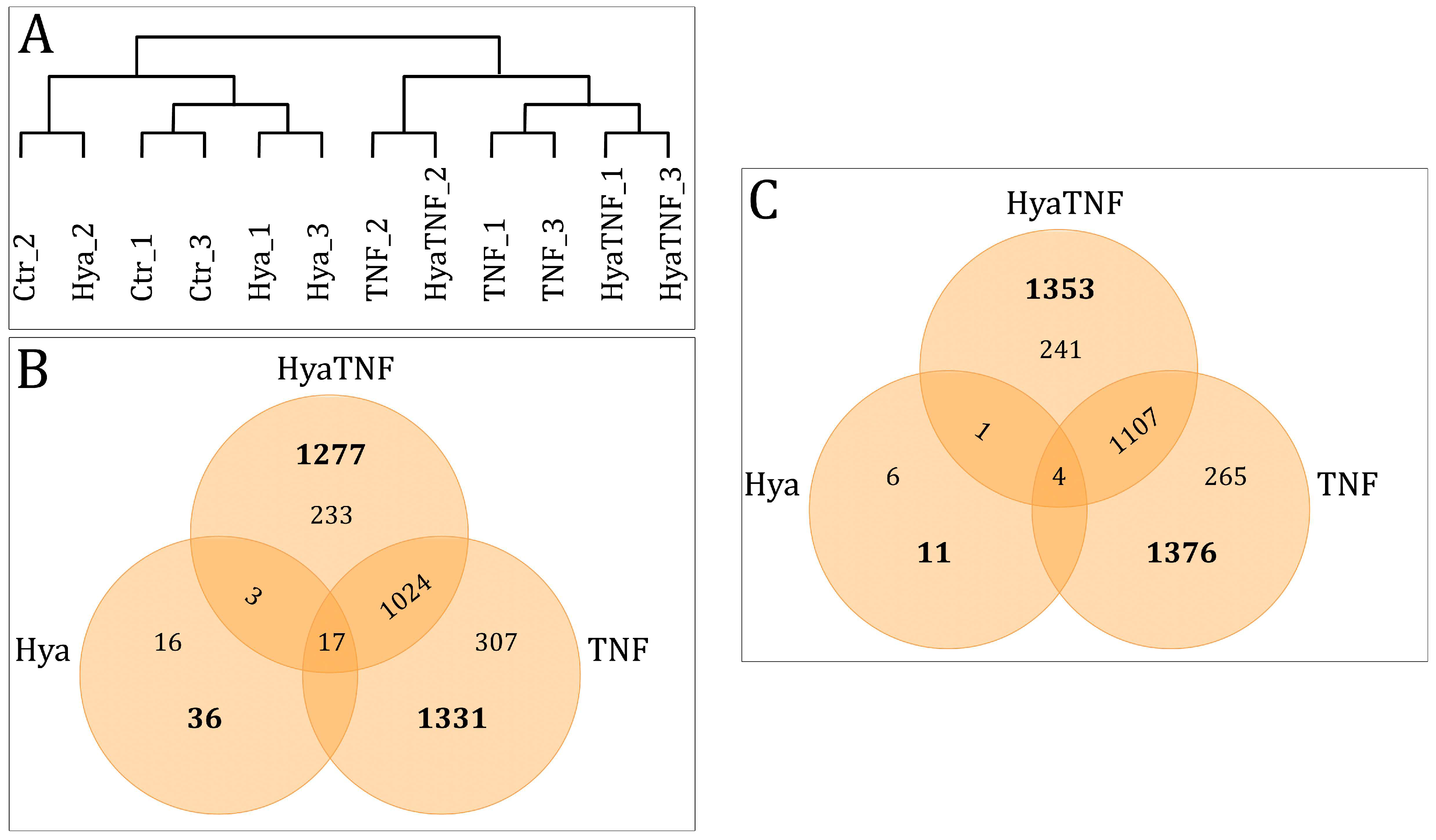
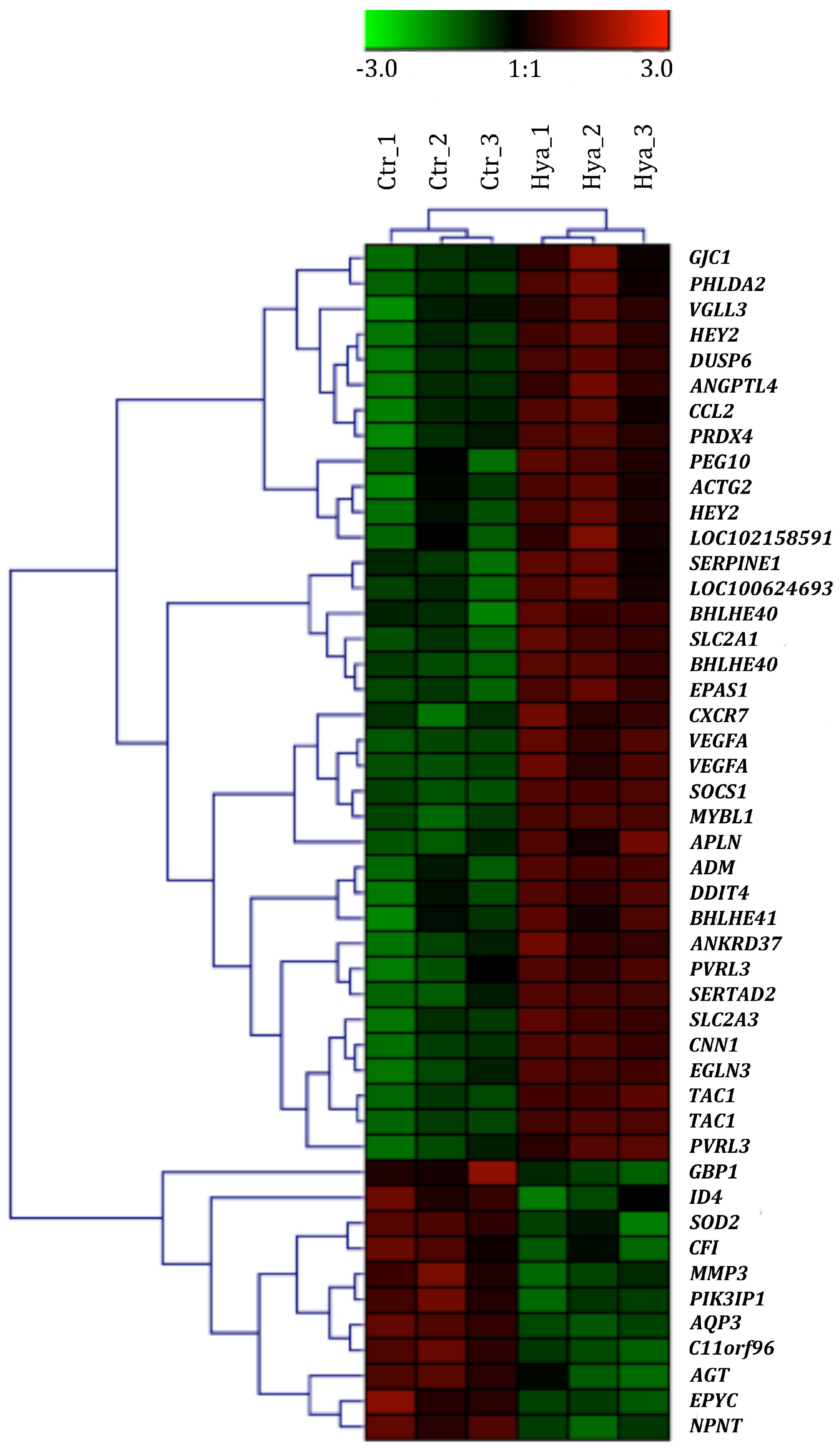
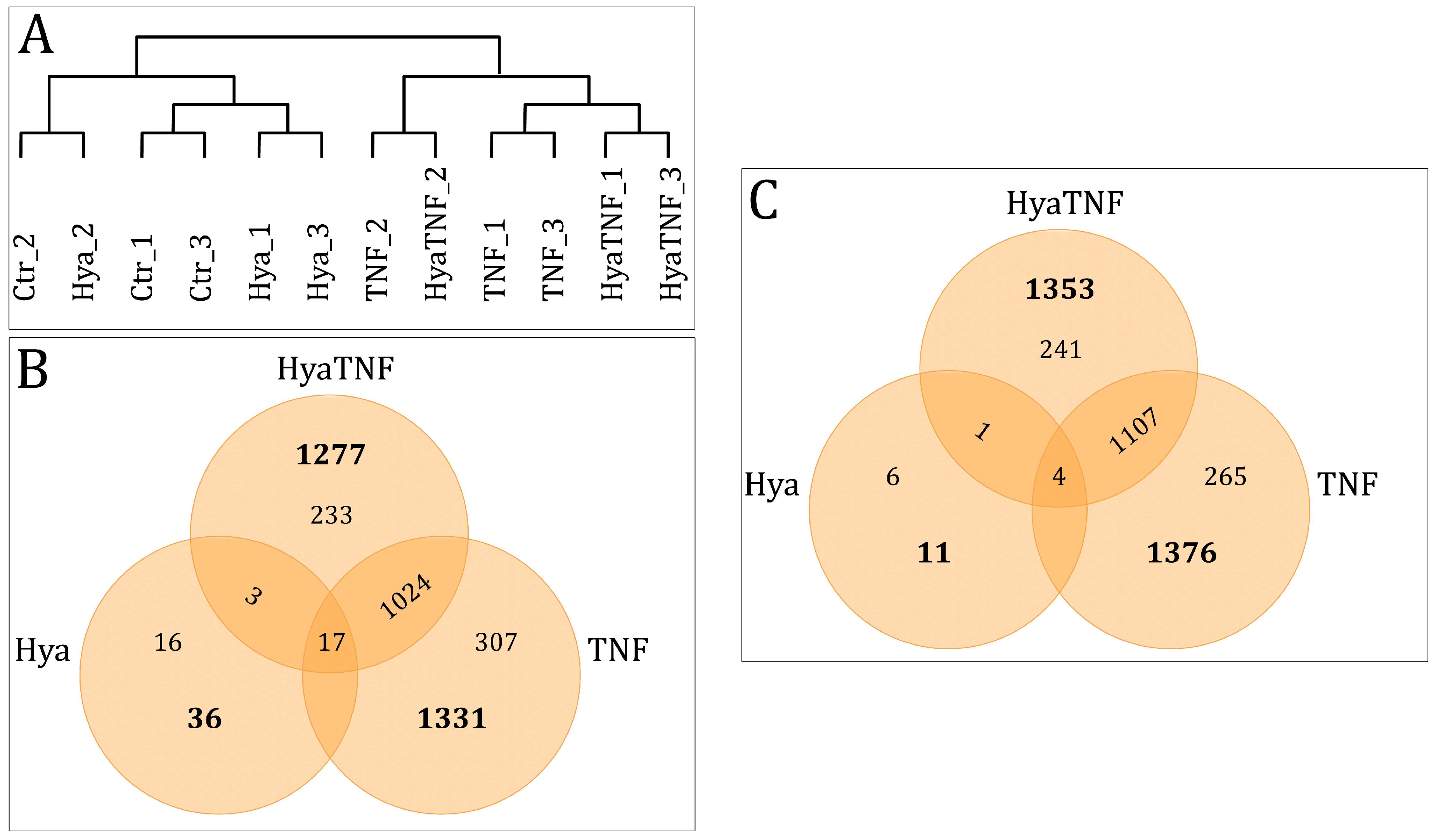
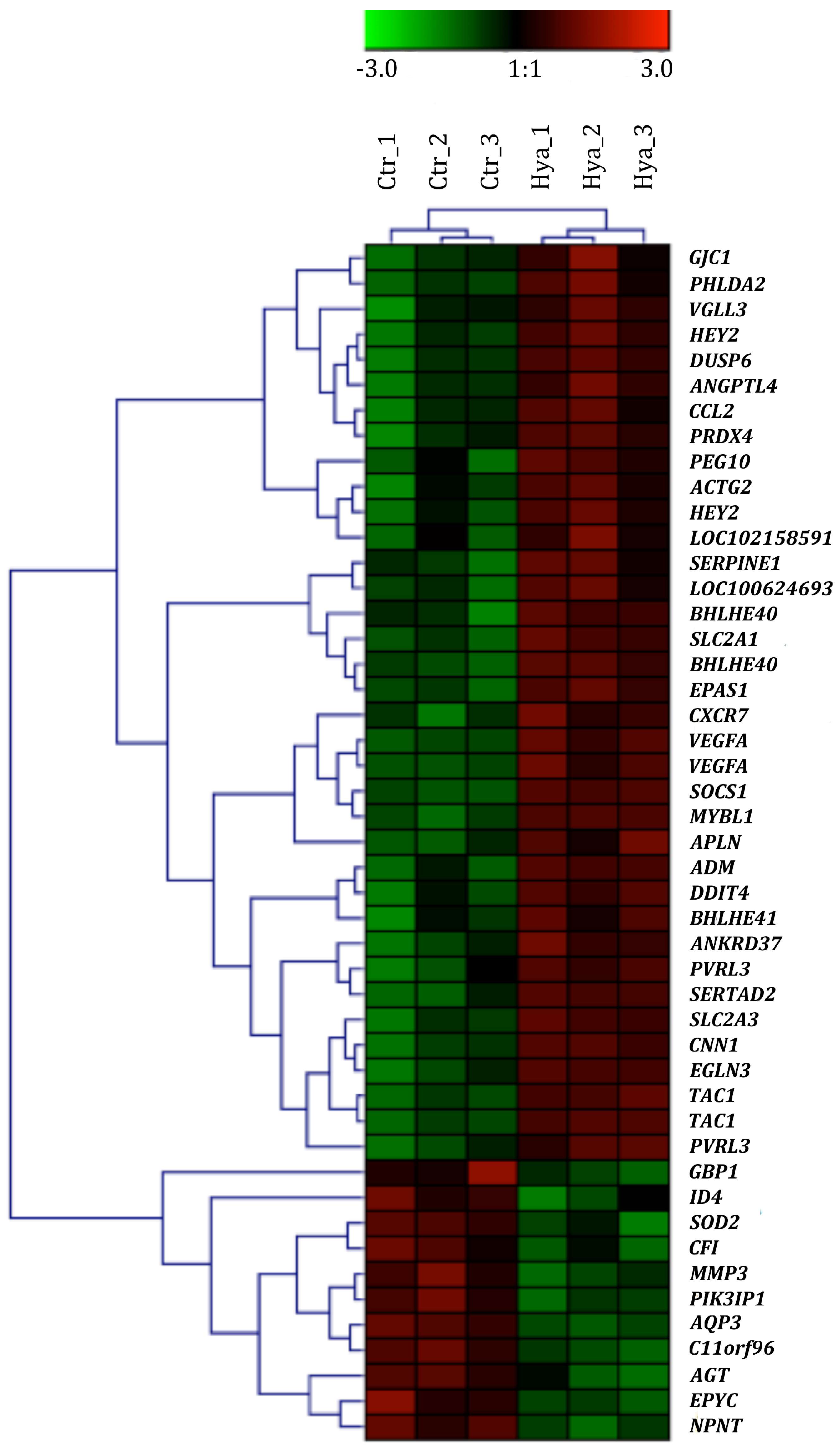
2.3. Gene Expression Profiling as Response to HA Treatment
2.4. Overview of Differentially Expressed Genes between Controls and Treated Groups
3. Discussion
4. Materials and Methods
4.1. Chondrocyte Isolation
4.2. Preparation of 3D-Chondrocyte Micromass Cultures
4.3. Live/Dead Assay
4.4. Histological and Immunohistochemical Staining
4.5. RNA Isolation
4.6. Microarray Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ACAN | aggrecan |
| AGT | angiotensinogen |
| ANGPTL4 | included angiopoietin-like 4 |
| ANKRD37 | ankyrin repeat domain 37 |
| APLN | apelin |
| BP | biological process |
| CC | cellular component |
| CCL2 | C-C motif chemokine 2 |
| CD44 | cluster of differentiation 44 |
| CILP | cartilage intermediate layer protein |
| COL2A1 | collagen type 2 alpha 1 chain |
| COL9A1 | collagen type IX alpha 1 chain |
| COMP | cartilage oligomeric compound |
| CSPG4 | chondroitin sulphate proteoglycan 4 |
| CXCL3 | C-X-C motif chemokine ligand 3 |
| DAVID | database for annotation, visualization and integrated discovery |
| ECM | extra cellular matrix |
| EPYC | epiphycan |
| FBS | foetal bovine serum |
| FC | fold change |
| FGF | fibroblast growth factor |
| FGFR | fibroblast growth factor receptor |
| GAG | glycosaminoglycan |
| GBP1 | guanylate binding protein 1 |
| GCOS | GeneChip operating software |
| GO | gene ontology |
| HA | hyaluronic acid |
| HAPLN1 | hyaluronan and proteoglycan link protein 1 |
| HCL | hierarchical clustering |
| IGFBP | insulin-like growth factor binding protein |
| IL | interleukin |
| KEGG | Kyoto encyclopaedia of genes and genomes |
| MF | molecular function |
| MMP | matrix metalloproteinase |
| OA | osteoarthritis |
| Pen | penicillin |
| PI/FDA | propidium iodide/fluorescein diacetate |
| PRELP | proline and arginine-rich end leucine-rich repeat protein |
| PTGDS | prostaglandin D2 synthase |
| SERPINA | serpin family A member |
| SFRP1 | secreted frizzled related protein 1 |
| SLC2A3 | solute carrier family 2 member 3 |
| SOX | SRY-Box |
| Strep | streptomycin |
| TIMP3 | TIMP metallopeptidase inhibitor 3 |
| TNF-α | tumour-necrosis factor-alpha |
| VEGFA | vascular endothelial growth factor A |
References
- Karna, E.; Miltyk, W.; Surażyński, A.; Pałka, J.A. Protective effect of hyaluronic acid on interleukin-1-induced deregulation of β1-integrin and insulin-like growth factor-I receptor signalling and collagen biosynthesis in cultured human chondrocytes. Mol. Cell. Biochem. 2008, 308, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Swann, D.; Radin, E.; Nazimiec, M.; Weisser, P.; Curran, N.; Lewinnek, G. Role of hyaluronic acid in joint lubrication. Ann. Rheum. Dis. 1974, 33, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Kosinska, M.K.; Ludwig, T.E.; Liebisch, G.; Zhang, R.; Siebert, H.C.; Wilhelm, J.; Kaesser, U.; Dettmeyer, R.B.; Klein, H.; Ishaque, B.; et al. Articular Joint Lubricants during Osteoarthritis and Rheumatoid Arthritis Display Altered Levels and Molecular Species. PLoS ONE 2015, 10, e0125192. [Google Scholar] [CrossRef] [PubMed]
- Elmorsy, S.; Funakoshi, T.; Sasazawa, F.; Todoh, M.; Tadano, S.; Iwasaki, N. Chondroprotective effects of high-molecular-weight cross-linked hyaluronic acid in a rabbit knee osteoarthritis model. Osteoarthr. Cartil. 2014, 22, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Moreland, L.W. Intra-articular hyaluronan (hyaluronic acid) and hylans for the treatment of osteoarthritis: Mechanisms of action. Arthritis Res. Ther. 2003, 5, 1–14. [Google Scholar] [CrossRef]
- Hunter, C.J.; Levenston, M.E. Maturation and integration of tissue-engineered cartilages within an in vitro defect repair model. Tissue Eng. 2004, 10, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, N.; Dehne, T.; Mans, K.; Endres, M.; Stuhlmüller, B.; Sittinger, M.; Kaps, C.; Ringe, J. Suitability of porcine chondrocyte micromass culture to model osteoarthritis in vitro. Mol. Pharm. 2014, 11, 2092–2105. [Google Scholar] [CrossRef] [PubMed]
- Haward, S.J.; Jaishankar, A.; Oliveira, M.; Alves, M.; McKinley, G. Extensional flow of hyaluronic acid solutions in an optimized microfluidic cross-slot device. Biomicrofluidics 2013, 7, 044108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.-F.; Chou, Y.-J.; Hsu, C.-W.; Hwang, C.-W.; Hsu, P.-T.; Wang, J.-L.; Hsu, Y.-W.; Chou, M.-C. Efficacy of intra-articular hyaluronic acid in patients with osteoarthritis of the ankle: A prospective study. Osteoarthr. Cartil. 2006, 14, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Pohlig, F.; Guell, F.; Lenze, U.; Lenze, F.W.; Muhlhofer, H.M.; Schauwecker, J.; Toepfer, A.; Mayer-Kuckuk, P.; von Eisenhart-Rothe, R.; Burgkart, R.; et al. Hyaluronic Acid Suppresses the Expression of Metalloproteinases in Osteoarthritic Cartilage Stimulated Simultaneously by Interleukin 1β and Mechanical Load. PLoS ONE 2016, 11, e0150020. [Google Scholar] [CrossRef] [PubMed]
- Responte, D.J.; Natoli, R.M.; Athanasiou, K.A. Identification of potential biophysical and molecular signalling mechanisms underlying hyaluronic acid enhancement of cartilage formation. J. R. Soc. Interface 2012, 9, 3564–3573. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Triantafillou, S.; Parker, A.; Youssef, P.; Coleman, M. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J. Rheumatol. 1997, 24, 365–371. [Google Scholar] [PubMed]
- Ozler, K.; Aktas, E.; Atay, C.; Yilmaz, B.; Arikan, M.; Gungor, S. Serum and knee synovial fluid matrix metalloproteinase-13 and tumour necrosis factor-alpha levels in patients with late-stage osteoarthritis. Acta Orthop. Traumatol. Turc. 2016, 50, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Balazs, E.A.; Watson, D.; Duff, I.F.; Roseman, S. Hyaluronic acid in synovial fluid. I. Molecular parameters of hyaluronic acid in normal and arthritic human fluids. Arthritis Rheumatol. 1967, 10, 357–376. [Google Scholar] [CrossRef]
- Smyth, N.A.; Ross, K.A.; Haleem, A.M.; Hannon, C.P.; Murawski, C.D.; Do, H.T.; Kennedy, J.G. Platelet-Rich Plasma and Hyaluronic Acid Are Not Synergistic When Used as Biological Adjuncts with Autologous Osteochondral Transplantation. Cartilage 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Pritzker, K.; Gay, S.; Jimenez, S.; Ostergaard, K.; Pelletier, J.-P.; Revell, P.; Salter, D.; van den Berg, W. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 2006, 14, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, D.; Stoker, A.; Kane, S.; Cockrell, M.; Cook, J. Biochemical effects of two different hyaluronic acid products in a co-culture model of osteoarthritis. Osteoarthr. Cartil. 2006, 14, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Foldager, C.B.; Nielsen, A.B.; Munir, S.; Ulrich-Vinther, M.; Søballe, K.; Bünger, C.; Lind, M. Combined 3D and hypoxic culture improves cartilage-specific gene expression in human chondrocytes. Acta Orthop. 2011, 82, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Zelzer, E.; Mamluk, R.; Ferrara, N.; Johnson, R.S.; Schipani, E.; Olsen, B.R. VEGFA is necessary for chondrocyte survival during bone development. Development 2004, 131, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.; Manjoo, A.; Fierlinger, A.; Niazi, F.; Nicholls, M. The mechanism of action for hyaluronic acid treatment in the osteoarthritic knee: A systematic review. BMC Musculoskelet. Disord. 2015, 16, 321. [Google Scholar] [CrossRef] [PubMed]
- Ramos, Y.F.; den Hollander, W.; Bovee, J.V.; Bomer, N.; van der Breggen, R.; Lakenberg, N.; Keurentjes, J.C.; Goeman, J.J.; Slagboom, P.E.; Nelissen, R.G.; et al. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; the RAAK study. PLoS ONE 2014, 9, e103056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julovi, S.M.; Ito, H.; Nishitani, K.; Jackson, C.J.; Nakamura, T. Hyaluronan inhibits matrix metalloproteinase-13 in human arthritic chondrocytes via CD44 and P38. J. Orthop. Res. 2011, 29, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Sahebjam, S.; Khokha, R.; Mort, J.S. Increased collagen and aggrecan degradation with age in the joints of TIMP3−/− mice. Arthritis Rheumatol. 2007, 56, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Black, R.; Castner, B.; Slack, J.; Tocker, J.; Eisenman, J.; Jacobson, E.; Delaney, J.; Winters, D.; Hecht, R.; Bendele, A. A14 Injected TIMP-3 Protects Cartilage in A Rat Meniscal Tear Model. Osteoarthr. Cartil. 2006, 14, S23–S24. [Google Scholar] [CrossRef]
- Andreas, K.; Lübke, C.; Häupl, T.; Dehne, T.; Morawietz, L.; Ringe, J.; Kaps, C.; Sittinger, M. Key regulatory molecules of cartilage destruction in rheumatoid arthritis: An in vitro study. Arthritis Res. Ther. 2008, 10, R9. [Google Scholar] [CrossRef] [PubMed]
- Leijten, J.C.; Emons, J.; Sticht, C.; van Gool, S.; Decker, E.; Uitterlinden, A.; Rappold, G.; Hofman, A.; Rivadeneira, F.; Scherjon, S.; et al. Gremlin 1, frizzled-related protein and Dkk-1 are key regulators of human articular cartilage homeostasis. Arthritis Rheumatol. 2012, 64, 3302–3312. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; di Battista, J.; Lajeunesse, D.; Pelletier, J.-P. IGF/IGFBP axis in cartilage and bone in osteoarthritis pathogenesis. Inflamm. Res. 1998, 47, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.; Dehne, T.; Lindahl, A.; Brittberg, M.; Pruss, A.; Sittinger, M.; Ringe, J. Genome-wide expression profiling reveals new candidate genes associated with osteoarthritis. Osteoarthr. Cartil. 2010, 18, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Fundel, K.; Saas, J.; Gebhard, P.M.; Haag, J.; Weiss, T.; Zien, A.; Obermayr, F.; Zimmer, R.; Bartnik, E. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheumatol. 2006, 54, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Von Lospichl, B.; Hemmati-Sadeghi, S.; Dey, P.; Dehne, T.; Haag, R.; Sittinger, M.; Ringe, J.; Gradzielski, M. Injectable hydrogels for treatment of osteoarthritis–A rheological study. Colloids Surf. B 2017, 159, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Lübke, C.; Ringe, J.; Krenn, V.; Fernahl, G.; Pelz, S.; Kreusch-Brinker, R.; Sittinger, M.; Paulitschke, M. Growth characterization of neo porcine cartilage pellets and their use in an interactive culture model. Osteoarthr. Cartil. 2005, 13, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L. Chemical basis for the histological use of safranin O in the study of articular cartilage. J. Bone Jt. Surg. 1971, 53, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Hemmati-Sadeghi, S.; Dey, P.; Ringe, J.; Haag, R.; Sittinger, M.; Dehne, T. Biomimetic sulphated polyethylene glycol hydrogel inhibits proteoglycan loss and tumour necrosis factor-α-induced expression pattern in an osteoarthritis in vitro model. J. Biomed. Mater. Res. Part B 2018. [Google Scholar] [CrossRef] [PubMed]
- Sturn, A.; Quackenbush, J.; Trajanoski, Z. Genesis: Cluster analysis of microarray data. Bioinformatics 2002, 18, 207–208. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization and integrated discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Anderson, T.W.; Darling, D.A. Asymptotic theory of certain “goodness of fit” criteria based on stochastic processes. Ann. Math. Stat. 1952, 23, 193–212. [Google Scholar] [CrossRef]
- Box, G.E. Non-normality and tests on variances. Biometrika 1953, 40, 318–335. [Google Scholar] [CrossRef]
- Ahmed, E.M. Hydrogel: Preparation, characterization and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Name | d14_Ref * | (d14) Start Point | (d21) Ctr | FC Trend | |
|---|---|---|---|---|---|---|
| Collagens | COL1A2 | collagen type I α2 | + | + | + | −2.7 |
| COL2A1 | collagen type II α1 | + | + | + | −25.8 | |
| COL9A2 | collagen type IX α1 | + | + | + | −11.0 | |
| ECM connectors | FMOD | fibromodulin | + | + | + | −1.3 |
| HAPLN1 | hyaluronan and proteoglycan link protein 1 | + | + | + | −9.5 | |
| LGALS3 | carbohydrate-binding protein 35 | + | + | + | 1.2 | |
| PCOLCE2 | C-endopeptidase enhancer 2 | + | + | + | / | |
| PRELP | prolargin | + | + | + | −1.9 | |
| Enzymes | MMP3 | matrix metallopeptidase 3 | + | + | + | 19.9 |
| SERPINA1 | serpin peptidase inhibitor clade A member 1 | + | + | + | −31.3 | |
| SERPINA3 | serpin peptidase inhibitor clade A member 3 | - | + | + | / | |
| Growth factors | BMP2 | bone morphogenetic protein 2 | + | + | + | / |
| BMP4 | bone morphogenetic protein 4 | + | + | + | −1.8 | |
| BMP7 | bone morphogenetic protein 7 | - | - | - | 3.2 | |
| FGF18 | fibroblast growth factors 18 | - | - | - | −2.6 | |
| FGF2 | fibroblast growth factors 2 | + | + | + | 4.6 | |
| FGF9 | fibroblast growth factors 9 | - | - | - | −3.6 | |
| IGF1 | insulin-like growth factor 1 | + | + | + | −2.8 | |
| IGFBP3 | insulin-like growth factor binding protein 3 | + | + | + | 4.2 | |
| TGFB1 | transforming growth factor beta 1 | + | + | + | −1.6 | |
| TGFB2 | transforming growth factor beta 2 | + | + | + | 1.6 | |
| TGFB3 | transforming growth factor beta 3 | + | + | + | −2.6 | |
| THRA | thyroid hormone receptor α | + | + | + | −1.7 | |
| Proteoglycan | ACAN | aggrecan | + | + | + | −13.1 |
| BGN | biglycan | + | + | + | −2.6 | |
| CSPG4 | chondroitin sulphate proteoglycan 4 | + | + | + | −3.8 | |
| DCN | decorin | + | + | + | −2.4 | |
| HSPG2 | heparan sulphate proteoglycan 2 | + | + | + | / | |
| VCAN | versican | + | + | + | / | |
| Receptors | FGFR1 | fibroblast growth factor receptor 1 | + | + | + | −1.2 |
| FGFR2 | fibroblast growth factor receptor 2 | + | + | + | −1.7 | |
| FGFR3 | fibroblast growth factor receptor 3 | + | + | + | −2.5 | |
| Structural integrity of cartilage | CHI3L1 | cartilage intermediate layer protein | + | + | + | 1.8 |
| CILP | cartilage oligomeric protein | + | + | + | 7.9 | |
| COMP | extracellular matrix protein 1 | + | + | + | −8.6 | |
| ECM1 | fibrillin 1 | + | + | + | 4.0 | |
| FBN1 | fibronectin 1 | + | + | + | 2.4 | |
| FN1 | matrix gla protein | + | + | + | / | |
| MGP | cartilage intermediate layer protein | + | + | + | −2.1 | |
| Transcription factors | SOX6 | SRY (Sex Determining Region Y)-Box 6 | + | + | + | −3.3 |
| SOX9 | SRY (Sex Determining Region Y)-Box 9 | + | + | + | / |
| Category | GO ID | GO Name | Gene Nr. | p-Value | Genes |
|---|---|---|---|---|---|
| CC | GO:0005615 | extracellular space | 16 | 3.0 × 10−4 | CXCL3, IGFBP6, LMCD1, PLBD1, ECM1, TIMP3, COL14A1, PTGDS, SFRP1, HIST2H2BE, GPX3, VEGFA, IGFBP3, APLN, ANGPTL4, IGFBP5 |
| MF | GO:0001968 | fibronectin binding | 3 | 2.3 × 10−4 | VEGFA, IGFBP3, IGFBP5 |
| MF | GO:0031994 | insulin-like growth factor I binding | 3 | 3.4 × 10−4 | IGFBP6, IGFBP3, IGFBP5 |
| MF | GO:0031995 | insulin-like growth factor II binding | 3 | 3.4 × 10−4 | IGFBP6, IGFBP3, IGFBP5 |
| BP | GO:0043567 | regulation of insulin-like growth factor receptor signalling pathway | 3 | 6.9 × 10−4 | IGFBP6, IGFBP3, IGFBP5 |
| CC | GO:0005604 | basement membrane | 4 | 1.8 × 10−3 | P3H2, COL4A1, ITGA6, TIMP3 |
| BP | GO:0071456 | cellular response to hypoxia | 4 | 2.1 × 10−3 | PTGS2, SFRP1, VEGFA, ANGPTL4 |
| BP | GO:0045663 | positive regulation of myoblast differentiation | 3 | 3.7 × 10−3 | CDON, BOC, IGFBP3 |
| BP | GO:0045892 | negative regulation of transcription, DNA-templated | 6 | 9.5 × 10−3 | CRY2, SFRP1, BEND5, CCDC85B, BASP1, HMGA1 |
| CC | GO:0005576 | extracellular region | 8 | 9.7 × 10−3 | FGF7, PTGDS, PAPPA, AGT, NMB, FGF13, CFD, GHR |
| BP | GO:0001558 | regulation of cell growth | 3 | 1.4 × 10−2 | IGFBP6, IGFBP3, IGFBP5 |
| BP | GO:0017148 | negative regulation of translation | 3 | 1.7 × 10−2 | BTG2, ENC1, IGFBP5 |
| CC | GO:0042567 | insulin-like growth factor ternary complex | 2 | 1.9 × 10−2 | IGFBP3, IGFBP5 |
| CC | GO:0070062 | extracellular exosome | 18 | 2.2 × 10−2 | SCPEP1, IGFBP6, NPR3, ECM1, TIMP3, ARG1, COL14A1, BTG2, SFRP1, PTGDS, RAB19, HIST2H2BE, AGT, PCBP2, BLVRB, GPX3, IGFBP3, MEST |
| BP | GO:0044342 | type B pancreatic cell proliferation | 2 | 3.5 × 10-2 | IGFBP3, IGFBP5 |
| BP | GO:0014912 | negative regulation of smooth muscle cell migration | 2 | 3.5 × 10−2 | IGFBP3, IGFBP5 |
| BP | GO:0006979 | response to oxidative stress | 3 | 3.8 × 10−2 | PTGS2, GPX3, SRXN1 |
| BP | GO:0043568 | positive regulation of insulin-like growth factor receptor signalling pathway | 2 | 4.0 × 10−2 | IGFBP3, IGFBP5 |
| BP | GO:0045893 | positive regulation of transcription, DNA-templated | 5 | 4.0 × 10−2 | FGF7, SFRP1, AGT, SERTAD3, HMGA1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hemmati-Sadeghi, S.; Ringe, J.; Dehne, T.; Haag, R.; Sittinger, M. Hyaluronic Acid Influence on Normal and Osteoarthritic Tissue-Engineered Cartilage. Int. J. Mol. Sci. 2018, 19, 1519. https://doi.org/10.3390/ijms19051519
Hemmati-Sadeghi S, Ringe J, Dehne T, Haag R, Sittinger M. Hyaluronic Acid Influence on Normal and Osteoarthritic Tissue-Engineered Cartilage. International Journal of Molecular Sciences. 2018; 19(5):1519. https://doi.org/10.3390/ijms19051519
Chicago/Turabian StyleHemmati-Sadeghi, Shabnam, Jochen Ringe, Tilo Dehne, Rainer Haag, and Michael Sittinger. 2018. "Hyaluronic Acid Influence on Normal and Osteoarthritic Tissue-Engineered Cartilage" International Journal of Molecular Sciences 19, no. 5: 1519. https://doi.org/10.3390/ijms19051519
APA StyleHemmati-Sadeghi, S., Ringe, J., Dehne, T., Haag, R., & Sittinger, M. (2018). Hyaluronic Acid Influence on Normal and Osteoarthritic Tissue-Engineered Cartilage. International Journal of Molecular Sciences, 19(5), 1519. https://doi.org/10.3390/ijms19051519